

Abstract
Development of the telencephalon involves the coordinated growth of diversely patterned brain structures. Previous studies have demonstrated the importance of β-catenin-mediated Wnt signaling in proliferation and fate determination during cerebral cortical development. In this paper, we present novel evidence that β-catenin-mediated Wnt signaling also critically maintains progenitor proliferation in the subcortical (pallidal) telencephalon of mice. Targeted deletion of β-catenin severely impairs proliferation in the medial ganglionic eminence without grossly altering differentiated fate. Several lines of evidence suggest that this phenotype is primarily due to loss of canonical Wnt signaling. As previous studies have suggested that the ventral patterning factor Shh also stimulates dorsal telencephalic proliferation, we propose a model whereby Wnt and Shh signaling promote distinct dorsal-ventral patterning, while also having broader effects on proliferation that serve to coordinate the growth of telencephalic subregions.
Introduction
Generation of telencephalic structures during embryonic development requires the parallel and coordinated occurrence of two general events, patterning/fate determination and proliferation/tissue growth. Patterning of the diverse telencephalic structures occurs via the actions of multiple molecules including Sonic Hedgehog (Shh), Wnts, bone morphogenetic proteins (BMPs), and fibroblast growth factor 8 (Fgf8)1. Several of these factors, along with CNTF/LIF from the choroid plexus, have also been implicated in the regulation of growth of the telencephalon2–6. While distinct mechanisms regulate the generation of distinct structures, additional regulatory machinery should exist to orchestrate the relative growth of these structures that will eventually connect to form a functioning brain.
One factor that may coordinate growth of the dorsal and ventral telencephalon is Shh. Shh, expressed in the preoptic ventricular zone and in the mantle region of the medial ganglionic eminence (MGE), regulates initial patterning of the subcortical (pallidal) telencephalon as well as the maintenance of the fate-determining transcription factor Nkx2.1 during neurogenesis7,8 (for review see3). Shh also has mitogenic effects on the MGE9,10, as well as on dorsal telencephalic progenitors2,8,11,12.
Another candidate for regulating the growth of telencephalic structures is the Wnt family of secreted signaling molecules13–15. In the “canonical” Wnt signaling pathway, activation by Wnt ligands results in stabilization of cytoplasmic β-catenin, which translocates to the nucleus to bind to Tcf/Lef transcription factors, converting them from repressors to activators of their target genes16. Gain of function studies in vivo support a critical role of β-catenin-mediated Wnt signaling in stimulating cortical proliferation17,18. Loss of Wnt signaling in the telencephalon in vivo results in reduced cortical proliferation and disruption of cortical identity as well as defects in neuronal migration6,14,15,19,20. In the spinal cord Wnts function to coordinate ventral and dorsal neural tube proliferation21, but in the telencephalon canonical Wnt activity and function has been suggested to be restricted to dorsal (pallial) regions20,22.
To address the question of whether the regulation of ventral telencephalic growth may involve Wnt/β-catenin signaling, we used the Cre-loxP system to eliminate β-catenin from subcortical (pallidal) telencephalic Nkx2.1-expressing progenitors. Loss of β-catenin dramatically reduced the growth of the MGE due to premature cell cycle exit, without grossly altering initial differentiated fate. Several lines of evidence suggest that this phenotype results primarily from disruption of canonical Wnt signaling. As studies have suggested that the ventral patterning factor Shh also stimulates dorsal telencephalic proliferation2,8,11,12, mitogenic influences from Shh and Wnt signaling could provide a mechanism for the coordinated growth of dorsal and ventral telencephalic regions despite their divergent patterning and structure.
Results
Loss of β-catenin impairs growth of the MGE
To reduce β-catenin in the MGE we used the Cre-loxP approach, in which transgenic mice expressing Cre under the control of NKX2.1 promoter elements23 were crossed with “floxed” β-catenin mice (β-catFl/Fl)24. As the mutation causes perinatal lethality, we focused our analysis on embryonic ages (E11.5-E19). Nkx2.1 is first detected in the basal telencephalon around E8.75-E9 and is strongly expressed in progenitor cells of the MGE by E10.5–E11.525,26. Nkx2.1Cre-mediated recombination is apparent in the MGE at E10.523. To confirm that recombination has occurred in the MGE of Nkx2.1Cre;β-catFl/Fl brains, coronal forebrain sections of E11.5–E14.5 embryos were co-immunolabeled for β-catenin and Cre (an example of an E12.5 brain is shown in Fig. 1a–d). The expression of β-catenin is normally concentrated at the apical surface of neuroepithelial cells surrounding the lateral ventricles (see insert in Fig. 1b)27, but was greatly reduced in Cre-expressing cells of the mutant MGE (see insert in Fig. 1d). Residual β-catenin expression was present in the dorsal-most MGE (Fig. 1d) due to the weak or absent expression of the Nkx2.1Cre transgene in this region (Fig. 1a)23.
Figure 1. Loss of β-catenin expression in the ventral pallidum impairs growth of the MGE.
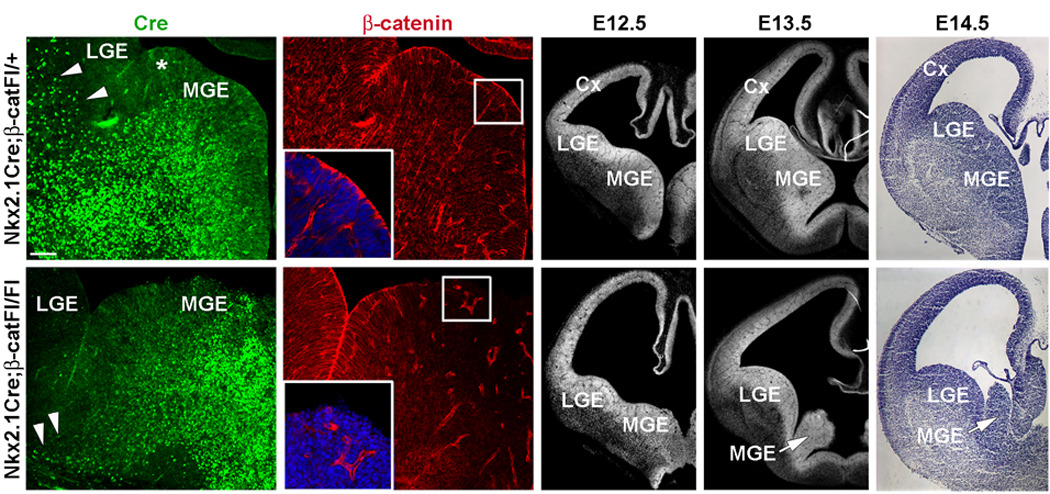
(a–d) Coronal sections from E12.5 Nkx2.1Cre;β-catFl/+ control (top row) and Nkx2.1Cre;β-catFl/Fl mutants (bottom row) co-labeled by immunofluorescence for Cre (a, c) and β-catenin (b, d). In Nkx2.1Cre mice Cre is strongly expressed through most of the MGE, with the exception of the dorsal most region (asterisk in a). Cre is also detected in cells that appear to be streaming from the MGE dorsally toward the developing striatum and cortex (arrowheads in a). This stream is greatly reduced in the mutant section (arrowheads in c). (b) Apical localization of β-catenin expression in neuroepithelial cells surrounding the lateral ventricle (insert shows higher magnification of the boxed area). (d) Consistent with the expression of Cre, β-catenin is greatly reduced in the ventral two-thirds of the mutant MGE (insert shows higher magnification of the boxed area). (e–j) DAPI (e, f, h, i) and Nissl (g, j) staining of coronal sections at E12.5, E13.5 and E14.5. After E12.5, little growth of the MGE (arrows in i and j) occurs. MGE-medial ganglionic eminence, LGE-lateral ganglionic eminence, Cx-cortex. Scale bar: 50 µm (a–d); 200 µm (e–j).
To examine the development of the MGE in Nkx2.1Cre;β-catFl/Fl mutant embryos, coronal sections were evaluated between E11.5 and E14.5 (Fig. 1e–j). The MGE in Nkx2.1Cre;β-catFl/Fl mutant was comparable in size with the control at E11.5 (Supplementary Fig. 2), however it appeared smaller than control by E12.5–E13.5 (Fig. 1e,h and 1f,i) and was greatly diminished by E14.5 (Fig. 1g,j). These findings suggest that loss of β-catenin expression in the ventral telencephalon impairs the growth of the MGE.
Loss of β-catenin reduces proliferation in the MGE
Wnt proteins possess stage-specific functions during development, maintaining cortical (pallial) patterning prior to the neurogenic period (E8.5–11)20, acting as mitogens at early neurogenic stages (≤E13.5)13,14,28,29 and promoting neural differentiation of cortical progenitors later in the neurogenic period (>E13.5)13,28. To investigate whether loss of β-catenin results in impaired proliferation of the MGE, sections from control and Nkx2.1Cre;β-catFl/Fl mutant embryos were immunolabeled for the S-phase marker BrdU (Fig. 2a,b) and for the cell cycle marker Ki67 (Fig. 2c,d). Results were quantified in E12.5 brains since this stage is prior to the point when the dramatic reduction in the size of the MGE occurred (Fig. 1e,h), although modest reductions in the same markers appeared to be present at E11.5 (Supplementary Fig. 2a,b). Deletion of β-catenin in the MGE caused a dramatic decrease in the number of cell profiles in S-phase (Fig. 2a,b and 2m: 192.7±11.9/0.01 mm2 vs 27.7±2.2/0.01 mm2, areal density of BrdU+ profiles in control vs mutant MGE, n=3, P=.007). Since differences in BrdU incorporation could reflect a reduced number of cycling progenitors, or a greatly extended cell cycle time (or both), proliferating cells were identified by immunofluorescence labeling for Ki67 (Fig. 2c,d). Loss of β-catenin expression leaded to a greatly decreased number of cycling progenitors (Fig. 2c,d and 2n: 236±7.6/0.01 mm2 vs 44.7±3.2/0.01 mm2 areal density of Ki67+ profiles in control vs mutant MGE, n=3, P=.002). In addition, the number of cell profiles in M-phase was also significantly lower in the mutant MGE than in control (Supplementary Fig. 2g,h). Reduced expression of the radial glial progenitor marker RC2, further supports the notion that loss of β-catenin reduces the number of progenitors in the mutant MGE (Supplementary Fig. 2e,f). Finally, and consistent with effects of loss of β-catenin on the cortical (pallial) telencephalon6,14, the MGE of Nkx2.1Cre;β-catFl/Fl mutants did not appear to have increased apoptotic cell death (Supplementary Fig. 2k,l).
Figure 2. Deletion of β-catenin in MGE progenitors causes decreased proliferation and precocious neuronal differentiation.
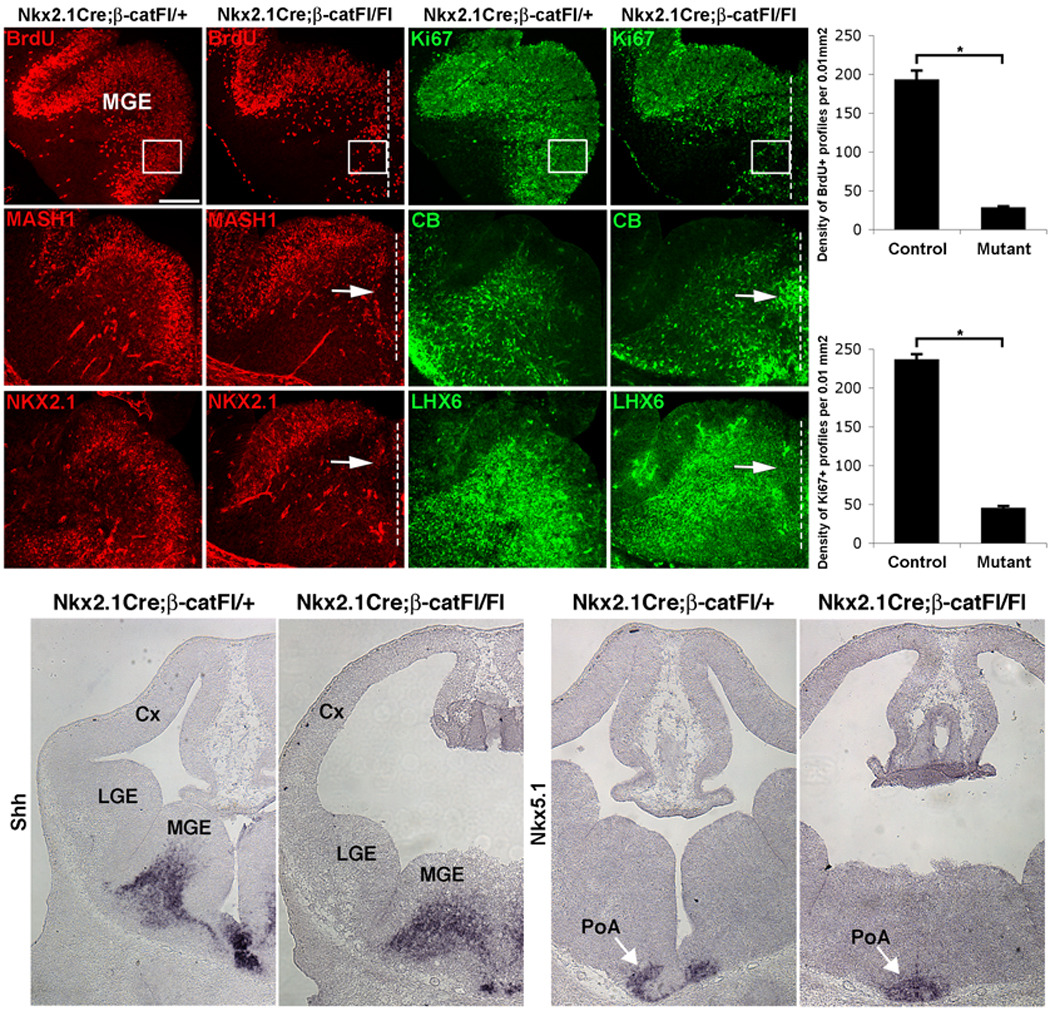
Coronal sections through the medial ganglionic eminence (MGE) from E12.5 control and Nkx2.1Cre;β-catFl/Fl mutants show double-labeling for (a–d) BrdU, incorporated during S-phase (a, b) and Ki67, detectable in most of cell cycle (c, d). The dashed lines indicate the midline which, although devoid of a ventricular space, is evident by the maintained symmetry of the mutant ventral pallidum (e.g. panel q). S-phase and cell cycle markers are significantly reduced in the mutant ventral MGE quantified in m and n, respectively. (e–l) Immunodetection of MASH1 (e, f) or NKX2.1 (i, j), markers of subcortical progenitors and calbindin (CB; g, h) or LHX6 (k, l), markers of differentiating neurons. In the control MGE MASH1+ and NKX2.1+ progenitors line the lateral ventricles (e, i), while CB+ and LHX6+ neurons primarily occupy the mantle zone (g, k). In mutant sections, the neuroepithelium and ventricular space are replaced by a fused region of mantle zone, where MASH1 and NKX2.1 are diminished (arrows in f, j) while CB and LHX6 are ectopically present (arrows in h, l).
(o–r) In situ hybridization labeling on sections from E12.5 control (o, q) and mutant (p, r) embryos. The mantle zone expression of Shh in the MGE (o, p), and Nkx5.1 in the preoptic area (PoA; arrows in q and r) are maintained in the Nkx2.1Cre;β-catFl/Fl mutants (o, p). Cx-cortex, LGE-lateral ganglionic eminence. Scale bar: 100 µm (a–l); 200 µm (o–r).
Grossly normal patterning in Nkx2.1Cre; β-catFl/Fl mutants
The reduced MGE proliferation without increased cell death in Nkx2.1Cre;β-catFl/Fl mutants raises the question of whether this premature differentiation of MGE progenitors is accompanied by an alteration of cell fate. First, sections were examined for markers of proliferating cells of the MGE ventricular zone (VZ), NKX2.1, or subventricular zone (SVZ), NKX2.1 and MASH1. Consistent with the reduction of proliferating cells in the mutant MGE, both the VZ and the SVZ progenitor populations were depleted (Fig. 2e,f,i,j).
The reduction of NKX2.1 and MASH1 in Nkx2.1Cre;β-catFl/Fl mutants, while consistent with the loss of proliferating cells, could also indicate a change in the patterning of the ventral pallidal neuroepithelium. In fact, studies have suggested that canonical Wnt activator signal may influence patterning in the ventral spinal cord21,30. To examine this possibility, sections were examined for markers of the MGE mantle zone, including calbindin31, LHX632 (also expressed in a minority of proliferating cells of the MGE SVZ)33 and Shh25. All three of these markers were strongly expressed in the mutant ventral pallidum, although their domains were expanded towards the ventral midline that would normally be occupied by proliferating cells (Fig. 2h,l,p). Both calbindin and LHX6 were ectopically present at the ventral midline (Fig. 2h,l), where the neuroepithelium and ventricular space were replaced by what appeared to be a fused region of mantle zone. Co-labeling of NKX2.1 and LHX6 in the mutant ventral pallidum at E11.5, when a substantial number of mitotic cells still exist in this region, revealed the normal complimentary pattern of expression of these genes despite the patchy presence of this pattern that extended into the fused, midline region. Triple-labeling of cells in this region for NKX2.1, LHX6 and the nuclear stain DAPI revealed that essentially all nuclei in the fused region at E11.5 expressed either NKX2.1 or LHX6 (DAPI labeling not shown but see Supplementary Fig. 2d). Since these genes are also expressed in portions of the hypothalamus, mutant and control sections were examined for the telencephalic marker FoxG1 (Supplementary Fig. 2i,j). FoxG1 was densely expressed through the mutant fused region (Supplementary Fig. 2j), strongly suggesting that the neuroepithelium of the mutant ventral pallidum has not been replaced by a non-telencephalic tissue.
Since NKX2.1 and LHX6 are also expressed in the preoptic zone at the base of the telencephalon, it is possible that the fused region contains an expansion of this domain. Nkx5.1 was expressed in the preoptic mantle region, and like the more dorsally placed MGE-mantle markers Shh, LHX6, and calbindin, Nkx5.1 expression was shifted towards the midline (Fig. 2q,r). However, Nkx5.1 expression was not shifted dorsally within the fused region, suggesting that there is no dorsal expansion of the most ventral pallidal domain.
We also found that in Nkx2.1Cre;β-catFl/Fl mutants patterning of the MGE-LGE sulcus is intact, as shown by the expression of Gli1, Patched1 and NKX6.2 (Supplementary Fig. 3g–j and Supplementary Fig. 3b,e). Moreover, the LGE mantle marker Islet1 was not expanded into the MGE mantle, and the dorsal pallidal boundaries of PAX6 and GSH2 expression were also unchanged in Nkx2.1Cre;β-catFl/FL mutants (Supplementary Fig. 3a–f). Together, these results suggest that the removal of β-catenin in the ventral MGE causes cells to exit the cell cycle prematurely, resulting in the replacement of most ventral pallidal neuroepithelium by telencephalic mantle zone that is patterned grossly normally as MGE and preoptic mantle.
Reduced MGE-derived neurons in Nkx2.1Cre; β-catFl/Fl mice
Progenitors of the MGE give rise to a variety of cells later inhabiting the cerebral cortex, the striatum, the globus pallidus and the basal magnocellular complex (BMC)34, 25, 26, 23, 35. To determine whether failed growth of the ventral pallidum (the Nkx2.1-expressing domain of the pallidum) results in reduction in these cell populations, coronal sections from E19 control and littermate Nkx2.1Cre;β-catFl/Fl mutant forebrains were analyzed for selected markers of Nkx2.1-lineage cells (Fig. 3). Immunolabeling for choline acetyltransferase (ChAT) revealed reduced density of cholinergic projection neurons of the BMC in these mutants (Fig. 3e,f, n=3). In addition, although the E19 analysis precludes examination of some interneuron markers such as parvalbumin, labeling for cortical interneuron markers calbindin (Calb; Fig. 3c,d), neuropeptide Y (NPY; Supplementary Fig. 4c,d), and somatostatin (SOM; Supplementary Fig. 4e,f), was also reduced in the Nkx2.1Cre;β-catFl/Fl mutants. The density of Calb+ profiles in sections from control vs mutant superficial cortical layers was 339.7±30.7/mm2 vs 174±1.26/mm2 (n=3, P=.03) and in deep cortical layers was 279±9.9/mm2 vs 95±3.2/mm2 (n=3, P=.002). Like the cholinergic neurons of the ventral pallidum, these cortical interneuron markers have previously been demonstrated to depend on expression of Nkx2.1 in the MGE36, 37, 25. These reductions of cortical interneurons and subcortical (pallidal) cholinergic neurons appear to result mainly from reduced neuronal production rather than from defects produced by loss of β-catenin in postmitotic subcortical neurons, since Dlx5/6Cre;β-catFl/Fl nulls were grossly healthy with no apparent alteration in forebrain histology, including PV, SOM, or Calretinin-expressing cortical interneurons (data not shown).
Figure 3. Loss of β-catenin expression in MGE progenitors reduces the detection of Nkx2.1-lineage cells at E19.
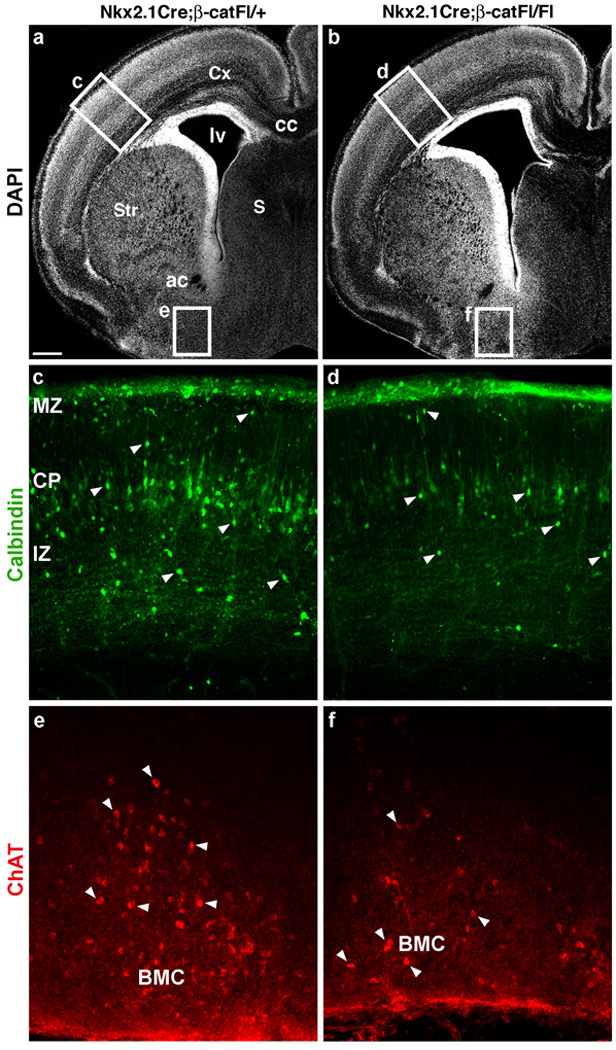
(a, b) DAPI-stained hemisections of E19 control (a) and mutant (b) brains demonstrating the rostral-caudal level at which sections were chosen for immunolabeling for selected markers of MGE-derived cells. The enlarged lateral ventricle (lv) is seen in all mutant samples examined at E19 (n=3). (c, d) Calbindin (Calb), expressed in layer V pyramidal neurons as well as interneuron populations (arrowheads) at this age, is decreased in cells with interneuron-morphology in the mutant section (compare c and d). The density of Calb+ profiles was quantified in the primary somatosensory area (see text), boxed area in a and b shown at higher magnification in c and d. (e, f) Immunolabeling for choline acetyltransferase (ChAT) reveals reduced density of cholinergic projection neurons (arrowheads) of the basal magnocellular complex (BMC) in the mutant section. Cx-cortex, Str-striatum, S-septum, cc-corpus callosum, ac-anterior commissure, MZ-marginal zone, CP-cortical plate, IZ-intermediate zone, VZ/SVZ-ventricular and subventricular zones. Scale bar: 300 µm (a, b); 200 µm (c, d); 100 µm (e, f).
Canonical Wnt activity in the ventral telencephalon
β-catenin functions in both canonical Wnt signaling and cell adhesion. To address the question of whether impaired growth of the MGE in Nkx2.1Cre;β-catFl/Fl mutants results from defective Wnt signaling, we first tested whether canonical Wnt signaling occurs in the Nkx2.1-expressing domain. The possibility of this activity was suggested by the expression of the transcription factor Tcf4, an effector of canonical Wnt signaling, within the proliferative zone of the MGE (Fig. 4a), but previous reports have suggested that canonical Wnt signaling is restricted to the dorsal (pallial) telencephalon20, 22. To re-examine this issue we used a transgenic Wnt reporter line in which β-galactosidase expression is driven by a minimal promoter containing six TCF binding sites (TCF-LacZ)38. As shown previously with different canonical Wnt activity reporter lines, strong lacZ reporter signal was present in the medial telencephalic wall, while lower level of lacZ signal was evident in the dorso-lateral cortex (Fig. 4c,d,f and Supplementary Fig. 5b; and 20). However, TCF reporter activity was also detected in the LGE and MGE of the subcortical (pallidal) telencephalon (Fig. 4c,d,f and Supplementary Fig. 5b; n=9 embryos). Despite the presence of background lacZ staining in the choroid plexus of TCF-LacZ– mice, no lacZ signal was found in the telencephalic neuroepithelium of these control mice (Fig. 4b,e , Supplementary Fig. 5a; n=5 embryos).
Figure 4. Canonical Wnt activity is present in the ventral telencephalon.
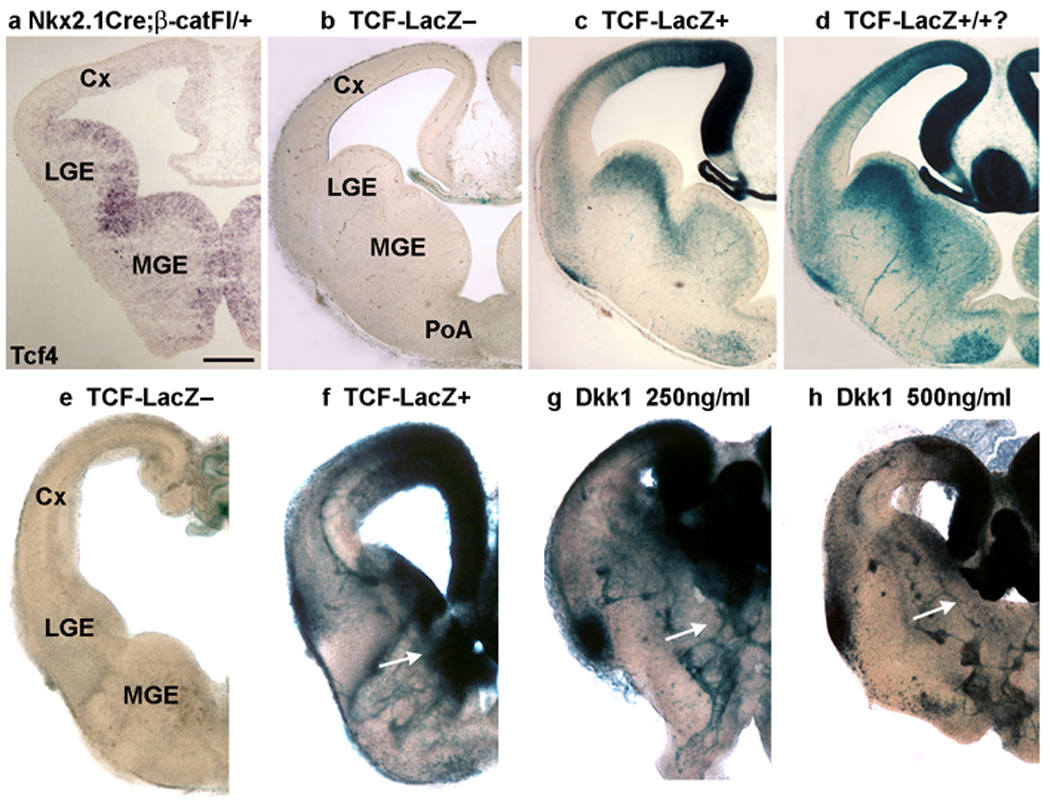
(a) Tcf4, a transcription factor and effector of canonical Wnt signaling, is expressed in the ventricular zone of the medial ganglionic eminence (MGE) in this section from an E12.5 embryo. (b–h) Analysis of canonical Wnt activity in the developing telencephalon using a transgenic Wnt reporter line (TCF-LacZ)38. The samples shown in (b–d) or in (e–h) were maintained in the same staining solution for equivalent lengths of time. (b) Apart from a small level of background activity in the choroid plexus, no lacZ activity is evident in the telencephalon of mice not containing the TCF-LacZ transgene. (c, d) Two examples of reporter activity from different TCF-LacZ embryos. The stronger reporter signal in d may indicate that it is homozygous for the TCF-LacZ transgene. In addition to the strong signal present in the medial cortex, the ventral telencephalon, including the lateral and medial ganglionic eminences and the preoptic area also show TCF-LacZ activity. (e–h) Coronal slices of E12.5 TCF-LacZ– (e) and TCF-LacZ+ (f–h) embryos were cultured for 1 day in vitro (div) in the absence (e, f) or presence (g, h) of the Wnt inhibitor Dkk1 before staining with X-gal to reveal reporter activity. (e) No signal is detected in the slice not containing the TCF-LacZ transgene. (f) Reporter activity is robust in a TCF-LacZ+ slice, and this activity is reduced within the mid to lateral cortex, as well as in the MGE (arrows), by Dkk1 (g, h). Cx-cortex, LGE-lateral ganglionic eminence, MGE-medial ganglionic eminence, PoA-preoptic area. Scale bar: 200 µm.
To confirm that this novel finding of TCF-LacZ reporter activity in the ventral telencephalon indicates the presence of Wnt signaling, coronal slices of E12.5 TCF-LacZ+ embryos were cultured for 1 day in vitro (DIV) in the absence or presence of the Wnt signaling inhibitor Dkk1. LacZ activity was revealed with X-gal staining. E12.5 telencephalic slices from TCF-LacZ+ embryos cultured 1 DIV without Dkk1 showed strong lacZ activity (Fig. 4f, Supplementary Fig. 5b; 11/12 slices displayed lacZ activity in the MGE). Application of Dkk1 (250 ng/ml or 500 ng/ml) to the culture medium consistently and extensively reduced lacZ activity in the MGE, LGE, and also in the lateral cortex (250 ng/ml Dkk1 decreased lacZ activity in 9/10 slices, Fig. 4g, Supplementary Fig. 5d; administration of 500 ng/ml Dkk1 reduced lacZ activity in 4/4 slices, Fig. 4h, Supplementary Fig. 5e). These results suggest that canonical Wnt signaling is indeed present in the ventral (pallidal) telencephalon.
To determine whether in vivo deletion of β-catenin in the MGE affects the activity of the Wnt/β-catenin pathway, coronal sections through the forebrain of E12.5 Nkx2.1Cre;β-catFl/+;TCF-LacZ+ and Nkx2.1Cre;β-catFl/Fl;TCF-LacZ+ mouse embryos were stained with X-gal. Targeted deletion of β-catenin reduced lacZ activity in the MGE of Nkx2.1Cre;β-catFl/Fl;TCF-LacZ+ embryos (Fig. 5a,b,d; n=3 embryos). This effect was most apparent in the ventral MGE, where expression of the Nkx2.1Cre transgene was stronger (Fig. 1a and 23), and where the loss of β-catenin expression was most pronounced (Fig. 5c). These results indicate that loss of β-catenin in MGE progenitors in vivo reduces TCF-LacZ signal and therefore Wnt activity in the MGE.
Figure 5. Canonical Wnt activity is reduced in the MGE of Nkx2.1Cre;β-catFl/Fl mutants.
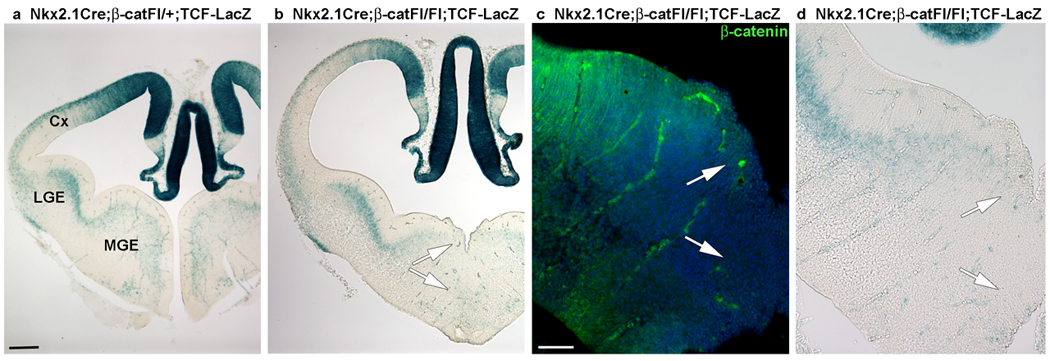
(a) A coronal section through an E12.5 Nkx2.1Cre;β-catFl/+ ;TCF-LacZ+ embryo shows lacZ activity in the MGE. (b, d) This activity is greatly reduced in the ventral MGE in a section from an Nkx2.1Cre;β-catFl/Fl;TCF-LacZ+ embryo (b; shown at higher magnification in d). (c) Immunofluorescence labeling of a nearby section for β-catenin shows the tight correspondence of the β-catenin loss in the ventral MGE with the loss of TCF-LacZ-reporter activity (compare area between arrows in c and d). This result suggests that Tcf activator function in the MGE is dependent on β-catenin signaling. Cx-cortex, LGE-lateral ganglionic eminence, MGE-medial ganglionic eminence. Scale bar: 200 µm (a, b); 100 µm (c, d).
Inhibition of TCF activity reduces MGE proliferation
Since the TCF-LacZ signal is mainly detectable in the proliferative zone of the MGE, the reduction of TCF-LacZ signal in the MGE of Nkx2.1Cre;β-catFl/Fl embryos is supportive but does not necessarily indicate that a direct functional link exists between the loss of this signal and the proliferation phenotype. Any reduction of proliferation, Wnt-mediated or not, would likely reduce TCF-LacZ signal as a consequence of reducing the proliferating population. To further examine the role of Wnt/β-catenin signaling in ventral telencephalon development, coronal telencephalic slices were electroporated with either a control plasmid (p-GFP) or with a vector co-expressing GFP and a dominant-repressive form of Lef1 (drLef1-GFP). drLef1 is unable to bind β-catenin, and therefore acts as a constitutive repressor of Lef1 target genes. Slices were cultured for 1 DIV before being re-sectioned and immunolabeled for GFP (to identify electroporated cells) (Fig. 6a,d) and proliferating cell nuclear antigen (PCNA; to detect proliferating cells) (Fig. 6b,e). The proportion of GFP+ cells co-expressing PCNA was determined in corresponding regions of the MGE of p-GFP- and drLef1-GFP-electroporated slices (boxes in Fig. 6a,d). Expression of drLef1-GFP in the MGE significantly reduced the proportion of GFP+ cells co-expressing PCNA (Fig. 6g: 50.7±2.4% vs 29.5±2.6%, n=4, P=.03). Consistent with a reduction in MGE progenitors, expression of drLef1-GFP also reduced NKX2.1 expression (data not shown). In summary, focal inhibition of β-catenin-mediated TCF activity mimics the effects of in vivo loss of β-catenin expression on the proliferation of MGE progenitors, suggesting that β-catenin/Tcf signaling regulates proliferation in the subcortical (pallidal) telencephalon.
Figure 6. Inhibition of β-catenin-mediated Tcf activation down-regulates proliferation in MGE progenitors.
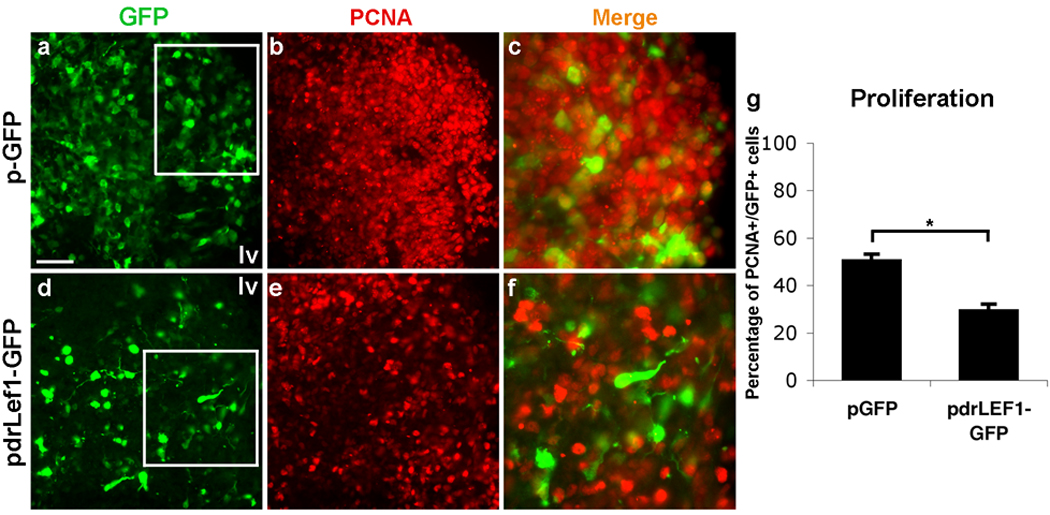
Shown are sections of E12.5 coronal slices that were electroporated with either p-GFP (a–c) or drLef1-GFP (d–f) vectors. Slices were cultured for 1 day before being re-sectioned at 14 µm and immunolabeled for GFP (a, d) and proliferating cell nuclear antigen (PCNA; b, e). The boxed areas in (a) and (d) are enlarged in (c) and (f). Expression of drLef1-GFP causes a significant reduction in the expression of PCNA (compare b with e, and c with f; quantified in g: 50.7±2.4% vs 29.5±2.6% (s.e.m.) of GFP+ cells in the proliferative region of the MGE are immunoreactive for PCNA in p-GFP vs drLef1-GFP electroporated slices, n=4, P=.03). Scale bar: 50 µm (a, b, d, e); 25 µm (c, f); 100 µm.
Inhibition of Wnt signaling reduces MGE proliferation
Taken together, the above results suggest that β-catenin mediated activation or disinhibition of Tcf activity regulates proliferation in the MGE. To examine whether Wnt-mediated stabilization of β-catenin is at least partially responsible for this effect, the effect of Wnt inhibitors on proliferation of MGE progenitors was studied. First, telencephalic slices from E11.5 embryos were exposed to Dkk1 (500 ng/ml) for 72 hours. Relative to controls, Dkk1 treatment resulted in a 40% reduction in Ki67+ cell profiles within the MGE (Fig. 7a–d, quantified in 7e: 113.4±2.9/0.01 mm2 vs 68.3±8.1/0.01 mm2, areal density of Ki67+ profiles in the MGE of untreated vs Dkk1 treated slices, n=3, P<.04). Treatment with the general Wnt signaling inhibitor Frizzled8-Fc39 (Fz8-Fc, 500 ng/ml) or with both Dkk1 and Fz8-Fc, resulted in similar reductions in MGE proliferation [Fig. 7e; areal density of Ki67+ profiles in the MGE, control: 113.4±2.9/0.01 mm2; Fz8-Fc: 74.9±5.4/0.01 mm2, P<.04; Fz8-Fc+Dkk1: 64.5±5.1/0.01 mm2, P<.004, n=3 of each treatment condition].
Figure 7. Inhibition of Wnt signaling suppresses proliferation of MGE progenitors.
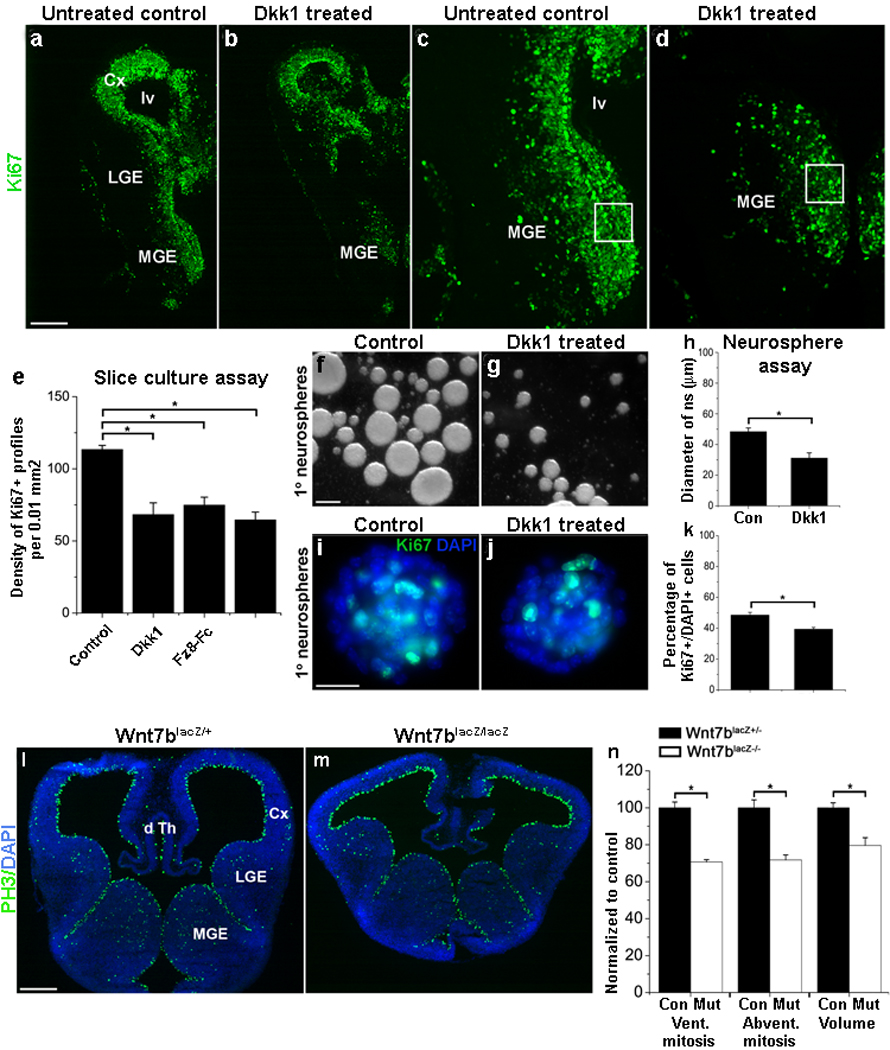
(a–e) Immunofluorescence analysis of proliferation by MGE progenitors in untreated control (a, c) and Dkk1 treated (b, d) E11.5 telencephalic slices. Application of Dkk1 (500 ng/ml; 72 hr) significantly reduces the number of Ki67+ progenitors (boxes in c and d show regions quantified in e). (Dkk1 alone: n=3, P<.04). (e) Exposure of slices to the general Wnt signaling inhibitor Frizzled 8-Fc (Fz8-Fc) alone or to a combination of Dkk1 and Fz8-Fc results in decreased proliferation similar to the effect of Dkk1 alone (Fz8-Fc alone: n=3, P<.04; Dkk1 and Fz8-Fc together: n=3, P<.004).
(f–k) Shown are primary neurospheres generated from E12.5 wild-type MGE cells after mitogenic stimulation with 10 ng/ml FGF2 (f, g, i, j). Neurospheres grown for 4 days in the presence of Dkk1 (250 ng/ml) are significantly smaller than neurospheres grown in the absence of Dkk1 (compare g and f; data is quantified in h) and contain fewer Ki67+ cells (compare j and i, quantified in k).
(l–n) Immunofluorescence analysis of proliferation of MGE progenitors in coronal sections from E12.5 Wnt7blacZ+/− controls and Wnt7blacZ−/− mutants. There is a significant reduction in the M-phase marker PH3 in the mutant MGE (compare l and m). Panel (n) shows quantification of these counts expressed as the percentage of control. Ventricular mitoses: n=4, P<.04; abventricular mitoses: n=4, P<.005; estimated volume: n=4, P<.01. Cx-cortex, LGE-lateral ganglionic eminence, MGE-medial ganglionic eminence, lv-lateral ventricle, dTh-dorsal thalamus. Scale bar: 250 µm (a–d); 50 µm (f, g); 25 µm (i, j); 200 µm (l, m).
Second, dissociated MGE progenitors were cultured at single cell density and allowed to form primary neurospheres in the presence or absence of Dkk1 (250 ng/ml). After 4 days in vitro, Dkk1 treatment significantly reduced neurosphere growth (Fig. 7f–h; 45.8±1.7µm vs 32.3±2.6µm; untreated vs Dkk1 treated neurospheres, n=4, P<.02). Application of Dkk1 also significantly decreased the proportion of Ki67+/DAPI+ cells both in intact and acutely dissociated neurospheres (Fig. 7i–k; intact ns culture: 46.5±4.3 vs 33±3.4 Ki67+ cells/ns in untreated vs Dkk1 treated neurosphere cultures, n=4; P<.02; dissociated ns culture: 48.6±1.7% vs 39.4±1.2% Ki67+/DAPI+ cells in control vs Dkk1 treated neurosphere cultures, n=4, P<.02). These results are consistent with a role for canonical Wnt signaling in MGE progenitor proliferation.
Since exogenous Wnt ligands were not added to the medium of neurosphere cultures, a reduction in MGE proliferation upon Dkk1 treatment also suggests that a Wnt ligand or a set of Wnt ligands is produced by MGE progenitors. Wnt7a and Wnt7b are both expressed in the proliferative regions of the MGE40. As Wnt7a mutant mice, despite infertility and limb phenotypes, are fully viable41, we analyzed proliferation in Wnt7b mutants42. To evaluate whether Wnt7b stimulates the proliferation of MGE progenitors, telencephalic sections from E12.5 Wnt7b heterozygous controls (Wnt7blacZ+/−) and mutant embryos (Wnt7blacZ−/−) were collected and immunolabeled for the M-phase marker PH3 (Fig. 7l,m). Lack of Wnt7b resulted in a 30% reduction in both ventricular and abventricular mitoses in the MGE (Fig. 7n: ventricular mitoses: 29.4±3.1 vs 20.8±1.2 PH3+ cells/500 µm; control vs Wnt7blacZ−/−, n=4, P<.04; abventricular mitoses: 78.6±4.3 vs 56.4±2.7 PH3+ cells/MGE; control vs Wnt7blacZ−/−, n=4, P<.005). Also, volumetric analysis at E12.5 revealed a 20% reduction in MGE volume in the Wnt7b mutant (Fig. 7n; estimated volume: 0.11mm3 vs 0.08mm3, control vs Wnt7blacZ−/−, n=4, P<.01). This phenotype, together with the results from slice or neurosphere treatment with Wnt inhibitors, suggests that Wnt signaling stimulates proliferation of MGE progenitors. However, analysis at E14.5 did not show a significant effect of the Wnt7b mutant on MGE proliferation or volume (data not shown), suggesting that in addition to Wnt7b, other Wnt ligands are likely to be stimulating proliferation in the MGE.
Discussion
Loss of β-catenin impairs proliferation in the MGE
While previous studies have mainly investigated the role of β-catenin regulating progenitor expansion and growth within the dorsal telencephalon, here we focused on the function of β-catenin in ventral telencephalic development. Loss of β-catenin expression in MGE progenitors dramatically impaired the growth of the MGE (Fig. 1). This effect is due to premature cell cycle exit of Nkx2.1-expressing progenitors, as indicated by a dramatic decline in markers for cycling progenitors (Fig. 2 and Supplementary Fig. 2). These findings are consistent with the previously reported role of β-catenin in regulating the proliferation of cortical progenitors14, 17, 18, 6, 28. Thus, our study indicates that in addition to controlling proliferation in the cerebral cortex, β-catenin also plays a role in regulating progenitor expansion in the ventral telencephalon.
Canonical Wnt signaling supports proliferation in the MGE
β-catenin is known to function in canonical Wnt signaling, cell adhesion, and could have other functions in mammalian cells that influence proliferation. Thus, linking β-catenin loss to canonical Wnt signaling requires demonstration that upstream of β-catenin signaling, inhibition of Wnt activity reduces proliferation. In addition, one must demonstrate that downstream of β-catenin signaling, Tcf activator function is present and its inhibition also reduces proliferation. We provide evidence for both of these effects of canonical Wnt signaling on proliferation, as well as evidence from published data and the current study that a cell adhesion function of β-catenin is unlikely to contribute to the Nkx2.1Cre;β-catFl/Fl phenotype.
First, Tcf4 expression as well as Tcf activator signal is present within the proliferative zone of the MGE (Fig. 4). Remarkably, this activator signal is reduced both in Nkx2.1Cre;β-catFl/Fl mutants (Fig. 5), and in slices treated with the Wnt inhibitor Dkk1 (Fig. 4 and Supplementary Fig. 5). Expression of a dominant repressor form of Tcf/Lef1 in the MGE of slice cultures reduces proliferation (Fig. 6), suggesting, in conjunction with the results from Fig. 4 and Fig. 5, that Tcf activator function stimulates MGE progenitor proliferation downstream of Wnt mediated stabilization of β-catenin.
Second, to demonstrate that inhibition of Wnt signaling reduces proliferation in the MGE upstream of β-catenin signaling, we used Wnt inhibitor application in vitro as well as in vivo Wnt ligand loss of function approaches (Fig. 7). In addition to reducing Tcf activator signal (Fig. 4), application of the Wnt inhibitor Dkk1 to slice cultures significantly reduces proliferation in the MGE (Fig. 7). Treatment of MGE progenitors dissociated and grown as primary "neurospheres" with Dkk1 has a similar effect. Finally, mouse mutants for Wnt7b, that along with Wnt7a is expressed in MGE progenitors40, have a significant loss of both ventricular and abventricular mitoses in the MGE (Fig. 7). Taken individually, each of these experiments comes with caveats that complicate their interpretation. Taken together, they provide good evidence that canonical Wnt signaling drives proliferation by MGE progenitors, and that Wnt7b is likely to participate in this process.
Third, support for the notion that the MGE phenotype in Nkx2.1Cre;β-catFl/Fl mutants is highly unlikely to result from the cell adhesion function of β-catenin can be found through comparisons to other transgenic mouse lines. Several studies show that deletion of β-catenin drives dorsal telencephalic progenitors out of the cell cycle18,6, 14. In contrast, disruption of the adherens junctions formed by radial glial endfeet, by deleting αE-catenin43, N-cadherin44, or numb (that also disrupts cell polarity)45, disrupts the organization of the proliferative zone but does not rapidly end proliferation. In fact, loss of the connection of radial glial endfeet to the apical (ventricular) surface actually appears to enhance proliferation. Since these studies focused on the role of cell adhesion and cell polarity in cortical development, it is feasible that loss of cell adhesion has different effects on progenitor proliferation in the subcortical (pallidal) telencephalon. However, mouse mutants for the Par6 complex-interacting protein Lgl1, also result in the loss of apical-basal polarity, formation of ectopic rosettes of proliferating cells away from the ventricular surface, and a hyperproliferation phenotype in both the cortical (pallial) and subcortical (pallidal) telencephalon53.
Based on these studies, and on our demonstration that disruption of N-cadherin in slice cultures does not appear to affect proliferation in the MGE (Supplementary Fig. 6), it seems unlikely that the Nkx2.1Cre;β-catFl/Fl phenotype, that did not result in ectopic abventricular proliferation as occurs in the mutants for cell adhesion and cell polarity genes, results from loss of β-catenin function at the adherens junction or on cell polarity per se. In addition, the reduction of proliferation by MGE progenitors transfected with the drLef1-GFP construct independently supports a role of canonical Wnt signaling in MGE proliferation. However, we cannot rule out the possibility that the Nkx2.1Cre:β-catFl/Fl phenotype also reflects some unknown function of β-catenin on progenitor proliferation.
In sum, multiple lines of evidence suggest that canonical Wnt signaling is largely responsible for the dramatic loss of proliferation seen in the MGE of Nkx2.1Cre:β-catFl/Fl mutants. So which Wnt ligand(s) are responsible for this effect? As discussed above, Wnt7b nulls have reduced mitoses in the MGE (Fig. 7). However, the far more dramatic influence on MGE proliferation seen in the Nkx2.1Cre;β-catFl/Fl mutants suggest that Wnt7b does not act alone in this process. As both Wnt7a and Wnt7b can increase proliferation by cortical progenitors13, one possibility is that Wnt7a can partially compensate for the loss of Wnt7b. Another possibility is that Wnts from the dorsal midline, such as Wnt3a that can increase proliferation by ventral spinal cord progenitors21, can support MGE proliferation via the CSF.
Finally, the Nkx2.1Cre;β-catFl/Fl mutant phenotype may reflect synergistic functions of Wnt and Shh signaling. Although previous publications and our own findings do not suggest that the Nkx2.1Cre;β-catFl/Fl mutant phenotype results from the role of β-catenin on cell polarity, we do find a loss of the cilia marker acetylated α-tubulin along the ventricular surface of the MGE in these mutants (Gulacsi and Anderson, unpublished data). As Shh responsiveness requires intact cilia46, and Shh signaling is known to have a moderate effect on MGE proliferation at this stage10, 8, the dramatic loss of MGE proliferation in Nkx2.1Cre;β-catFl/Fl embryos may reflect the combined loss of both Wnt and Shh signaling.
Functions of Wnt and Shh in telencephalon development
Regardless of whether the Nkx2.1Cre;β-catFl/Fl proliferation phenotype partially reflects a loss of Shh signaling in affected progenitors, other manipulations of canonical Wnt signaling, both upstream and downstream of β-catenin (Fig. 4–Fig. 7), support an important role for this signaling in MGE proliferation. So why should a fate determination and mitogenic signaling system of the dorsal telencephalon also serve to regulate proliferation in the ventral telencephalon? In the chick spinal cord, Wnt signaling mediates a “growth gradient” that coordinates the increased growth of dorsal versus ventral regions21. In the mouse telencephalon, Wnts may play a similar role, only both dorsal and ventral proliferation are critically stimulated. Concomitantly, Shh more moderately drives proliferation ventrally and dorsally 2,8,10,11. This raises the possibility that Wnt and Shh signaling, that are initially involved in fate determination in the dorsal and ventral telencephalon, respectively, during the period of neurogenesis also regulate proliferation by the apposing field. The net effect could be to coordinate growth of these highly fate-divergent but interconnected regions that develop within the finite compartment of the rostral skull (Supplementary Fig. 1).
In sum, this paper provides evidence that β-catenin-mediated Wnt signaling exists in the Nkx2.1-expressing region of the ventral telencephalon where, like its previously established function in the dorsal telencephalon, it critically regulates proliferation. A purpose of this activity may be to, in a reciprocal fashion with the ventral fate-determining factor Shh, connect regionalized patterning with coordinated growth. An additional implication of this work is that alterations in Wnt and β-catenin signaling can impact the generation of MGE-derived interneurons that critically participate in forebrain function.
Methods
Animals, BrdU labeling and tissue preparation
β-catenin-loxP (β-catFl/Fl)24, were crossed with an Nkx2.1-Cre transgenic line23. The morning of vaginal plug discovery was considered embryonic day 0.5 (E0.5). We immersion-fixed embryonic brains in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS; 0.1 M pH7.4) for 2–8 hr at 4°C prior to being processed for cryosectioning. For in vivo labeling of S-phase cells, dams received BrdU (100 mg/kg; i.p.) 1 hr prior to sacrifice. We obtained coronal telencephalic sections at 14 µm and stored them at −80 °C. E18.5–E19 embryos were collected the night before expected birth, anesthetized by cooling, perfused transcardially with 4% PFA and 0.1% glutaraldehyde in PBS and postfixed in the same fixative for 2 hr. We obtained 40 µm coronal sections using a vibrating microtome. Both Nkx2.1Cre(+);β-catFl/+ and Nkx2.1Cre(−);β-catFl/Fl genotypes were used as controls. We crossed mice expressing Cre under the control of the Dlx5/6Cre promoter47 with β-catFl/Fl mice to generate Dlx5/6Cre;β-catFl/Fl mutants. Postnatal day 75 (P75) mice were perfused and brains were processed as described above.
We crossed Nkx2.1Cre;β-catFl/+ with TCF-LacZ(+) mice38 to generate Nkx2.1Cre;β-catFl/+;TCF-LacZ(+) mice, which were then crossed with β-catFl/Fl to obtain Nkx2.1Cre;β-catFl/Fl;TCF-LacZ(+) embryos. LacZ expression was revealed with X-gal staining (see below) and confirmed by PCR analysis for lacZ38.
We used Wnt7b-lacZ embryos (Wnt7blacZ+/− and Wnt7blacZ−/−)42 to examine the role of Wnt7b in MGE proliferation. Mice were treated in accordance with the guidelines set by the Weill Cornell Medical College and the National Institutes of Health.
Detection of lacZ activity
We immersion-fixed E13.5 TCF-LacZ and Nkx2.1Cre;β-cat;TCF-LacZ transgenic brains in 2% PFA + 2% glutaraldehyde in PBS for 30 min at room temperature. After rinsing in PBS, they were sectioned on a vibrating microtome at 30 µm (VF-300; Precision Instruments Inc.). Coronal telencephalic sections were incubated in X-gal23 overnight at 37°C and rinsed in PBS the next day. We generated 250 µm coronal slices from E12.5 TCF-LacZ transgenic mice and cultured them in the absence or presence of different concentrations of Dkk1 (R&D Systems). Slices were maintained for 24 hr and then processed for X-gal staining.
Immunohistochemistry
Cryosections obtained from E11.5–E14.5 Nkx2.1Cre;β-cat brains were re-fixed on slides in 4% PFA in PBS for 10 min at room temperature. This step was followed by antigen retrieval in 1 mM EDTA at 65°C for 10 min for the antibodies against Nkx2.1, Lhx6 and Cre or in methanol at −20°C for 10 min for anti-proliferating cell nuclear antigen (PCNA) labeling. The list of primary antibodies used and details on dilutions are shown in Supplementary Table 1. We used the following fluorescent secondary antibodies: goat anti-mouse or goat anti-rabbit Alexa488 or Alexa568 (1:500, Molecular Probes). We counter-stained sections with 0.3 µM 4′-6-Diamidino-2-phenylindole (DAPI, Molecular Probes) applied together with the secondary antibodies.
Floating sections generated from E18.5–E19 Nkx2.1Cre;β-cat and P75 Dlx5/6Cre;β-cat brains were incubated for 2 nights in primary antibodies and for 1 night in secondary antibodies. The primary antibodies used and details on dilutions are shown in Supplementary Table 1.
Slice electroporation and cultures
We generated telencephalic slices at 250 µm thickness and electroporated them as described8. Slices were maintained in vitro for 24 hr in serum-free medium and then processed for immunolabeling as above.
We treated E11.5 telencephalic slices with either Dkk1 (R&D, 500 ng/ml), Fz8-Fc (R&D, 500 ng/ml) or the combination of Dkk1 and Fz8-Fc (500 ng/ml each) for 72 hr (Wnt inhibitors were added to the culture medium every day). For the data summarized in Fig. 7e, a counting box (100µm × 100µm) was placed 100 µm ventral to the dorsal-most region of NKX2.1 expression, that generally occurred at or just ventral to a residual MGE-LGE sulcus.
For the data shown in Supplementary Fig. 6, we treated E12.5 slices with a mouse N-cadherin blocking antibody (mNCD-2, DSHB, 1:200 in medium) for 48 hr.
Neurosphere assay
MGE cells from E12.5 CD1 embryos were enzymatically dissociated, plated at single cell density (3×104 cells per well in a 24-well plate) and cultured in serum-free medium with 10 ng/ml FGF2 (Promega) with or without Dkk1 (R&D; 250 ng/ml). After 4 days in these culture conditions MGE progenitors generated primary “neurospheres”. Intact and acutely dissociated neurospheres were plated on poly-D-lysine-coated glass slides and immunolabeled for Ki67.
Data collection and statistical analysis
The areal density of cycling progenitors and cells in S-phase in the MGE of E12.5 Nkx2.1Cre;β-cat mice were determined by counting Ki67+ and BrdU+ nuclear profiles in a 100×100 µm box placed over the ventral MGE in region-matched control and mutant sections from three pairs of littermate embryos. In Wnt7b mutants and heterozygous controls (n=4 of each genotype, with both hemispheres of two sections counted) ventricular mitoses were estimated by counting PH3 immunolabeled cell profiles along a continuous 500-µm stretch of the MGE, starting from the ventricular surface of the MGE-LGE sulcus. Profiles were counted as “ventricular” when present within one cell body of the ventricular surface.
Blinded quantitative stereologic analysis of volume of the MGE of Wnt7b control and mutant brains (n=3–4 per genotype) was performed on 14 µm coronal sections. For each brain a series of every fifth section was randomly selected and processed for DAPI stain to identify all cells, NKX2.1 immunolabeling to identify the MGE region and phospho-histone H3 (PH3) immunolabeling to determine the extent of the proliferative region within the MGE. Using NKX2.1 and PH3 immunolabeling, the relevant structure was traced using a 4x objective. MGE volume estimates were calculated using the Cavalieri principle and StereoInvestigator software (Microbrightfield).
Quantification of markers of interneuron subgroups in the primary somatosensory cortex of E19 Nkx2.1Cre;β-cat brains (3 brains per genotype) was performed using profile the fractionator probe and systematic random sampling (Microbrightfield StereoInvestigator software). Student’s t-test on group means±s.e.m was performed using Microsoft Excel and Statview software. The proportion of GFP+ cells co-expressing NKX2.1 or PCNA was determined in corresponding regions of the MGE of p-GFP and drLef1-GFP-electroporated slices (n=4 slices/condition, data was pooled from both hemispheres of 2–3 non-consecutive sections for each slice).
Supplementary Material
Acknowledgement
We thank Michael P. Matise for helpful discussions, the following people for the gifts of mice: Qing Xu (Nkx2.1Cre), Daniel Dufort (TCF-LacZ), Rolf Kemler (β-catenin-Jackson Laboratory), Kenny Campbell (Dlx5/6Cre), Edward Morrisey (Wnt7b-lacZ embryos), for probes or reagents: Michael Matise (TCF4 riboprobe), Oscar Marin (Nkx5.1 riboprobe), Samuel Pleasure (drLef1-GFP construct). We also thank Susan LoCurto for her technical assistance. This work was supported by research grants to S.A.A. from the NIH P01 NS048120 and K02MH070031.
References
- 1.Guillemot F. Cellular and molecular control of neurogenesis in the mammalian telencephalon. Curr Opin Cell Biol. 2005;17:639–647. doi: 10.1016/j.ceb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Dahmane N, et al. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 3.Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- 4.Gutin G, et al. FGF signalling generates ventral telencephalic cells independently of SHH. Development. 2006;133:2937–2946. doi: 10.1242/dev.02465. [DOI] [PubMed] [Google Scholar]
- 5.Gregg C, Weiss S. CNTF/LIF/gp130 receptor complex signaling maintains a VZ precursor differentiation gradient in the developing ventral forebrain. Development. 2005;132:565–578. doi: 10.1242/dev.01592. [DOI] [PubMed] [Google Scholar]
- 6.Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience. 2003;122:129–143. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- 7.Chiang C, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 8.Xu Q, Wonders CP, Anderson SA. Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development. 2005;132:4987–4998. doi: 10.1242/dev.02090. [DOI] [PubMed] [Google Scholar]
- 9.Britto J, Tannahill D, Keynes R. A critical role for sonic hedgehog signaling in the early expansion of the developing brain. Nat Neurosci. 2002;5:103–110. doi: 10.1038/nn797. [DOI] [PubMed] [Google Scholar]
- 10.Machold R, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 11.Gulacsi A, Lillien L. Sonic hedgehog and bone morphogenetic protein regulate interneuron development from dorsal telencephalic progenitors in vitro. J Neurosci. 2003;23:9862–9872. doi: 10.1523/JNEUROSCI.23-30-09862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komada M, et al. Hedgehog signaling is involved in development of the neocortex. Development. 2008;135:2717–2727. doi: 10.1242/dev.015891. [DOI] [PubMed] [Google Scholar]
- 13.Viti J, Gulacsi A, Lillien L. Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J Neurosci. 2003;23:5919–5927. doi: 10.1523/JNEUROSCI.23-13-05919.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. J Neurosci. 2006;26:12620–12630. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou CJ, Borello U, Rubenstein JL, Pleasure SJ. Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience. 2006;142:1119–1131. doi: 10.1016/j.neuroscience.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Behrens J, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 17.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 18.Zechner D, et al. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–418. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 19.Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24:121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backman M, et al. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279:155–168. doi: 10.1016/j.ydbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- 22.Junghans D, Hack I, Frotscher M, Taylor V, Kemler R. Beta-catenin-mediated cell-adhesion is vital for embryonic forebrain development. Dev Dyn. 2005;233:528–539. doi: 10.1002/dvdy.20365. [DOI] [PubMed] [Google Scholar]
- 23.Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- 24.Brault V, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 25.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 26.Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. Journal of Neuroscience. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chenn A, Zhang YA, Chang BT, McConnell SK. Intrinsic polarity of mammalian neuroepithelial cells. Mol Cell Neurosci. 1998;11:183–193. doi: 10.1006/mcne.1998.0680. [DOI] [PubMed] [Google Scholar]
- 28.Hirabayashi Y, et al. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development. 2004;131:2791–2801. doi: 10.1242/dev.01165. [DOI] [PubMed] [Google Scholar]
- 29.Solberg N, Machon O, Krauss S. Effect of canonical Wnt inhibition in the neurogenic cortex, hippocampus, and premigratory dentate gyrus progenitor pool. Dev Dyn. 2008;237:1799–1811. doi: 10.1002/dvdy.21586. [DOI] [PubMed] [Google Scholar]
- 30.Lei Q, et al. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic shh-gli signaling in the neural tube. Dev Cell. 2006;11:325–337. doi: 10.1016/j.devcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997b;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 32.Grigoriou M, Tucker AS, Sharpe PT, Pachnis V. Expression and regulation of Lhx6 and Lhx7, a novel subfamily of LIM homeodomain encoding genes, suggests a role in mammalian head development. Development. 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
- 33.Du T, Xu Q, Ocbina PJ, Anderson SA. NKX2.1 specifies cortical interneuron fate by activating Lhx6. Development. 2008;135:1559–1567. doi: 10.1242/dev.015123. [DOI] [PubMed] [Google Scholar]
- 34.Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- 35.Kessaris N, et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- 37.Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohamed OA, Clarke HJ, Dufort D. Beta-catenin signaling marks the prospective site of primitive streak formation in the mouse embryo. Dev Dyn. 2004;231:416–424. doi: 10.1002/dvdy.20135. [DOI] [PubMed] [Google Scholar]
- 39.Hsieh JC, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci U S A. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grove EA, Tole S, Limon J, Yip L, Ragsdale CW. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- 41.Parr BA, McMahon AP. Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature. 1995;374:350–353. doi: 10.1038/374350a0. [DOI] [PubMed] [Google Scholar]
- 42.Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129:4831–4842. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- 43.Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science. 2006;311:1609–1612. doi: 10.1126/science.1121449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kadowaki M, et al. N-cadherin mediates cortical organization in the mouse brain. Dev Biol. 2007;304:22–33. doi: 10.1016/j.ydbio.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 45.Rasin MR, et al. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci. 2007;10:819–827. doi: 10.1038/nn1924. [DOI] [PubMed] [Google Scholar]
- 46.Huangfu D, et al. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 47.Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ericson J, et al. Sonic hedgehog induces the differentiation of ventral forebrain neurons: a common signal for ventral patterning within the neural tube. Cell. 1995;81:747–756. doi: 10.1016/0092-8674(95)90536-7. [DOI] [PubMed] [Google Scholar]
- 49.Ganzler-Odenthal SI, Redies C. Blocking N-cadherin function disrupts the epithelial structure of differentiating neural tissue in the embryonic chicken brain. J Neurosci. 1998;18:5415–5425. doi: 10.1523/JNEUROSCI.18-14-05415.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.