

Abstract
In this paper we report a new method to prepare and characterize a contrast agent based on a fourth-generation (G4) polyamidoamine (PAMAM) dendrimer conjugated to the gadolinium complex of the bifunctional diethylenetriamine pentaacetic acid derivative (1B4M-DTPA). The method involves pre-forming the metal-ligand chelate in alcohol prior to conjugation to the dendrimer. The dendrimer-based agent was purified by a Sephadex® G-25 column and characterized by elemental analysis. The analysis and SEHPLC data gave a chelate to dendrimer ratio of 30:1 suggesting conjugation at approximately every other amine terminal on the dendrimer. Molar relaxivity of the agent measured at pH 7.4 displayed a higher value than that of the analogous G4 dendrimer based agent prepared by the post-metal incorporation method (r1 = 26.9 vs. 13.9 mM-1s-1 at 3T and 22°C). This is hypothesized to be due to the higher hydrophobicity of this conjugate, and the lack of available charged carboxylate groups from non-complexed free ligands that might coordinate to the metal and thus also reduce water exchange sites. Additionally, the distribution populations of compounds that result from the post-metal incorporation route are eliminated from the current product simplifying characterization as quality control issues pertaining to the production of such agents for clinical use as MR contrast agents. In vivo imaging in mice showed a reasonably fast clearance (t1/2 = 24 min) suggesting a viable agent for use in clinical application.
Introduction
The quest for the development of a more efficient contrast agent for magnetic resonance imaging (MRI) is an ongoing process. Although clinically used low molecular weight paramagnetic chelates, such as Gd(III)-diethylenediaminepentaacetic acid (GdDTPA) (Magnevist) and Gd(III)-N,N',N",N"'-tetracarboxymethyl-1,4,7,10-tetraazacyclododecane (Gd-DOTA),(1, 2) are thermodynamically stable within the context of their usage, they suffer from rapid clearance from vascular circulation and renal excretion.
Dendrimer-based macromolecular MR contrast agents in which numerous chelated gadolinium ions (Gd(III)) are covalently attached to a multivalent dendritic architecture are a promising class of diagnostic agents for medical imaging applications.(3-6) There are also growing applications of dendrimers in preparation of macromolecules for radioimmunotherapy.(7-9) Due to their macromolecular size and weight, dendrimer-based MR agents feature higher molar relaxivities along with longer retention and circulation times as compared to the clinically approved Magnevist® (Gd(III)-DTPA), thereby resulting in greatly enhanced sensitivity and operational window.(10) Extensive efforts have been expanded on the preparation and evaluation of these agents in our laboratory along with several other research groups worldwide to study their in vivo properties and to eventually reach the goal of producing viable imaging agents that might be translated into clinical use.(4, 11-14) Pre-clinical results have been promising in animal studies and by virtue of choosing generation size, core elements, and other exterior modifications dendrimer-based MR agents have been successfully employed for the imaging blood pool,(14) specific organs such as kidney(4, 5) and liver, lymphatics,(6) and tumor vasculature(12, 13) in mice with acceptable clearance rates.
The typical procedure for preparation of these agents usually involves 3 key steps. The first step is conjugation of an isothiocyanate or N-hydroxysuccinimidyl active ester form of either 2-(4-isothiocyanatobenzyl)-6-methyl-diethylenetriaminepentaacetic acid (1B4M-DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) derivatives to the multiple periphery functional groups (e.g. -NH2) of various types of polyamino terminated dendrimers in either aqueous or organic solution. The second step has been complexation of paramagnetic metal Gd(III) ions with the conjugated DTPA or DOTA chelates attached on the dendrimers. The final and highly important step has been the purification of the product to completely remove “free” Gd(III) ions usually by an extensive dialysis technique using appropriate filtration membranes.(3, 5, 6, 10, 15)
Unlike small molecules, macromolecular agents are usually difficult to define with accurate and precise chemical structures and to characterize their purities by traditional means. This limitation generates real challenges to establishing quality control for reproducible preparation of agents. For instance, even when the same protocol is followed for preparation of these macromolecular dendrimer-based agents, it is exceedingly difficult to insure that both the same number of conjugated chelates and the same number of Gd(III) ions for each batch will be achieved. This is particularly so when considering that there are potentially hundreds of reactive sites in higher generation dendrimers, e.g., a generation 8 PAMAM dendrimer potentially has 1024 terminal primary amines.(16, 17) One has to be aware that these products are also effectively a distribution of chelate conjugate products that has been carried forward into another distribution of Gd(III) complexes and that while characterization reports numbers of chelates and Gd(III) that those numbers represent average numbers. For clinical use, however, these agents need to have a narrow molecular weight range and the same chemical composition to insure reproducible expected in vivo behavior. The Gd(III) metal content in these dendrimer-based MR contrast agents is a critical factor not only to the level of contrast efficacy, but also the biological properties (e.g., PD/PK). In our prior synthetic approaches, and to the best of our knowledge of all all others, the Gd(III) metal incorporation has always been performed as the last step, following conjugation of bifunctional chelating agents to the dendrimer. In general, by combustion analysis data, this sequence generally results in ~ 80% of the bifunctional chelating agents to be complexed with Gd(III) ions while the remaining ~ 20% of those chelates remain vacant of Gd(III).(18, 19)
These 20% “open” chelates possess a significant number of carboxyl groups, which may impact overall hydrophilicity and charge of the agents at the physiological conditions. Accurate determination of what role these vacant or “open” chelates play is an extremely difficult task to perform. Additionally, concerns pertaining to chelation of exogenous biologically relevant metal ions in vivo impacting toxicity may be valid when taking into account absolute molar injected amounts versus clearance rates. Lastly, as noted above, these products are actually complex mixtures of products and elimination of some aspect of that character should strengthen the potential of their clinical translation. With that impetus in hand, we felt it necessary to explore a means to simplify the process while arriving at a more defined product by limiting these variables as much as possible.
With this note, we report a refined synthetic procedure for the preparation of dendrimer-based MR contrast agents, wherein (1B4M-DTPA) is first used to sequester Gd(III) and thereafter the resulting metal complexes are covalently attached to the terminal -NH2 groups in a PAMAM G4 dendrimer. Such an approach allows creation of a simplified standard procedure making small molecule Gd(III) complexes thereby eliminating a manipulation step in preparation of the macromolecular dendrimer molecules while also insuring full saturation of all of the conjugated chelates with Gd(III) to simultaneously eliminate an entire array of product distribution mixture. The PAMAM generation 4 dendrimer (G4D) with amine surface groups (-NH2) is a convenient building block or core for the conjugation of metal chelates, and the G4D Gd(III) chelate conjugate is known to have moderate blood circulation time and relatively fast excretion via the kidney.(18)
In order to validate the agent, the molar relaxivity of the generation 4 1B4M-DTPA Gd(III) conjugate, (G4-(1B4M-Gd)30(CS)) was determined for comparison to the analogous generation 4 dendrimer-based agent, G4-(1B4M)60[Gd42](CS), prepared in our laboratory using the previously established published synthesis route.(18) Additionally, sets of mice (n = 2) were imaged with both G4-(1B4M-Gd)30(CS) or G4-(1B4M)60[Gd42](CS) to establish what impact, if any, elimination of the vacant chelators on the exterior of the dendrimer product has on the quality of MR imaging.
Experimental
Materials and Methods
PAMAM dendrimer generation-4 (G4 dendrimer) with an ethylenediamine core in MeOH (10% w/v) was obtained from Dendritech. Gadolinium nitrate pentahydrate (Gd(NO)3·5H2O) was purchased from Aldrich (St. Louis, MO). G4-(1B4M)60[Gd42](CS) prepared and purified as previously reported was employed in these studies.(18) Phosphate buffered saline (1X PBS) at pH 7.4 was obtained from Digene (Gaithersburg, MD). Size exclusion HPLC (SE-HPLC) was performed using a Beckman System Gold (Fullerton, CA) equipped with model 126 solvent delivery module and a model 168 UV detector (λ 254 and 280 nm) controlled by 32 Karat software. Size exclusion chromatography was performed on a TSK-gel G2000SW 6 μm, 7.8 mm × 300 mm (Tosoh Bioscience, Montgomeryville, PA), with a TSK-gel 10 μm guard column (Tosoh Bioscience, Montgomeryville, PA) using phosphate buffered saline (1X PBS) solution as the eluent at 0.5 mL/min and 1.0 mL/min, respectively. Reverse phase HPLC was performed by using a Beckman System Gold HPLC equipped with 165 UV-Vis detector with peak detection at 254 and 280 nm and a C18 Varian microsorb™-mv column (250 × 4.6 mm; 5 um). A gradient system composed of a pH 9.0 buffer (50 mM triethylamine/acetic acid) and methanol (25 min linear gradient from 0% to 100% methanol) with a flow rate of 1 mL/min was employed. All water used was purified using a Hydro Ultrapure Water Purification system (Rockville, MD). The Sephadex® G-25 resin was purchased from Pharmacia (Sweden). The resin was pre-treated with 1X PBS and loaded to a Pharmacia Biotech column 2.6 × 39.7 cm (Uppsala, Sweden). Elemental analyses were performed by Galbraith Laboratories, Inc. (Knoxville, TN) using combustion analysis method for C, H, N and inductively coupled plasma-atomic emission spectroscopy (ICP-OES) method for determining the S, Gd. The Bio-Rad's gel filtration standard used to compare the molecular weight of the dendrimer conjugate was purchased from Bio-Rad (Hercules, CA).
Syntheses
2-(4-nitrobenzyl)-6-methyl-diethylenetriaminepentaacetic acid · 5HCl
The HCl salt of the bifunctional ligand, 2-(4-nitrobenzyl)-6-methyldiethylenetriaminepentaacetic acid, 1B4M from this point on, was prepared as previously described with a brief modification.(20) Briefly, the pentaester form of the ligand (8.1g, 9.8 mmol) was treated with HCl (g) saturated 1,4-dioxane (600 mL) and the mixture was stirred for 18 h. The product was precipitated with ethyl ether (400 mL), filtered and dried to afford a whitish solid (4.8 g, 90%). (See the reference for NMR data). ESI m/e: 543 (M+1). Calcd for C22H30N4O12·5HCl: C, 36.44; H, 4.83; N, 7.72. Found: C, 36.57; H, 5.05; N, 7.89.
2-(4-nitrobenzyl)-6-methyl-diethylenetriaminepentaacetic acid gadolinium complex (p-NO2-1B4M-[Gd] (1)
1B4M·5HCl ligand (0.50 g, 0.69 mmol) and Gd(NO3)3.5H2O (0.31 g, 0.72 mmol) in 20 mL MeOH were refluxed for 6 h. The solvent was reduced to 1.5 mL and CH2Cl2 was added until a precipitate formed. The supernatant was decanted and the solid was dried. The precipitation process was repeated one more time to yield a pale yellow solid (0.65 g, 80 %). ESI m/e: 700 (M+1). Reverse phase HPLC tR = 13.77 min. Calcd for C22H30N4O12Gd·3NO3·5HCl·2CH3OH: C, 25.33; H, 3.78; N, 8.62. Found: C, 25.21; H, 3.67; N, 8.67.
2-(4-aminobenzyl)-6-methyl-diethylenetriaminepentaacetic acid gadolinium complex (p-NH2-1B4M-[Gd] (2)
A solution of compound 1 (0.50 g, 0.42 mmol) and 10% Pd/C (0.10 g) in H2O (10 mL) was placed in a Parr hydrogenator at 15 psi. The reaction was allowed to go until the consumption of H2 gas was completed (~3 h), which was confirmed by HPLC. The mixture was filtered on a glass frit through a pad of Celite (Celite® 535 Coarse; Fluka) and washed with H2O (20 mL). The solvent was evaporated under high vacuum to afford amine 2 (0.41 g, 87 %) as a yellowish solid. ESI m/e: 670 (M+1). Reverse phase HPLC tR = 11.32 min. Calcd for C22H32N4O10Gd·3NO3·5HCl: C, 25.44; H, 3.57; N, 9.44. Found: C, 25.32; H, 3.59; N, 9.57.
2-(4-isothiocyanatobenzyl)-6-methyl-diethylenetriaminepentaacetic acid gadolinium complex (SCN-1B4M-[Gd] (3))
A solution of 0.5 mL of thiophosgene in CH2Cl2 (3 mL) was added drop wise into a solution of 2 (1.0 g, 0.96 mmol) in H2O (5 mL). The mixture was continued to stir for 1 h. The aqueous was separated and washed with CH2Cl2 (5 mL) and dried to afford compound 3 as a yellow solid (0.81 g, 75 %). ESI m/e: 712 (M+1). Reverse phase HPLC tR = 19.17 min. Calcd for C23H30N4SO10Gd·3NO3·5HCl·2H2O: C, 24.69; H, 3.67; N, 8.77. Found: C, 24.61; H, 3.65; N, 8.78.
Conjugation of G4 with 3 - (G4-(1B4M-Gd)30(CS))
G4 PAMAM dendrimer (1.34g, 0.0145 mmol; 15.35 % w/w) in a 500 mL round bottom flask was dried under vacuum to remove MeOH. The flask was charged with 350 mL 1X conjugation buffer with EDTA (0.48M NaHCO3 + 0.02 M Na2CO3 + 1.5 M NaCl + 0.005 M EDTA) (pH ~ 8.5) (250 mL). Compound 3 (1.51 g, 1.85 mmol) was added in portions while the pH was adjusted to 8.5 with 1M NaOH. The mixture was stirred at room temperature for 5 d and 35 °C for 1 d. The solution was cooled to room temperature and filtered. The solvent was reduced under high vacuum to ~30 mL at room temperature. The dendrimer-chelate was purified by a Sephadex® G-25column eluted with water (pH 7.5; the pH was adjusted with NaOH). The first band was collected and lyophilized to yield the conjugate as a yellow spongy solid (65% based on dendrimer). SE-HPLC tR =14.0. Calcd for C622H1250N250O125·30(C23H30N4SO10Gd)·49Na·50H2O [G4-(1B4MGd)30(CS)Na49(H2O)50]: 41.01 (C), 5.87 (H), 13.51 (N), 2.50 (S), 12.29(Gd). Found: 40.77, 5.94, 13.79, 2.49, 12.03.
Molar Relaxivity Measurements
The molar relaxivity of G4-(1B4M)60[Gd42](CS) was measured as reported elsewhere.(18) Stock solutions of the G4 dendrimer agents, G4-(1B4M-Gd)30 (5.0 Mm in Gd), were diluted to concentrations of 0.1, 0.25, 0.50, 0.75 and 1.0 mM in 1X PBS (300 μL). Solutions of Gd(III)-DTPA (Magnevist) at 0.25, 0.50, 0.75, 1.0, 1.25, 1.5 and 2.0 mM were prepared in 1X PBS (300 μL) and used as a reference standard. Relaxivity measurements were obtained at ~22° C using a 3-Tesla clinical scanner (Signa Excite, GE Medical System, Waukesha, WI) equipped with a rectangular single loop receiver coil (84 × 126 × 6 mm). A series of single slice 2D inversion recovery (IR) fast spin echo images of all the solutions were acquired at the same time with a TE around 9ms and using different inversion recovery times (TI = 50, 100, 350, 750, 1250, 2500, and 5000 ms) followed by a single slice 8-echo SE image (TE = 9 ms). The R1 values for each dilution were determined by fitting ROI intensity values from variable IR images using Igor Pro (http://www.wavemetrics.com). R2 values were measured from ROI values from T2 maps, which were calculated from the multi-echo images in ImageJ (http://rsb.info.nih.gov/ij) using the MRI analysis plug-in (http://rsb.info.nih.gov/ij/plugins/mri-analysis.html). The molar relaxivities, r1 and r2, were obtained from the slope of 1/T1 or 1/T2 vs. [Gd(III)] plots determined from region of interest measurements.
In Vivo Magnetic Resonance Imaging (MRI)
Mice were imaged two at a time to increase throughput using a 3T clinical scanner (Achieva, Philips Healthcare, Best, Holland) equipped with a dual mouse coil comprised of two parallel receive-only coils (saddle-shaped Helmholtz coils, 38 mm in diameter × 70 mm in length) connected to independent receiver channels and spaced 43 mm apart. Each mouse was placed in a physical restraint while a catheter line (30 gauge needle on a 0.010” ID × 6” long Tygon tubing) filled with 1X PBS was inserted into the tail vein, anesthetized using gas mixtures of 3% isoflurane in O2, and then carefully placed in a mouse bed equipped with a nose cone, respiratory pad, and fiber optic temperature sensor for physiology monitoring. The mice were positioned in the dual mouse coil which was heated with air to maintain the mice at 34°C. The anesthesia gas was adjusted between 1.5-2.5% isoflurane to maintain a respiration rate of ~ 30 bpm during the acquisition of all images. After acquiring a tri-planar gradient echo survey, a coronal view T1-weighted 3D-fast spoiled gradient echo image with a low flip angle (repetition time of 14 ms, echo time of 2.4 ms, flip angle of 8°, field of view of 80 mm, matrix size of 512 × 512 pixels, 40 slices, slice thickness of 0.6 mm and 1 average; scan time of 2.3 min) was acquired followed by a dynamic series using a higher flip angle of 24° repeated every 2.5 min for one hour. The contrast agent was injected (50 uL of 12 mM Gd(III) in 1 X PBS pushed with 50 uL of 1 X PBS) after the first dynamic image at a rate of 150 ul/min using a syringe pump (BS-9000-8, Braintree Scientific, Braintree, MA). Blood clearance rates were determined from ROI intensity measurements of the jugular vein in the low flip angle image and high flip angle dynamic images using Image J. The intensity values during the dynamic scans were then converted to gadolinium concentration and the resulting [Gd] time curves were fitted to a single exponential function using an Igor Pro (Wavemetrics) macro.
Results and Discussion
The preparative scheme leading to the formation of the title conjugate G4-(1B4MGd)30(CS) is shown in Figure 1. The Gd(III) complex 1 was prepared by treatment of the ligand 1B4M with Gd(NO3)3·5H2O in methanol. Complex 1 could also be prepared by incubation of the ligand with Gd(NO3)3·5H2O in H2O with the pH maintained at 5.5, but preparation of the Gd(III) complex in methanol was chosen as a more convenient route, especially for ligands that have limited solubility in water. Previous studies showed that complexes formed in methanol were stable.(21, 22). ESI-MS spectrum of the predominant ion for the three complexes showed the presence of carboxylate arm protons. This is in line with the ESI-MS data reported for the commercially available products such as Gd(DTPA)-2 and Gd(DOTA)-1.(23) Other authors have also reported ESI-MS data of lanthanide complexes with numbers of carboxylate protons ranging from 1 to 3.(24-26) This observation might be due to the nature of the initial electrostatic interaction, instead of covalent bonding, between the cationic metal ion and the anionic carboxylate ion, however having protonated complexes is not unreasonable. Additionally, the complex was isolated as the HCl salt as the starting ligand was in theHCl form also with nitrate as the counter ion for the metal ion. Nitrate is known to be a better counter ion for lanthanide ions than chloride.(27-29) Lastly, for all the complexes the amount of the free metal ion determined by uv/vis spectroscopic method was 0.002% (0.2uM/10 mM) of the total.(30) Indeed, if one examines the synthesis of the DTPA[Gd(III)] complex directly from DTPA and Gd2O3, this route produces a diprotonated complex that requires the addition of two equivalents of base to make this complex physiologically acceptable.(31)
Figure 1.

Synthetic scheme of the G4-(1B4M-Gd)30(CS).
For the purpose of comparison, the Gd(III) complex was formed in methanol by refluxing the ligand and the metal salt in the presence of seven equivalents of a solid base (NaOH) for 18 h. The Gd(III) complex was also formed under aqueous condition as previously reported.(26) The ESI-MS data of both complexes indicated the presence of the complex with two less protons (697). However, these two latter complexes were also less stable than that formed in methanol alone. As soon as thiophosgene in CH2Cl2 was added to form complex 3 (for the third step) an insoluble precipitate formed when using the Gd(III) complexes formed by the latter means. This result was not observed for the complex formed in methanol without base. The reason for that behavior may reside with those carboxylic acid protons or that the complex is protonated. Once deprotonated by base, the complex becomes vulnerable to attack by acidic protons generated by the thiophosgene reaction that could perturb the M-O coordination and cause the metal to dissociate; a protonated complex eliminates this problem. It is also worth noting that the reaction with thiophosgene brought the pH of the solution down to ~2. For that very reason, we chose to form the complex in methanol without a base. A full and thorough comparative study of the fundamental coordination chemistry of these three complexes will be reported in due course.
The reduction of the nitro group to amine was done using a Parr hydrogenator at 15 psi within a short period of time (~3 h). The ESI-MS and combustion analysis data did not indicate any sign of the cleavage of the nitrobenzyl group (over-reduction product) or any dissociation of the metal ion from the ligand cavity. For preparation of complex 3, a mixture of thiophosgene in dichloromethane was added drop wise and the reaction was allowed to stir for 1 hour thereafter, which was sufficient to complete the reaction. This also limits the exposure of the metal complex to low pH (1-2) over an extended period of time, which might induce the dissociation of the Gd(III) from the complex.
The conjugation reaction was done in 1X conjugation buffer (see experimental) with EDTA at pH of 8.5. EDTA was used to sequester the free metal ion (Gd3+), in case it dissociates from the chelate, which is rarely the case under this experimental condition. The dendrimer-based conjugate was purified by a Sephadex® G-25column. This method proved to be more efficient as compared to the exhaustive and time consuming transverse-flow filtration (TFF) method.(18) No reasonable MALDI-TOF could be obtained due to extreme broadening of the peak. However, we utilized SE-HPLC to determine the approximate molecular weight of the dendrimer conjugate by comparing with the BIO-Rad's gel filtration standard combined with the elemental analysis data. Based on the analysis data and the SE-HPLC results the formula of [G4-(1B4MGd)30Na49(H2O)50] was obtained.
To the best of our knowledge, the number of chelates conjugated to dendrimer is sensitive to the method used to append them to the dendrimer. The method used in a previous report to prepare the G4-(1B4M-Gd)X(CS) conjugate, which is under aqueous condition, managed to occupy all of the available sites yielding G4-(1B4MGd)64(CS)).(32) However, a later report, where the conjugation reaction was also done in aqueous condition, but with different buffer, yielded a lower number of chelates (G4-(1B4M60-Gd42)(CS).(18) An even lower number conjugated was achieved when the conjugation reaction was done in methanol followed by the metal chelation under aqueous condition (G4-(1B4M57-Gd41)(CS)).(18) It is worth noting that all these preparations focused on inserting the metal ion (Gd3+) in the final step. In this report, we have inserted the metal ion early on to simplify the solution chemistry and the distribution of the species of the products.
We noticed in these studies that it was not possible to achieve the number of chelates higher than the reported value even in an extended period of time, or higher temperature (ca. 45°C) or even when using large excesses of the Gd(III) complex. Based on the number of chelates that we obtained, G4-(1B4M-Gd)30(CS)), we hypothesized that the chelate with the metal ion in it may be less soluble, and that the inter-metal or protonated complex interactions might limit the numbers of complexes adjacent to one another, or that an equilibrium reaction with the solvent water limits the actual concentration of reactive isothiocyanate species present. It is known that the isothiocyanide slowly reacts with water. Nevertheless, the relaxivity of this conjugate prepared by the reported means here is higher than any of the above mentioned, which is one of the most important factors to be a viable agent. This is clear evidence that higher number of chelates, greater numbers of Gd(III), and thus higher molecular weight does not always translate to higher relaxivity.
Figure 2 shows a plot of inverse longitudinal relaxation time (1/T1) versus gadolinium concentration for G4-(1B4M-Gd)30(CS) conjugate (0.1-1 mM) and magnevist (0.25-2mM). Although G4-(1B4M)60[Gd42](CS) has a higher dendrimer to metal chelate ratio (1:42), the molar relaxivity of the G4-(1B4M-Gd)30(CS) conjugate reported here is almost twice the value reported for the G4-(1B4M)60[Gd42](CS) (13.9 vs 26.9 mM-1s-1) (Table 1).(3,18) There are three factors that might have contributed to this higher value. First, the conjugate prepared here does not have any free uncomplexed chelate (chelate without Gd) population of dendrimer conjugation products unlike former method, G4-(1B4M)60[Gd42](CS) (Figure 3). This condition eliminates the possibility of a negatively charged carboxylate group from an unoccupied adjacent DTPA to coordinate to the metal ion and thus delete the availability of the water binding site. Reduced number of water exchangeable sites results in lower relaxivity. Reduced water exchange site through anion binding has been well documented with protein binding,(33) human serum albumin,(25) carbonate(34) and phosphate(35) anions. Secondly, the conjugate prepared here is more hydrophobic than the G4-(1B4M)60[Gd42](CS), attributable to the lack of charged noncomplexed chelates. Hydrophobic interaction slows down the tumbling time (τR) which results in increasing relaxivity. The rotational correlation time (τR) is one of the key parameters contributes to the efficacy of the agent. Such phenomenon has been reported by Wiener,(11) Kellar(36) and Caravan.(37) Thirdly, the impact of steric hindrance needs to be taken into consideration, which also contributes to the availability of second sphere water molecules. Because the agent here is not fully saturated, that is not all the available amino sites (64) are occupied, or lack free non-complex chelating agents that could either replace water binding site or hinder the path of exchanging water molecules, more second sphere water molecules may be readily available for exchange.(38) This also helps to explain why previously prepared dendrimer conjugates with high chelate numbers, which are also then more sterically hindered, also have low relaxivity. This is additional evidence that greater numbers of chelates and thus also higher molecular weight may not automatically translate to higher relaxivity.
Figure 2.
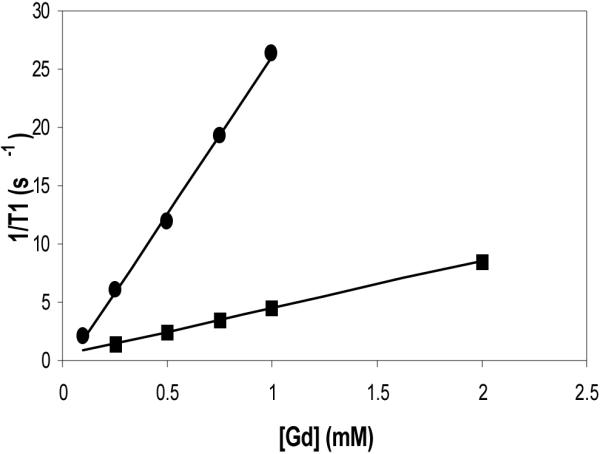
Molar relaxivity plots of G4-(1B4M-Gd)30(CS) (● 26.9 mM-1s-1) and magnevist (■; 4.2 mM-1s-1).
Table 1.
Comparison of results between G4-(1B4M-Gd)30(CS) (A) and G4-(1B4M)60[Gd42] (CS) (B)
| Agent | G4:chelate | r1 (mM-1s-1)a | r2 (mM-1s-1)a | Half life (min)b | Rate of clearance (min-1)c |
|---|---|---|---|---|---|
| A | 1:30 | 26.9 ± 1.2 | 56.1 ± 1.4 | 24.4 ± 4.74 | 0.029 ± 0.0047 |
| B | 1:41 | 13.9 ± 1.6 | 33.6 ± 3.1 | 72.7 ± 17.31 | 0.010 ± 0.0026 |
Molar relaxivity values obtained from phantom measurements.
Blood clearance half-life as measured from R1 map of the jugular vein in dynamic MR angiographic images.
Blood clearance rates. Reported numbers are the average values and errors are reported as standard deviations.
Figure 3.

A representation of dendrimers with (left) and without (right) free noncomplexed chelate.
Table 1 also compares the half-life and rates of clearance between G4(1B4MDTPA[Gd])30(CS) and G4-(1B4M)60[Gd42](CS). The clearance data were fit to a single exponential decay (Figure 4). The rate of clearance of G4-(1B4M)60[Gd42] (CS) is 2.9 times higher than G4-(1B4M-Gd)30(CS). We hypothesize that the agent was cleared mainly through kidneys. The data also indicate that there was a higher tissue uptake for G4-(1B4M-Gd)30(CS) that can be useful as a targeting agent. Overall G4-(1B4MGd)30(CS) has a faster rate of clearance and a shorter half-life, which is expected for a more positively charged hydrophobic agent. One might be tempted to think that G4-(1B4M-Gd)30(CS) is behaving like a small molecule based on the data presented, but the SE-HPLC data clearly indicates that the two agents have about the same size under the same conditions (tR = 13.2 vs 14.0). This indicates that the charge is the contributing factor in high relaxivity and fast clearance.
Figure 4.

Average blood clearance rates measured at the jugular vein of G4-(1B4MGd)30(CS) and G4-(1B4M)60[Gd42](CS).
Dynamic contrast-enhanced MR images shown in Figure 5 illustrate differences in circulation properties between G4-(1B4M-Gd)30(CS) and G4-(1B4M)60[Gd42](CS). We found that the signal intensity in the kidney increased gradually 10 min after injection due to the high blood flow through this area and fast clearance of the agent. It is also observed that after 40 min. post-injection images have no observable signal indicating the total clearance of the agent from the body. The signal intensity is low in kidneys for G4-(1B4M)60[Gd42](CS) when compared at the same acquisition time. This is expected due to the slow clearance (Table 1) and possible lower tissue uptake.
Figure 5.

Whole body 3D-micro-MR images of mice injected with 0.03 mmol/kg of G4-(1B4M-Gd)30(CS) (above) and G4-(1B4M)60[Gd42](CS) (below). Both images were acquired at 30 min post-injection.
In conclusion, we herein report an improved synthesis to prepare PAMAM G4 dendrimer-based MR contrast agents, which features full saturation of all of the conjugated chelates with Gd(III) by using pre-formed Gd(III) complexes as reagents. This better defined chemistry provides less complex products compared to our established published syntheses, which routinely had formed the chelating agent dendrimer conjugate first followed by addition of Gd(III). We believe that this new chemistry will strengthen the potential of translating these dendrimer-based MR agents into clinical trials by simplifying characterization as quality control issues pertaining to the production of such agents for clinical use as MR contrast agents.
Acknowledgement
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- (1).Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- (2).Lauffer RB. Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: theory and design. Chem. Rev. 1987;87:901–927. [Google Scholar]
- (3).Xu H, Regino CAS, Koyama Y, Hama Y, Gunn AJ, Bernardo M, Kobayashi H, Choyke PL, Brechbiel MW. Preparation and Preliminary Evaluation of a Biotin-Targeted, Lectin-Targeted Dendrimer-Based Probe for Dual-Modality Magnetic Resonance and Fluorescence Imaging. Bioconjugate Chem. 2007;18:1474–1482. doi: 10.1021/bc0701085. [DOI] [PubMed] [Google Scholar]
- (4).Dear JW, Kobayashi H, Jo S-K, Holly MK, Xuzhen H, Yuen PST, Brechbiel MW, Star RA. Dendrimer-enhanced MRI as a diagnostic and prognostic biomarker of sepsis-induced acute renal failure in aged mice. Kidney Int. 2005;67:2159–2167. doi: 10.1111/j.1523-1755.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- (5).Kobayashi H, Jo S-K, Kawamoto S, Yasuda H, Hu X, Knopp MV, Brechbiel MW, Choyke PL, Star RA. Polyamine dendrimerbased MRI contrast agents for functional kidney imaging to diagnose acute renal failure. J. Magn. Reson. Imaging. 2004;20:512–518. doi: 10.1002/jmri.20147. [DOI] [PubMed] [Google Scholar]
- (6).Kobayashi H, Kawamoto S, Choyke PL, Sato N, Knopp MV, Star RA, Waldmann TA, Tagaya Y, Brechbiel MW. Comparison of dendrimer-based macromolecular contrast agents for dynamic micro-magnetic resonance lymphangiography. Magn. Reson. Med. 2003;50:758–766. doi: 10.1002/mrm.10583. [DOI] [PubMed] [Google Scholar]
- (7).WÃngler C, Moldenhauer G, Eisenhut M, Haberkorn U, Mier W. Antibody−Dendrimer Conjugates: The Number, Not the Size of the Dendrimers, Determines the Immunoreactivity. Bioconjugate Chem. 2008;19:813–820. doi: 10.1021/bc700308q. [DOI] [PubMed] [Google Scholar]
- (8).Kobayashi H, Sato N, Saga T, Nakamoto Y, Ishimori T, Toyama S, Togashi K, Konishi J, Brechbiel MW. Monoclonal antibodydendrimer conjugates enable radiolabeling of antibody with markedly high specific activity with minimal loss of immunoreactivity. Eur. J. Nucl. Med. 2000;27:1334. doi: 10.1007/s002590000293. [DOI] [PubMed] [Google Scholar]
- (9).Wu C, Brechbiel MW, Kozak RW, Gansow OA. Metalchelate-dendrimer-antibody constructs for use in radioimmunotherapy and imaging. Bioorg. Med. Chem. Lett. 1994;4:449–454. [Google Scholar]
- (10).Lebduskova P, Sour A, Helm L, Toth E, Kotek J, Lukes I, Merbach AE. Phosphinic derivative of DTPA conjugated to a G5 PAMAM dendrimer: an 17O and 1H relaxation study of its Gd(iii) complex. Dalton Trans. 2006:3399–3406. doi: 10.1039/b517847a. [DOI] [PubMed] [Google Scholar]
- (11).Wang SJ, Brechbiel MW, Weiner EC. Invest. Radiol. 2003;38:662–668. doi: 10.1097/01.rli.0000084887.47427.75. [DOI] [PubMed] [Google Scholar]
- (12).Zhu W, Okollie B, Bhujwalla ZM, Artemov D. PAMAM dendrimer-based contrast agents for MR imaging of Her-2/neu receptors by a three-step pretargeting approach. Magn. Reson. Med. 2008;59:679–685. doi: 10.1002/mrm.21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Konishi J, Togashi K, Brechbiel MW. Micro-MR angiography of normal and intratumoral vessels in mice using dedicated intravascular MR contrast agents with high generation of polyamidoamine dendrimer core: Reference to pharmacokinetic properties of dendrimer-based MR contrast agents. J. Magn. Reson. Imaging. 2001;14:705–713. doi: 10.1002/jmri.10025. [DOI] [PubMed] [Google Scholar]
- (14).Wiener E, Brechbiel MW, Brothers H, Magin RL, Gansow OA, Tomalia DA, Lauterbur PC. Dendrimer-based metal chelates: A new class of magnetic resonance imaging contrast agents. Magn. Reson. Med. 1994;31:1–8. doi: 10.1002/mrm.1910310102. [DOI] [PubMed] [Google Scholar]
- (15).Laus S, Sour A, Ruloff R, Tóth E, Merbach AE. Rotational Dynamics Account for pH-Dependent Relaxivities of PAMAM Dendrimeric, Gd-Based Potential MRI Contrast Agents. Chem. Eur. J. 2005;11:3064–3076. doi: 10.1002/chem.200401326. [DOI] [PubMed] [Google Scholar]
- (16).Yang H, Kao WJ. Dendrimers for pharmaceutical and biomedical applications. J. Biomater. Sci., Polymer Edn. 2006;17:3–19. doi: 10.1163/156856206774879171. [DOI] [PubMed] [Google Scholar]
- (17).Diallo MS, Balogh L, Shafagati A, Johnson JH, Goddard WA, Tomalia DA. Poly(amidoamine) Dendrimers: A New Class of High Capacity Chelating Agents for Cu(II) Ions. Environ. Sci. Tech. 1999;33:820–824. [Google Scholar]
- (18).Xu H, Regino CAS, Bernardo M, Koyama Y, Kobayashi H, Choyke PL, Brechbiel MW. Toward Improved Syntheses of Dendrimer-Based Magnetic Resonance Imaging Contrast Agents: New Bifunctional Diethylenetriaminepentaacetic Acid Ligands and Nonaqueous Conjugation Chemistry. J. Med. Chem. 2007;50:3185–3193. doi: 10.1021/jm061324m. [DOI] [PubMed] [Google Scholar]
- (19).Ali MM, Woods M, Caravan P, Opina ACL, Spiller M, Fettinger JC, Sherry AD. Synthesis and Relaxometric Studies of a Dendrimer-Based pH-Responsive MRI Contrast Agent. Chem. Eur. J. 2008;14:7250–7258. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brechbiel MW, Beitzel PM, Gansow OA. Purification of pnitrobenzyl C-functionalized diethylenetriamine pentaacetic acids for clinical applications using anion-exchange chromatography. J Chromatogr. A. 1997;771:63–69. doi: 10.1016/s0021-9673(97)00108-8. [DOI] [PubMed] [Google Scholar]
- (21).Nwe K, Andolina CM, Morrow JR. Tethered Dinuclear Europium(III) Macrocyclic Catalysts for the Cleavage of RNA. J. Am. Chem. Soc. 2008 doi: 10.1021/ja8037799. [DOI] [PubMed] [Google Scholar]
- (22).Nwe K, Richard JP, Morrow JR. Direct excitation luminescence spectroscopy of Eu(iii) complexes of 1,4,7-tris(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane derivatives and kinetic studies of their catalytic cleavage of an RNA analog. Dalton Trans. 2007:5171–5178. doi: 10.1039/b710072h. [DOI] [PubMed] [Google Scholar]
- (23).KuÌ^nnemeyer J, Terborg L, Nowak S, Scheffer A, Telgmann L, Tokmak F, GuÌ^nsel A, WiesmuÌ^ller G, Reichelt S, Karst U. Speciation Analysis of Gadolinium-Based MRI Contrast Agents in Blood Plasma by Hydrophilic Interaction Chromatography/Electrospray Mass Spectrometry. Anal. Chem. 2008;80:8163–8170. doi: 10.1021/ac801264j. [DOI] [PubMed] [Google Scholar]
- (24).Chang CA, Francesconi LC, Malley MF, Kumar K, Gougoutas JZ, Tweedle MF, Lee DW, Wilson LJ. Synthesis, characterization, and crystal structures of M(DO3A) (M = iron, gadolinium) and Na[M(DOTA)] (M = Fe, yttrium, Gd) Inorg. Chem. 1993;32:3501–3508. [Google Scholar]
- (25).Hamblin J, Abboyi N, Lowe MP. A binaphthyl-containing Eu(iii) complex and its interaction with human serum albumin: a luminescence study. Chem. Commun. 2005:657–659. doi: 10.1039/b415464a. [DOI] [PubMed] [Google Scholar]
- (26).Woods M, Aime S, Botta M, Howard JAK, Moloney JM, Navet M, Parker D, Port M, Rousseaux O. Correlation of Water Exchange Rate with Isomeric Composition in Diastereoisomeric Gadolinium Complexes of Tetra(carboxyethyl)dota and Related Macrocyclic Ligands. J. Am. Chem. Soc. 2000;122:9781–9792. [Google Scholar]
- (27).Khopkar PK, Narayanankutty P. Effect of ionic media on the stability constants of chloride, nitrate and thiocyanate complexes of americium(III) and europium(III) J. Inorg. Nuc. Chem. 1971;33:495–502. [Google Scholar]
- (28).Bansal BML, Patil SK, Sharma HD. Chloride, nitrate and sulphate complexes of europium (III) and americium (III) J. Inorg. Nuc. Chem. 1964;26:993–1000. [Google Scholar]
- (29).Ikeda A, Itoh K, Suzuki T, Aida M, Fujii Y, Mitsugashira T, Hara M, Ozawa M. Effect of counter-anions on the adsorption of trivalent actinides and lanthanides on tertiary pyridine resin in alcoholic chloride and nitrate solutions. J. Alloys Compd. 2006;408-412:1052–1055. [Google Scholar]
- (30).Barge A, Cravotto G, Gianolio E, Fedeli F. How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Med. Mol. Imaging. 2006;1:184–188. doi: 10.1002/cmmi.110. [DOI] [PubMed] [Google Scholar]
- (31).Weinmann HJ, Brasch RC, Press WR, Wesbey GE. Characteristics of gadolinium-DTPA complex: a potential NMR contrast agent. Am. J. Roentgenol. 1984;142:619–624. doi: 10.2214/ajr.142.3.619. [DOI] [PubMed] [Google Scholar]
- (32).Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Ishimori T, Akita Y, Mamede MH, Konishi J, Togashi K, Brechbiel MW. Novel liver macromolecular MR contrast agent with a polypropylenimine diaminobutyl dendrimer core: Comparison to the vascular MR contrast agent with the polyamidoamine dendrimer core. Mag. Res. Med. 2001;46:795–802. doi: 10.1002/mrm.1259. [DOI] [PubMed] [Google Scholar]
- (33).Aime S, Gianolio E, Terreno E, Giovenzana GB, Pagliarin R, Sisti M, Palmisano G, Botta M, Lowe MP, Parker D. Ternary Gd(III)LHSA adducts: evidence for the replacement of inner-sphere water molecules by coordinating groups of the protein. Implications for the design of contrast agents for MRI. J. Biol. Inorg. Chem. 2000;5:488–497. doi: 10.1007/pl00021449. [DOI] [PubMed] [Google Scholar]
- (34).Messeri D, Lowe MP, Parker D, Botta M. A stable, high relaxivity, diaqua gadolinium complex that suppresses anion and protein binding. Chem. Commun. 2001:2742–2743. [Google Scholar]
- (35).Nonat A, Fries PH, Pécaut J, Mazzanti M. Structure, Stability, Dynamics, High-Field Relaxivity and Ternary-Complex Formation of a New Tris(aquo) Gadolinium Complex. Chem. Eur. J. 2007;13:8489–8506. doi: 10.1002/chem.200601856. [DOI] [PubMed] [Google Scholar]
- (36).Kellar KE, Henrichs PM, Hollister R, Koenig SH, Eck J, Wei D. High relaxivity linear Gd(DTPA)-polymer conjugates: The role of hydrophobic interactions. Magn. Reson. Med. 1997;38:712–716. doi: 10.1002/mrm.1910380506. [DOI] [PubMed] [Google Scholar]
- (37).Zech SG, Eldredge HB, Lowe MP, Caravan P. Protein Binding to Lanthanide(III) Complexes Can Reduce the Water Exchange Rate at the Lanthanide. Inorg. Chem. 2007;46:3576–3584. doi: 10.1021/ic070011u. [DOI] [PubMed] [Google Scholar]
- (38).Avedano S, Tei L, Lombardi A, Giovenzana GB, Aime S, Longo D, Botta M. Maximizing the relaxivity of HSA-bound gadolinium complexes by simultaneous optimization of rotation and water exchange. Chem. Commun. 2007:4726–4728. doi: 10.1039/b714438e. [DOI] [PubMed] [Google Scholar]