

Abstract
Manganese is an essential nutrient, integral to proper metabolism of amino acids, proteins and lipids. Excessive environmental exposure to manganese can produce extrapyramidal symptoms similar to those observed in Parkinson’s disease (PD). We used in vivo and in vitro models to examine cellular and circuitry alterations induced by manganese exposure. Primary mesencephalic cultures were treated with 10–00µM manganese chloride (MnCl2) which resulted in dramatic changes in the neuronal cytoskeleton even at subtoxic concentrations. Using cultures from mice with red fluorescent protein (RFP) driven by the tyrosine hydroxylase (TH) promoter, we found that dopaminergic neurons were more susceptible to manganese toxicity. To understand the vulnerability of dopaminergic cells to chronic manganese exposure, mice were given IP injections of MnCl2 for 30 days. We observed a 20% reduction in TH-positive neurons in the substantia nigra pars compacta (SNpc) following manganese treatment. Quantification of Nissl bodies revealed a widespread reduction in SNpc cell numbers. Other areas of the basal ganglia were also altered by manganese as evidenced by the loss of GAD67 in the striatum. These studies suggest that acute manganese exposure induces cytoskeletal dysfunction prior to degeneration and that chronic manganese exposure results in neurochemical dysfunction with overlapping features to PD.
Keywords: manganese, neurotoxicity, dopamine, Parkinson’s disease, striatum, substantia nigra
INTRODUCTION
Manganese (Mn2+) is a naturally occurring essential element with an environmental prevalence second only to iron. Manganese is crucial for maintaining proper cellular function and contributes to biological processes including maintenance of redox status, ensuring appropriate protein conformation, modulating ion and energy homeostasis and signal transduction (Takeda 2003; Aschner and Aschner 2005; Keen et al. 2005; Liu et al. 2006). Dietary intake is the largest source of manganese in the human body under normal circumstances, but airborne manganese particulates comprise the most prevalent source of excessive manganese exposure (Dobson et al. 2004; Fitsanakis et al. 2006b). Manganese is used in numerous industries including welding, mining, steel production, and formulating gasoline additives. Chronic manganese overexposure results in a neurological irreversible phenomenon referred to as manganism (Dorman et al. 2002; Dobson et al. 2003; Dorman et al. 2004; Aschner et al. 2005; Fitsanakis et al. 2006b). The factors that influence vulnerability to manganese and onset of manganism remain ill defined. The motor symptoms of the disorder are, however, strikingly similar to those observed in PD (Aschner and Aschner 1991; Lee 2000; Finkelstein et al. 2007). Dystonia and movement disorders have also been described in case reports of adults and children receiving prolonged total parental nutrition when the gastrointestinal tract is nonfunctional because of an interruption in continuity or because absorptive capacity is impaired (Kafritsa et al. 1998; Nagatomo et al. 1999; Takagi et al. 2001; Hsieh et al. 2007).
Manganese is capable of having direct actions on neurons and glia within the central nervous system. Manganese is readily transported into the brain, either as a free ion species or as a nonspecific protein-bound species (Aschner and Gannon 1994). Transport into a variety of tissues and cells occurs by way of the non-specific divalent metal transporter-1 (Aschner et al. 1999; Fitsanakis et al. 2006a), which belongs to the family of natural resistance-associated macrophage proteins. This protein is expressed broadly across the brain early in postnatal development (Siddappa et al. 2002; Wang et al. 2002). When complexed with transferrin, manganese is transported by transferrin receptors (Aschner and Aschner 1991). The citrate transporter has also been invoked to transport Mn2+ (Crossgrove et al. 2003). Candidates for the transport of the Mn2+-citrate complex include members of the organic anion transporter polypeptide or ATP-binding cassette superfamilies. Finally, an additional Mn2+ transporter is ZIP-8, a member of the solute carrier-39 (He et al. 2006). ZIP-8 is a Mn2+/HCO3 − symporter; an HCO3 − gradient across the plasma membrane is invoked as the driving force for Mn2+ uptake.
We have previously shown that primary astrocytic cultures are highly vulnerable to manganese and undergo apoptotic cell death involving mitochondrial dysfunction (Yin et al. 2008), a finding which is consistent with the work of Maynard and Cotzias who demonstrated preferential sequestration of this element in the mitochondria (Maynard and Cotzias 1955; Gunter et al. 2006). Bioenergetic studies have shown that neurons are even more intensely dependent upon intact mitochondria for respiration. The majority of mitochondria are located in dendrites (Wongriley 1989; Hertz and Peng 1992) where the density of excitatory inputs necessitates a high respiratory capacity due to the need to maintain Na+/K+ gradients during neural activation (Erecinska and Silver 1989; Hertz 2008). If mitochondria are an essential target organelle of manganese, one would predict that changes in neural processes may proceed nuclear or somal dysfunction and thus may contribute to circuit level dysfunction. Indeed, cytoskeletal changes may be essential to mediating neurodegeneration in PD and other neurological disorders. Tau, tubulin and neurotransmitter releasing proteins have been well/extensively documented in these disorders (Masliah et al. 1996; Billingsley and Kincaid 1997; Ovadi et al. 2004; Siman et al. 2004; Andreadis 2005; Cappelletti et al. 2005; Cuadrado-Tejedor et al. 2005; Willis et al. 2005; Heredia et al. 2006; Esposito et al. 2007; Fulga et al. 2007).
The purpose of this work was to extend our previous studies in astrocytes to dopaminergic neurons and in vivo systems to address the neurotoxic potential of manganese and to determine if nigrostriatal pathways are uniquely vulnerable to manganese exposure. The ability to define mechanisms of toxicity and cellular features, which increase cellular vulnerability, would enhance our ability to treat manganism and potentially provide essential insight into the vulnerability of the basal ganglia in this and other disorders.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal tyrosine hydroxylase antibody (TH, Chemicon, 1:1000) was used to identify dopaminergic neurons in vitro. Antibodies to β-tubulin (Sigma, 1:1000), synapsin-1 and tau-1 (Chemicon, 1:1000) were used to identify cytoskeletal and synaptic protein distribution. For staining of brain sections, mouse monoclonal α-glutamic acid decarboxylase 67 (GAD67, Chemicon, 1:2000, #MAB5406) and mouse monoclonal α-tyrosine hydroxylase (TH) (1:8000, Sigma, St. Louis, MO) were used. All secondary antibodies for primary culture work were purchased from Molecular Probes (Eugene, OR), while the brain section secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Bisbenzimide (Hoechst 33342; Sigma) was used as a nuclear counterstain.
Cytotoxicity of Manganese in Primary Mesencephalic Cultures
To study the effect of manganese on dopaminergic neurons, primary mesencephalic culture cells from rats at embryonic day 14 were plated on poly-L-ornithine-covered coverslips. Cultures were prepared as previously described (McLaughlin et al. 1998b). In brief, mesencephalon was isolated and incubated in trypsin at room temperature for 30min. Tissue was dissociated in 10ml plating medium containing DMEM, 10% F12 nutrients, 10% bovine calf serum (Hyclone, Logan, UT), 100U/ml penicillin, and 100µg/ml streptomycin. Cells were counted with a hemocytometer, diluted to 300,000 cells/ml and 2 ml of this stock was placed in each well of a 6-well plate. Typically viability following dissociation is > 95% by trypan blue exclusion, and neuronal cell number is maintained during the in vitro period (<5% loss over 2 weeks, unpublished observations). After two days in vitro, cells were treated for 24hr with 1–2µM cytosine arabinoside to prevent glial proliferation, and then transferred to Neurobasal media containing B27 supplement and 10µM β-mercaptoethanol. Mesencephalic cells were maintained for two weeks in culture after which they were treated with 10µM – 800µM MnCl2 in low serum media free from antioxidants and phenol red for 24hr (Musiek et al. 2006). The chosen MnCl2 concentrations are based on estimates from the literature. For example, weekly injections of MnCl2 over a 3-month period in the caudate-putamen (CPu) and globus pallidus (GP) of monkeys (0, 2.25, 4.5, and 9g) produce dose-related clinical signs, which are more severe in the higher dose range (Suzuki et al. 1975). At the highest dose, the MnCl2 concentration is increased 12-fold in the CPu and 9-fold in the GP. The “physiological range” (no symptoms) is ~75–100µM, and clinical signs increase in frequency and severity above this level, suggesting this concentration is near or at the threshold of toxicity. Upon treatment termination, cells were returned to the incubator, and neuronal viability was determined 20–24hr after exposure using a lactate dehydrogenase (LDH)-based in vitro toxicology assay kit (Sigma-Aldrich, St. Louis, MO). Media samples (40µl) were analyzed spectrophotometrically (490:630), according to the manufacturer's protocol, to obtain a measure of cytoplasmic LDH release from dead and dying neurons (McLaughlin et al. 2003). Cells were derived from 7 distinct culturing sessions. Statistical differences between control and treated cells were determined with one-way randomized ANOVA design. When the overall test of significance (p<0.05) leads to a rejection of the null hypothesis, post hoc Newman-Keuls comparison was performed. Statistical analyses were performed with Graphpad Instat software (GraphPad Software, La Jolla, CA).
Fluorescent Staining
Following MnCl2 treatment, cells were fixed in 3% paraformaldehyde for 30min. Non-specific binding of antigens was inhibited by incubating cells in blocking buffer (3% bovine serum albumin with 0.2% NP-40) for 1hr at room temperature. After blocking buffer immersion, cells were incubated with primary antibodies made in blocking buffer at 4°C overnight, followed by a 1hr incubation with appropriate conjugated secondary antibodies in 1% bovine serum albumin. Hoescht staining was performed as previously described by placing coverslips in 0.1mg/ml bisbenzamide for 10min prior to mounting coverslips on microscope slides (McLaughlin et al. 1998a).
Image Analysis
The AxioVision imaging program (Carl Zeiss, Thornwood NY) was used for morphological analysis of tau, β-tubulin and synapsin. Quantification of fluorescence intensity, threshold areas, and the number of cells and nuclei were established using Metamorph 4.0 software (Universal Imaging Corporation). For statistical analyses, comparisons were performed by a one-way ANOVA, followed by Bonferroni comparison test. All data are derived from averages of multiple coverslips per condition derived from at least 3 separate culturing conditions.
Cell Counts of Cultures from TH:RFP Transgenic Animals
Mesencephalic cultures were generated from a transgenic mouse expressing red fluorescent protein (RFP) under the control of the tyrosine hydroxylase (TH) promoter (Zhang et al. 2004). Mice were a generous gift from D. McMahon (Vanderbilt University). RFP-expressing midbrain dopaminergic neurons were isolated from E13 mice initially identified as TH:RFP by visualizing retinal expression of TH:RFP construct using a dissecting microscope equipped with a DsRed filter set. Identification was confirmed by PCR amplification using published primer sets (Zhang et al. 2004).
Primary murine cultures were generated as described above except that they were plated on plastic coverslips with embossed labeled grids for repeated identification of neurons and pre- and post-exposure cell counts as previously described (McLaughlin et al. 1998b). Precounts were done 12–14 days after dissociation. The following day, cells were exposed to MnCl2 for 24hr as described above. The experiments were terminated when medium was removed, and phosphate-buffered saline (PBS; pH 7.4) containing 0.5% Trypan Blue was added to the cultures for 5min. Coverslips were then washed twice with PBS and fixed in 4% paraformaldehyde made in PBS. Adherent TH+ and TH− cells excluding Trypan Blue were counted and data was expressed as percentages of the pre-exposure numbers obtained in the same squares.
In Vivo Manganese Treatment
All experiments were run under the oversight of Vanderbilt University IACUC of the and NIH guidelines. Male mice (approximately eight weeks of age) were purchased from The Jackson Laboratory (Bar Harbor, ME) and acclimated to a Vanderbilt University vivarium for two weeks. Mice were fed with ad lib water and Purina Lab Diet 5001 (~10 ppm Mn and 35 ppm Fe). For immunohistochemistry studies, twelve C57BL/6J mice (six in each group) were given intraperitoneal (IP) injections of MnCl2 (5 mg/kg/day) or saline vehicle control daily for 30 days.
Immunohistochemistry on In Vivo MnCl2 Exposed Mice
On day 31 of MnCl2 or control treatment, mice were anesthetized with sodium pentobarbital and transcardially perfused with 60–75mL of 4% paraformaldehyde. Brains were then removed and post-fixed at 4°C overnight. Cryoprotection was performed using a series of sucrose solutions in phosphate-buffered saline (10%, 20%, and 30%). Series of coronal sections were cut on a freezing microtome at 40µm and stored at −20°C.
Sections were treated with Tris-glycine (0.1M glycine, pH 7.4) to reduce non-specific labeling, blocked in 4% Blotto (Nestlé Carnation dry milk) in phosphate-buffered saline (PBS, pH 7.5), and incubated for 72hr with the appropriate primary antibody using previously published protocols (Stanwood et al. 2001; Stanwood et al. 2005). Triton X-100 (0.2%) was added to blocking solutions for all antibodies except GAD67. Following primary antibody incubation, sections were washed five times, and then incubated in biotinylated secondary antibodies for 60 min a t R T (Jackson Immunoresearch, West Grove PA, 1:1000 dilution). Sections were washed again and then avidin-biotin-perioxidase amplification (Vectastain ABC, Vector Labs) and 3,3’-diaminobenzidine reactions were used to visualize protein labeling. Sections were mounted on poly-l-lysine-coated slides, dehydrated in an ethanol series, and then coverslipped using DPX (Electron Microscopy Sciences, Hatfield PA). Sections from MnCl2-treated and control mice were processed in parallel to minimize variability in the quantity of immunostaining between groups, and negative controls, which omitted incubation with primary antibody, presented no specific labeling. An additional series of sections were mounted onto slides and stained with 0.5% cresyl-violet for labeling of all cell bodies.
Cell Counts of Sections and Statistical Analyses
Slides were coded so that the investigator was blinded to the treatment groups and sections were imaged using a Zeiss Axioplan II microscope to capture color images of sections using a high-resolution color CCD camera (Axiocam HR). Cells were counted in the 8-bit format, using the Image J program (NIH), bilaterally, from at least 3 nonadjacent sections per region. Labeled cells were counted and diameters were measured in 20x images to allow for correction of profile size by the method of Abercrombie (Abercrombie 1946).
Profiles of Nissl body distribution were counted in the SNpc and SNpr. Profiles of TH-immunoreactive cells were counted in the VTA and in the SNpc. GAD67-immunoreactive cells were analyzed in the anterior cingulate cortex, dentate gyrus, dorsomedial caudate putamen, and SNpr. Brain regions were identified using regional, cytoarchitectural and laminar landmarks. Three to six images from areas of the ventral midbrain (including the VTA, SNpc, and SNpr) were merged together using the automerge feature of Adobe Photoshop (version 9.02). For all regions of interest, at least four images were analyzed for each animal derived from at least 2 non-adjacent sections. Immunopositive cell densities were calculated as mean ± S.E.M and statistical analyses were performed using Student’s t-tests.
RESULTS
Our first experiments examined the relative vulnerability of mesencephalic cells to manganese by exposing primary cultures to increasing concentrations of the metal continuously for 24hr. We used neuron-enriched cultures (50% neurons) that were dissected mid-gestation to promote survival of TH-positive cell populations. Our cultures typically had 5–10% TH-positive neurons with the other population of neurons (identified with MAP2 staining) expressing the GABAergic marker GAD67 (data not shown, but similar to (Zeevalk et al. 1995). Cell death was assessed by measuring LDH release by dead and dying cells into the exposure media and by visually inspecting cells for signs of cell death including loss of neurites, soma shrinkage and presence of cellular debris in media. We found that MnCl2 induced cell death in a concentration-dependent manner with an LD50 of 909µM (Figure 1A). Representative photomicrographs were taken at the end of the 24hr exposure period and demonstrate the many phase-bright neurons (Figure 1B) in control cultures, which grow on top of a bed of glia. With increasing concentrations of MnCl2, soma volume shrinkage resulting in a stippled, less smooth appearance (800µM; Figure 1C), and extensive neuronal cell death can be seen at 3mM MnCl2 (Figure 1D).
Figure 1. Chronic Manganese is Cytotoxic to Primary Mesencephalic Cultures.

Mature primary mesencephalic neurons were exposed to increasing concentrations of MnCl2 for 24hr, at which time cell death was assessed. A) Lactate dehydrogenase release by dead and dying cells was assessed in the media and revealed that manganese induces cell death in a dose-dependant manner. Data represent the mean ± S.E.M. of 7 independent experiments, each performed in triplicate wells. Representative photomicrographs were taken following 24hr exposure to B) control conditions, C) 800µM MnCl2 or D) 3mM MnCl2. *p<0.05 vs. control by one-way ANOVA. Scale bar = 200 µm.
Next, we chose to focus on the consequences of pathophysiologically relevant concentrations of chronic manganese exposure at 100–800µM (Aschner et al. 1999). As demonstrated above, these concentrations were not overtly neurotoxic, but could alter cell structure in ways that might influence the circuitry of the basal ganglia and contribute to manganese-induced motor dysfunction. For this work, we surveyed the changes in the structural protein tau, the synapse specific marker synapsin and the cytoskeletal marker tubulin. Neurons were treated with vehicle (Figure 2 A1–3), 100µM MnCl2 (Figure 2 B1–3) or 800µM MnCl2 (Figure 2 C1–3). We observed that increasing manganese concentrations lead to a progressive loss of cohesive tau staining and an increase in cytoskeletal abnormalities as reflected by the >60% decrease in tau-positive neurites (Figure 2D). Similarly, synapsin was altered with both 100µM MnCl2 (Figure 3 B1–4) and 800µM MnCl2 exposure (Figure 3 C1–4), resulting in a ~75% decrease in synapsin staining (Figure 3D). However, as in Figure 2, no qualitative changes in nuclei were evident from our Hoechst stain suggesting that preapoptotic asymmetric chromatin formations were not induced and changes were confined to regions outside the nucleus.
Figure 2. Profound Changes in Neural Structure Occur at Subtoxic Exposures to Manganese.

Mature primary mesencephalic neurons were exposed to increasing concentrations of MnCl2 for 24hr, at which time Immunofluoroscent detection of tau (red) and nuclei (blue) was performed. Cultures were treated with vehicle (A1–3), 100µM MnCl2 (B1–3) or 800µM MnCl2 (C1–3). Results show that while both concentrations of MnCl2 were not toxic, they did lead to a dose dependent increase in cytoskeletal abnormalities, including a significant loss of architectural complexity, process retraction and beading (D) following 24hr exposure. Data represent the mean ± StDev *p< 0.05 one way analysis of variance with Bonferroni correction. Scale bar = 10 µm.
Figure 3. Synapsin and Tubulin Staining are Decreased with Subtoxic Manganese Exposures.

Primary mesencephalic neurons were exposed to increasing concentrations of MnCl2 for 24hr, at which time immunofluoroscent detection of tubulin (green), synapasin (red) and nuclei (blue) was performed. Cultures treated with vehicle (A1–4), 100µM MnCl2 (B1–4) or 800µM MnCl2 (C1–4) had increasing cytoskeletal abnormalities as evidenced by lack of continuity of neural processes and failure to discreetly localize the synapsin phosphoproteins which regulate transmitter release and trafficking. The loss of synapsin-positive cells was statistically significant at both concentrations of manganese used (D) as quantified by cell counting in at least four independent experiments using Metamorph software. Data represent the mean ± StDev *p< 0.05 one way analysis of variance with Bonferroni correction. Scale bar = 10 µm.
Mesencephalic cultures contain both GABAergic and dopaminergic populations although based on the symptomology of manganism, there appears to be a stronger dopaminergic dysfunction. To test this hypothesis we used a combination of in vitro and in vivo systems. We first stained manganese-exposed cultures for TH, the rate-limiting step in dopamine biosynthesis and a specific marker of dopaminergic neurons. We observed that manganese exposure resulted in shortening of TH-positive neurites with increasing exposure (data not shown). We next used cultures derived from TH:RFP reporter mice and counted TH-positive and TH-negative neurons before and after exposure to MnCl2. Cells containing this dopamine synthetic enzyme comprised 12 ± 3% of the total neuronal population. Manganese induced a similar concentration-dependent toxicity (data not shown) as in the cultures from the non-transgenic animals. The TH-positive cells were, however, more vulnerable to MnCl2 at both 300µM and 800µM concentrations than the other neuron populations in the mesencephalic cultures. This was evidenced by cell counts performed before and after MnCl2 exposure where we observed a progressive increasing in death of TH-positive neurons between concentrations of 300µM to 1mM, which was significantly greater than that of TH-negative neurons (Figure 4). These data also confirm that the effects of manganese were not simply to reduced reporter expression (i.e. TH promoter activity), because in that event one would predict the number of non-dopaminergic cells to increase concomitantly with the loss of TH+ neurons.
Figure 4. Tyrosine Hydroxylase Expression Decreases with Increasing Manganese Exposures.
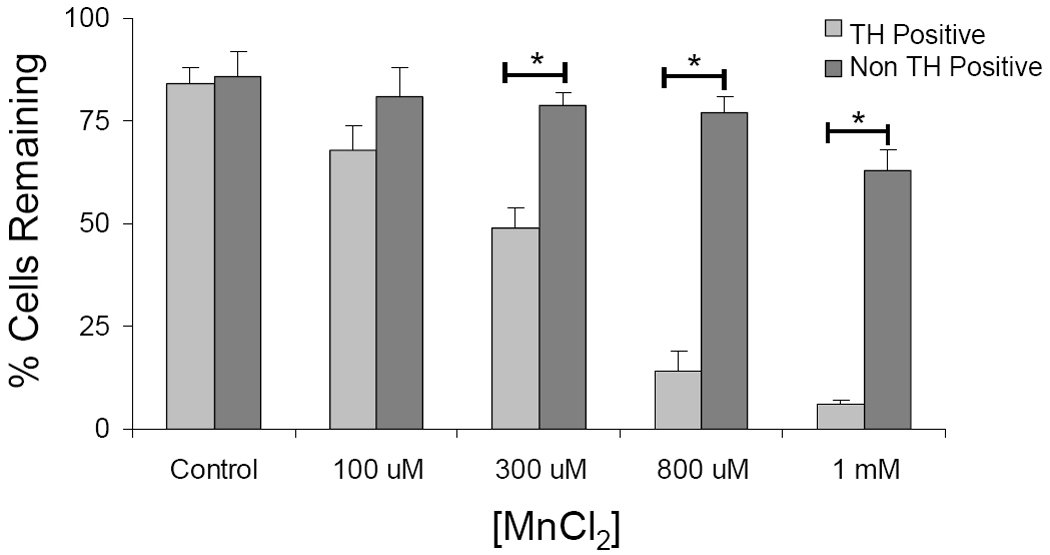
Mesencephalic cultures from transgenic TH:RFP mice were exposed to manganese and the number of TH-positive cells was counted. Data are expressed as a percent loss of TH-positive or TH-negative cells compared to preexposure cell counts in the same quadrants. Data represent the mean + SEM of 4 independent experiments, each performed in at least duplicate. Data was analyzed by two-tailed unpaired t-test. *p<0.05.
We next moved to an in vivo model of chronic manganese exposure to determine if systemic administration of manganese in any way recapitulated alterations in the direct and indirect circuits of the basal ganglia that have been observed in PD and PD models. Animals were given IP injections (5 mg/kg body mass, intraperitoneal) of MnCl2 or vehicle daily for 30 days. Coronal brain sections were stained with TH antibody and cell counts were performed. Representative photomicrographs of control (Figure 5A) or manganese treated (Figure 5B) animals are shown at the level of the SN. Cell counts revealed a significant decrease in TH-positive cells in the SN but not in the adjacent ventral tegmental area (VTA) (Figure 5C; n=6; p< 0.05 unpaired t test).
Figure 5. Chronic Manganese Exposure Decreases Tyrosine Hydroxylase Immunoreactive Neurons in Vivo.
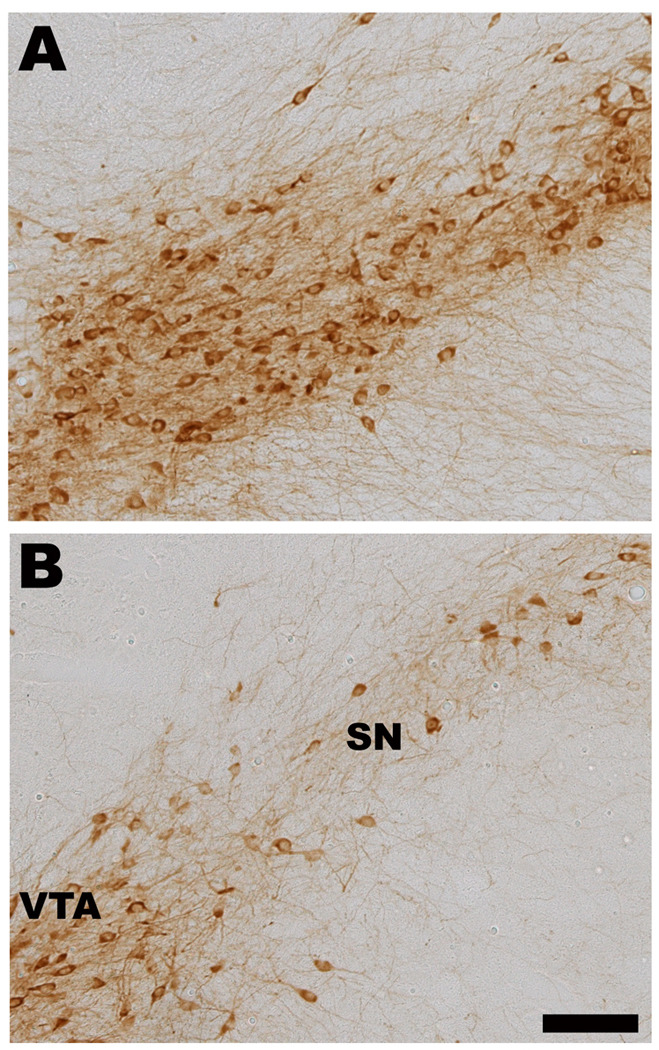
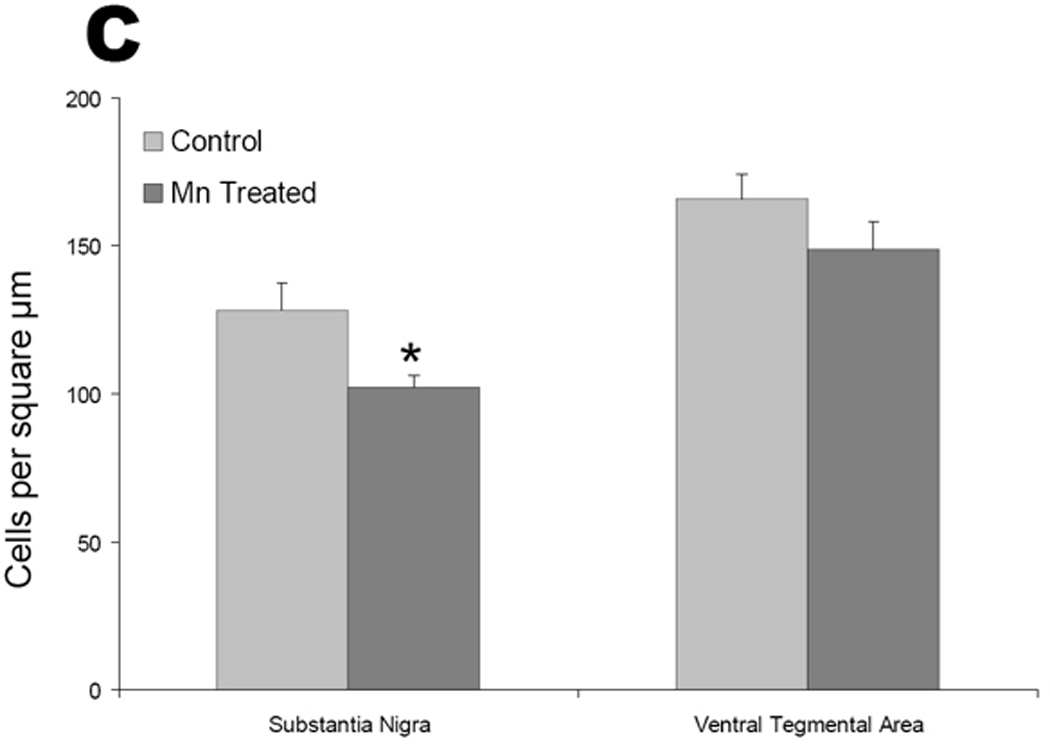
Mice were exposed to IP injections (5 mg/kg body mass) of MnCl2 or vehicle daily for 30 days. Coronal brain sections were stained with TH antibody and cell counts were performed. Photomicrographs of control (A) or MnCl2 treated (B) animals are shown at the level of the ventral midbrain. A particular robust example of TH cell loss following chronic MnCl2 is depicted in panel B. C) Cell counts revealed a significant decrease in TH positive cells in the SN but not in the VTA (n=6; p< 0.05, t test). Scale bar = 100 µm. SN = substantial nigra, VTA = ventral tegmental area, cp = cerebral peduncle, ml = medial lemniscus.
To determine if the loss of TH staining was caused by a loss of the dopamine synthetic enzyme itself or by a loss of cellular viability, we undertook cell counts of the SN and VTA (Figure 6). We found that the loss of TH staining was associated with cell loss in the SN as there was a 20% decrease in cresyl violet-stained cell numbers in this region, which was not evident in the VTA (Figure 6C).
Figure 6. Chronic Manganese Exposure Induces Cell Loss in the Substantia Nigra.

Mice were exposed to IP injections (5 mg/kg body mass) of MnCl2 or vehicle daily for 30 days. Coronal sections of brain were Nissl stained to identify cells and counts were performed within the SN and VTA. Representative photomicrographs of control (A) or manganese treated (B) animals are shown at the level of the SN. Note the lack of tightly compacted cell bodies (arrows in sections of vehicle-treated mice) in MnCl2 treated animals. Cell counts (C) reveal a 20% decrease in total cell density in the SN, which is not evident in the VTA. (n=6; * p< 0.05 unpaired t test.
Our final experiments were designed to evaluate the consequences of chronic manganese exposure on the basal ganglia by evaluating the GABA synthetic enzyme, glutamic-acid-decarboxylase (GAD67), staining within this circuit. We observed appreciable GAD67 loss in the CPu which receives the dopaminergic projections of the SNpc in manganese treated animals (Figure 7B) compared to controls (Figure 7A). GAD67 immunoreactivity was not altered in the SNpr, which was consistent with our in vitro findings suggesting that dopaminergic dysfunction occurs prior to GABAergic dysfunction in this region. We also observed a loss of GAD67 staining in the GP in manganese-treated animals compared to controls (Figures 7C and D). Cell counts of several other regions within and outside the basal ganglia showed no significant difference (Figure 7E), suggesting that the loss of GAD-positive cells was limited to the striatum (STR) and globus pallidus (GP), but not in the dentate gyrus or anterior cingulate cortex (n=6; p< 0.05 unpaired t test).
Figure 7. Chronic Manganese Exposure Decreases GAD Expression in Vivo.

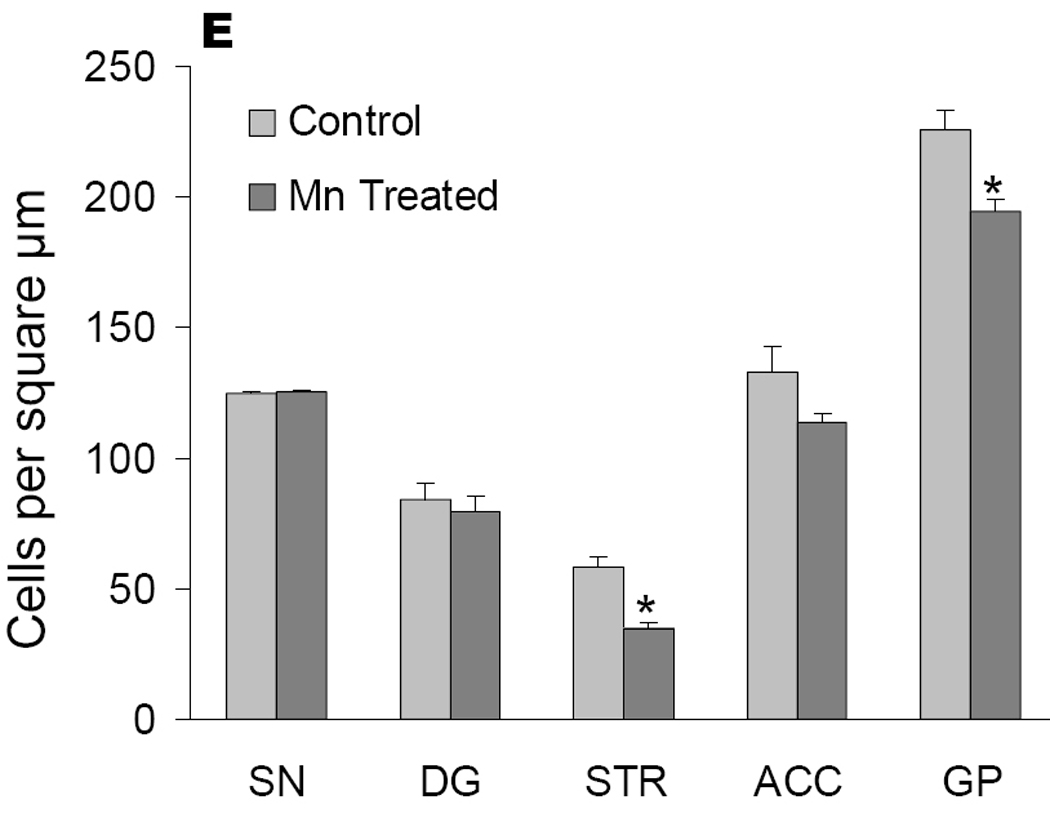
Mice were exposed to IP injections (5 mg/kg body mass) of MnCl2 or vehicle daily for 30 days. Coronal sections of brain were stained with a glutamic-acid-decarboxylase (GAD) reactive antibody and cell counts were performed. Representative photomicrographs from the striatum from control (A) or manganese treated (B) animals are shown. Immunoreactivity of GAD was also decreased in the globus pallidus (control, C) in manganese treated animals (D). Cell counts revealed that the loss of GAD-positive cells was limited to the striatum (STR) and globus pallidus (GP) and was not observed in the substantia nigra (SN), dentate gyrus (DG) or anterior cingulate cortex (ACC) (n=6; p< 0.05, unpaired t test).
DISCUSSION
Manganese concentrations present in mammalian cells can be estimated from the literature. In vivo chronic low-dose Mn exposure leads to accumulation of Mn in the striatal and globus pallidus at concentrations as high as 200 µM (Erikson and Aschner 2002). In addition, weekly injections of Mn over a 3-month period in striatum and GP of monkeys (0, 2.25, 4.5, and 9 gm), produce dose-related clinical signs, with increased severances at the higher dose range (Suzuki et al. 1975). At the highest dose (9 gm), the Mn concentration is increased 12-fold in the striatum and 9-fold in the GP compared to conrtrols. The “physiological range” (no symptoms) approximates Mn levels at 75–100 µM, and clinical signs increase in frequency and severity above this level. Accordingly, for the purpose of the present study, Mn at 100 µM was deemed to be represent a threshold level of toxicity. Concentrations of Mn below this level were deemed non-toxic, while Mn at 800 µM was deemed to represent a toxic levels. Accordingly, we used a low (10µM) and minimally toxic concentrations (100–800µM) in our acute studies. We note, however, that we did directly measure manganese concentrations within the cells. It has been shown previously that cultured cortical astrocytes and undifferentiated PC12 cells are capable of accumulating manganese intracellularly over a period of hours to days. In contrast, however, neuronally-derived NT2 cells and nerve growth factor-differentiated PC12 cells appear to only minimally accumulate Mn2+ above the media concentration (Gunter et al. 2005; Gunter et al. 2006).
Mn induces mitochondrial respiratory dysfunction in vitro, promotes free radical production and inhibits the antioxidant system by depleting glutathione and glutathione peroxidase (Liccione and Maines 1988; Gavin et al. 1992; Chen and Liao 2002; Weber et al. 2002; Stredrick et al. 2004; Erikson et al. 2006; Erikson et al. 2007; Zhang et al. 2007; Zhang et al. 2008). Activation of oxidative stress-sensitive kinases and transcription factors including NF-κB have also been observed in cell lines exposed to manganese (Zhang et al. 2007; Prabhakaran et al. 2008; Yin et al. 2008). Expression changes and inflammatory responses in glial cells can be observed following bath application of manganese at concentrations as low as 10 µM (Moreno et al. 2008).
In the present study, we sought to identify changes in neuronal cytoarchitecture associated with manganese exposure and to determine if these changes are more pronounced in mesencephalic cells. Neurites are rich in mitochondria, which produce the most reactive oxygen species in cells and thus are likely early targets of redox stress. Tau plays an essential role in the stabilization of the microtubule tracks and mutations in tau have been linked to PD pathology. Both tau and synapsin have been shown to be altered in PD and PD models as well as by environmental and genetic stress (Fortin et al. 2005; Ansari et al. 2008), but neither has been evaluated following manganese treatment. Our data support the hypothesis that acute manganese exposure induces early and profound changes in neurite length and integrity at concentrations that are not overtly neurotoxic (100µM). This would suggest that circuit level dysfunction caused by the loss of dopaminergic projections to the striatum may be an early consequence of manganese exposure. Mechanistic overlap between PD and manganese toxicity is also suggested by the recent observation that PARK9 can provide protection both to α-synuclein mutation and manganese exposure (Gitler et al. 2009) and that the divalent metal transporter-1 (DMT1), a transporter of manganese, contributes to neurodegeneration and effects inherent to PD-related cell death (Salazar et al. 2008).
Microtubules play obligatory functions in the maintenance of cellular transport, but overexpression of the tubulin gene or tubulin depolymerization by toxins cause significant damage to the cell, especially in projection neurons with long axons, such as the dopaminergic neurons of the nigrostriatal pathway (Ren et al. 2003). Failure to transport sufficient dopamine containing vesicles via microtubules to synaptic terminals early in PD decreases synaptic plasticity in the striatum (Brown et al., 2005). An inherited form of PD is caused by a mutation in the E3 ubiquitin ligase Parkin that binds to α- and β-tubulins and increases their ubiquitination and subsequent proteasomal degradation. Phosphorylation of Parkin results in the loss of Parkin’s ability to ubiquitinate damaged proteins thereby preventing their proteasomal degradation, leading to the accumulation of tubulin and other misfolded proteins. Parkin mutations also result in the formation of Lewy bodies consisting of β-tubulin and α-synuclein and are linked to the onset of autosomal recessive juvenile PD (Ren et al. 2003). The loss of synapsin staining observed with manganese treatment is consistent with data supporting an essential role for appropriate manganese content in maintaining neurotransmission and synaptic function (Takeda et al. 2002, 2003) and the role of PD associated proteins in regulating synaptic function (Murphy et al. 2000; Sidhu et al. 2004b; Sidhu et al. 2004a). While the underlying signaling pathways remain ill-defined, manganese has been shown to interact with dopamine-reactive quinones and other dopamine specific species, which may contribute to the in vitro pathology of manganese-induced damage in dopaminergic cells. Recent work has shown that manganese toxicity is indeed enhanced by dopamine preexposure in mesencephalic cultures (Prabhakaran et al. 2008), although specific changes in the dopaminergic vs. GABAergic cell populations in these cultures were not evaluated. Our data strongly support a preferential loss of TH-positive neurons in mesencephalic cultures at concentrations of manganese which are not overtly toxic to other populations.
The endogenous pools of manganese have been studied extensively in an effort to determine if the concentration of manganese in discreet anatomical sites correlates with regional injury. Magnetic resonance imaging has demonstrated preferential manganese sequestration in the striatum, GP, and SN of primates (Calne et al. 1994; Shinotoh et al. 1995). In rodent studies, however, there are discrepancies in the literature. Brenneman and colleagues reported that rat CPu and GP do not preferentially accumulate manganese after excess exposure (Brenneman et al. 1999). In other works, accumulation in these regions could be promoted by placing animals on diets deficient in iron implicating iron deficiency as a risk factor for manganese accumulation (Kim et al. 2005).
The neuropathological changes we observed using chronic exposure suggest that manganese toxicity is not limited to dopaminergic neurons, but also occurs in the globus pallidus and striatum. These findings overlap with the pathological changes observed in the scant number of post mortem human tissue samples from manganese cases, where morphological alterations range from no gross changes to massive atrophy and gliosis which appears to be most consistently observed in the internal segment of the globus pallidus. Loss of neurons in the SN has been reported albeit less consistently, but there is a clear need to obtain more specimens, correlate changes with the mode of Mn2+ exposure and resulting motor and cognitive dysfunction as well as measure the manganese levels in tissue (McKinney et al. 2004; Perl and Olanow 2007).
Regarding the overlap between our neurochemical and pathological changes and PD, there appears to be a larger discrepancy. GABA is the major inhibitory neurotransmitter in the brain and the highest concentrations of GABA are found in the internal segment of the globus pallidus and the substantia nigra pars reticulata, which receive inputs from the striatum (caudate nucleus and putamen). Changes in GAD or loss of GABA content are not present in the post mortem striatum in PD, although the changes at circuit level of GABAergic tone will be altered by the hallmark loss of dopamine in early stages of the disease. While the changes in GAD-immunoreactive neuron number in the STR and GP were relatively subtle in our animals chronically exposed to manganese, they were statistically significant. In this regard chronic Mn exposure in this model differs from PD. Similarities between PD and manganism include the presence of generalized bradykinesia and widespread rigidity. Dissimilarities between PD and manganism were also recognized, notably the following in manganism: (a) a less frequent resting tremor, (b) more frequent dystonia, (c) a particular propensity to fall backward, (d) failure to achieve a sustained therapeutic response to levodopa, and (e) failure to detect a reduction in fluorodopa uptake by positron emission tomography (PET; for further details see (Calne et al. 1994). Other rodent models of manganese exposure have also pointed to alterations in GABA and GAD in CPu and GP (Gwiazda et al. 2002; Tomás-Camardiel et al. 2002).
Based on our data, we conclude th at acute in vitro manganese exposure is neurotoxic and produces more profound cytoarchitectural dysfunction and death in TH-positive neurons than other neuronal populations. In our chronic in vivo manganese exposure paradigm, we also observed cell loss in the TH-rich SN, but damage was not solely restricted to this region. The loss of GAD-positive cells further downstream in the basal ganglia circuitry may be caused by the loss of chemical, electrical or physical support due to neurite dysfunction or may be an independent neurotoxic event. Given that culture studies require isolation of embryonic tissue, it is also possible that developmental issues contribute to selective vulnerabilities (see also Erecinska et al. 2005). On net, our data provide some explanation for the motor manifestations of manganese intoxication but do not support a model of selective dopaminergic dysfunction in vivo. These data suggest that circuit level influences are essential to mediating manganese induced degeneration in the adult brain.
Acknowledgements
The authors would like to thank Brent Evans, Joshua Parlaman and Evan Cohen for their assistance in preparing the manuscript and performing several valuable experiments. This work was supported in part by DAMD Grant 17-01-1-0685, NIEHS 10563 and statistical and financial support of NICHD Grant P30HD15052 to the Vanderbilt Kennedy Center for Research on Human Development (GDS, JW, MA and BAM) and for travel grant (JNS).
Abbreviations
- BSA
bovine serum albumin
- CPu
caudate putamen
- DMEM
Dulbecco’s Modified Eagle Medium
- GAD67
glutamic acid decarboxylase, MW 67kDa
- GP
globus pallidus
- LDH
lactate dehydrogenase
- Mn2+
manganese
- MnCl2
manganese chloride
- PD
Parkinson’s Disease
- RFP
red fluorescent protein
- SNpc
substantia nigra pars compacta
- SNpr
substantia nigra pars reticulata
- TH
tyrosine hydroxylase
- VTA
ventral tegmental area
REFERENCES
- Abercrombie M. Estimation of nuclear populations from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. European Journal of Neuroscience. 2005;22:247–256. doi: 10.1111/j.1460-9568.2005.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochimica et Biophysica Acta-Molecular Basis Of Disease. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radical Biology And Medicine. 2008;45:443–452. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Molecular Aspects of Medicine. 2005;26:353. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Manganese Neurotoxicity - Cellular Effects And Blood-Brain-Barrier Transport. Neuroscience And Biobehavioral Reviews. 1991;15:333–340. doi: 10.1016/s0149-7634(05)80026-0. [DOI] [PubMed] [Google Scholar]
- Aschner M, Gannon M. Manganese (Mn) transport across the rat blood-brain barrier: saturable and transferrin-dependent transport mechanisms. Brain Res Bull. 1994;33:345–349. doi: 10.1016/0361-9230(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Aschner M, Vrana KE, Zheng W. Manganese uptake and distribution in the central nervous system (CNS) Neurotoxicology. 1999;20:173–180. [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Dorman DC. Manganese dosimetry: species differences and implications for neurotoxicity. Crit Rev Toxicol. 2005;35:1–32. doi: 10.1080/10408440590905920. [DOI] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochemical Journal. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman KA, Cattley RC, Ali SF, Dorman DC. Manganese-induced developmental neurotoxicity in the CD rat: Is oxidative damage a mechanism of action? Neurotoxicology. 1999;20:477–487. [PubMed] [Google Scholar]
- Calne DB, Chu NS, Huang CC, Lu CS, Olanow W. Manganism and Idiopathic Parkinsonism - Similarities and Differences. Neurology. 1994;44:1583–1586. doi: 10.1212/wnl.44.9.1583. [DOI] [PubMed] [Google Scholar]
- Cappelletti G, Surrey T, Maci R. The parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilizing factor. FEBS Letters. 2005;579:4781–4786. doi: 10.1016/j.febslet.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Liao SL. Oxidative stress involves in astrocytic alterations induced by manganese. Experimental Neurology. 2002;175:216–225. doi: 10.1006/exnr.2002.7894. [DOI] [PubMed] [Google Scholar]
- Crossgrove JS, Allen DD, Bukaveckas BL, Rhineheimer SS, Yokel RA. Manganese distribution across the blood-brain barrier. I. Evidence for carrier-mediated influx of managanese citrate as well as manganese and manganese transferrin. Neurotoxicology. 2003;24:3–13. doi: 10.1016/s0161-813x(02)00089-x. [DOI] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M, Sesma MT, Gimenez-Amaya JM, Ortiz L. Changes in cytoskeletal gene expression linked to MPTP-treatment in mice. Neurobiology of Disease. 2005;20:666–672. doi: 10.1016/j.nbd.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Dobson AW, Erikson KM, Aschner M. Manganese neurotoxicity. Redox-Active Metals In Neurological Disorders. 2004;Vol. 1012:115–128. doi: 10.1196/annals.1306.009. [DOI] [PubMed] [Google Scholar]
- Dobson AW, Weber S, Dorman DC, Lash LK, Erikson KM, Aschner M. Oxidative stress is induced in the rat brain following repeated inhalation exposure to manganese sulfate. Biol Trace Elem Res. 2003;93:113–126. doi: 10.1385/BTER:93:1-3:113. [DOI] [PubMed] [Google Scholar]
- Dorman DC, Brenneman KA, McElveen AM, Lynch SE, Roberts KC, Wong BA. Olfactory transport: a direct route of delivery of inhaled manganese phosphate to the rat brain. J Toxicol Environ Health A. 2002;65:1493–1511. doi: 10.1080/00984100290071630. [DOI] [PubMed] [Google Scholar]
- Dorman DC, McManus BE, Parkinson CU, Manuel CAq, McElveen AM, Everitt JI. Nasal toxicity of manganese sulfate and manganese phosphate in young male rats following subchronic (13-week) inhalation exposure. Inhal Toxicol. 2004;16:481–488. doi: 10.1080/08958370490439687. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Silver IA. ATP and Brain Function. Journal of Cerebral Blood Flow and Metabolism. 1989;9:2–19. doi: 10.1038/jcbfm.1989.2. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Progress in Neurobiology. 2004;73:397–445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Cherian S, Silver IA. Brain development and susceptibility to damage; Ion levels and movements. Current Topics In Developmental Biology. 2005;Vol 69(69):139–186. doi: 10.1016/S0070-2153(05)69006-0. [DOI] [PubMed] [Google Scholar]
- Erikson K, Aschner M. Manganese causes differential regulation of glutamate transporter (GLAST) taurine transporter and metallothionein in cultured rat astrocytes. Neurotoxicology. 2002;23:595–602. doi: 10.1016/s0161-813x(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Dorman DC, Lash LH, Aschner M. Manganese inhalation by rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Toxicological Sciences. 2007;97:459–466. doi: 10.1093/toxsci/kfm044. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Dorman DC, Fitsanakis V, Lash LH, Aschner M. Alterations of oxidative stress biomarkers due to in utero and neonatal exposures of airborne manganese. Biological Trace Element Research. 2006;111:199–215. doi: 10.1385/BTER:111:1:199. [DOI] [PubMed] [Google Scholar]
- Esposito A, Dohm CP, Kermer P, Bahr M, Wouters FS. alpha-synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiology Of Disease. 2007;26:521–531. doi: 10.1016/j.nbd.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Finkelstein Y, Milatovic D, Aschner M. Modulation of cholinergic systems by manganese. NeuroToxicology. 2007;28:1003. doi: 10.1016/j.neuro.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Fitsanakis VA, Erikson KM, Aschner M. Manganese transport in the CNS. Neurotoxicology. 2006a;27:895–896. [Google Scholar]
- Fitsanakis VA, Zhang N, Avison MJ, Gore JC, Aschner JL, Aschner M. The use of magnetic resonance imaging (MRI) in the study of manganese neurotoxicity. NeuroToxicology. 2006b;27:798. doi: 10.1016/j.neuro.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. Journal Of Neuroscience. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nature Cell Biology. 2007;9:139. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Mn2+ Sequestration by Mitochondria and Inhibition of Oxidative-Phosphorylation. Toxicology And Applied Pharmacology. 1992;115:1–5. doi: 10.1016/0041-008x(92)90360-5. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet J-C, Lindquist S. [alpha]-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter KK, Aschner M, Miller LM, Eliseev R, Salter J, Anderson K, Hammond S, Gunter TE. Determining the oxidation states of manganese in PC12 and nerve growth factor-induced PC12 cells. Free Radical Biology And Medicine. 2005;39:164–181. doi: 10.1016/j.freeradbiomed.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gavin CE, Aschner M, Gunter KK. Speciation of manganese in cells and mitochondria: A search for the proximal cause of manganese neurotoxicity. Neurotoxicology. 2006;27:765–776. doi: 10.1016/j.neuro.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Gwiazda RH, Lee D, Sheridan J, Smith DR. Low Cumulative Manganese Exposure Affects Striatal GABA but not Dopamine. NeuroToxicology. 2002;23:69. doi: 10.1016/s0161-813x(02)00002-5. [DOI] [PubMed] [Google Scholar]
- He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, Nebert DW. ZIP8, Member of the Solute-Carrier-39 (SLC39) Metal-Transporter Family: Characterization of Transporter Properties. Mol Pharmacol. 2006;70:171–180. doi: 10.1124/mol.106.024521. [DOI] [PubMed] [Google Scholar]
- Heredia L, Helguera P, de Olmos S, Kedikian G, Vigo FS, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Caceres A, Lorenzo A. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: A potential mechanism of neuronal dystrophy in Alzheimer's disease. Journal Of Neuroscience. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L. Bioenergetics of cerebral ischemia: A cellular perspective. Neuropharmacology. 2008;55:289. doi: 10.1016/j.neuropharm.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L. Energy-Metabolism at the Cellular-Level of the CNS. Canadian Journal of Physiology and Pharmacology. 1992;70:S145–S157. doi: 10.1139/y92-256. [DOI] [PubMed] [Google Scholar]
- Hsieh CT, Liang JS, Peng SS, Lee WT. Seizure associated with total parenteral nutrition-related hypermanganesemia. Pediatr Neurol. 2007;36:181–183. doi: 10.1016/j.pediatrneurol.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Kafritsa Y, Fell J, Long S, Bynevelt M, Taylor W, Milla P. Long-term outcome of brain manganese deposition in patients on home parenteral nutrition. Arch Dis Child. 1998;79:263–265. doi: 10.1136/adc.79.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen CL, Ensunsa JL, Lönnerdal B, Zidenberg-Cherr S, Benjamin C. Encyclopedia of Human Nutrition. Oxford: Elsevier; 2005. Manganese; p. 217. [Google Scholar]
- Kim Y, Park JK, Choi Y, Yoo C-I, Lee CR, Lee H, Lee J-H, Kim S-R, Jeong T-H, Yoon CS, Park J-H. Blood Manganese Concentration is Elevated in Iron Deficiency Anemia Patients, Whereas Globus Pallidus Signal Intensity is Minimally Affected. NeuroToxicology. 2005;26:107. doi: 10.1016/j.neuro.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Lee JW. Manganese intoxication. Arch Neurol. 2000;57:597–599. doi: 10.1001/archneur.57.4.597. [DOI] [PubMed] [Google Scholar]
- Liccione JJ, Maines MD. Selective Vulnerability Of Glutathione Metabolism And Cellular Defense-Mechanisms In Rat Striatum To Manganese. Journal of Pharmacology And Experimental Therapeutics. 1988;247:156–161. [PubMed] [Google Scholar]
- Liu X, Sullivan KA, Madl JE, Legare M, Tjalkens RB. Manganese-Induced Neurotoxicity: The Role of Astroglial-Derived Nitric Oxide in Striatal Interneuron Degeneration. Toxicol. Sci. 2006;91:521–531. doi: 10.1093/toxsci/kfj150. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Veinbergs I, Miller A, Samuel W. Alterations in apolipoprotein E expression during aging and neurodegeneration. Progress In Neurobiology. 1996;50 doi: 10.1016/s0301-0082(96)00038-x. 493-&. [DOI] [PubMed] [Google Scholar]
- Maynard LS, Cotzias GC. The Partition of Manganese Among Organs and Intracellular Organelles of the Rat. J. Biol. Chem. 1955;214:489–495. [PubMed] [Google Scholar]
- McKinney AM, Filice RW, Teksam M, Casey S, Truwit C, Clark HB, Woon C, Liu HY. Diffusion abnormalities of the globi pallidi in manganese neurotoxicity. Neuroradiology. 2004;46:291–295. doi: 10.1007/s00234-004-1179-1. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Nelson D, Erecinska M, Chesselet MF. Toxicity of dopamine to striatal neurons in vitro and potentiation of cell death by a mitochondrial inhibitor. Journal of Neurochemistry. 1998a;70:2406–2415. doi: 10.1046/j.1471-4159.1998.70062406.x. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet M-F. Methylmalonate toxicity in primary neuronal cultures. Neuroscience. 1998b;86:279–290. doi: 10.1016/s0306-4522(97)00594-0. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in ischemic preconditioning. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Sullivan KA, Carbone DL, Hanneman WH, Tjalkens RB. Manganese potentiates nuclear factor-kappa B-dependent: Expression of nitric oxide synthase 2 in astrocytes by activating soluble guanylate cyclase and extracellular responsive kinase signaling pathways. Journal of Neuroscience Research. 2008;86:2028–2038. doi: 10.1002/jnr.21640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. Journal Of Neuroscience. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. Journal of Neurochemistry. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatomo S, Umehara F, Hanada K, Nobuhara Y, Takenaga S, Arimura K, Osame M. Manganese intoxication during total parenteral nutrition: report of two cases and review of the literature. J Neurol Sci. 1999;162:102–105. doi: 10.1016/s0022-510x(98)00289-5. [DOI] [PubMed] [Google Scholar]
- Ovadi J, Orosz F, Hollan S. Functional aspects of cellular micro-compartmentation in the development of neurodegeneration: Mutation induced aberrant protein-protein associations. Molecular And Cellular Biochemistry. 2004;256:83–93. doi: 10.1023/b:mcbi.0000009860.86969.72. [DOI] [PubMed] [Google Scholar]
- Perl DP, Olanow CW. The neuropathology of manganese-induced parkinsonism. Journal of Neuropathology And Experimental Neurology. 2007;66:675–682. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- Prabhakaran K, Ghosh D, Chapman GD, Gunasekar PG. Molecular mechanism of manganese exposure-induced dopaminergic toxicity. Brain Research Bulletin. 2008;76:361–367. doi: 10.1016/j.brainresbull.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin Binds to alpha /beta Tubulin and Increases their Ubiquitination and Degradation. J. Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. roceedings Of The National Academy Of Sciences Of The United States Of America. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinotoh H, Snow BJ, Hewitt KA, Pate BD, Doudet D, Nugent R, Perl DP, Olanow W, Calne DB. MRI And Pet Studies Of Manganese-Intoxicated Monkeys. Neurology. 1995;45:1199–1204. doi: 10.1212/wnl.45.6.1199. [DOI] [PubMed] [Google Scholar]
- Siddappa AJ, Rao RB, Wobken JD, Leibold EA, Connor JR, Georgieff MK. Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J Neurosci Res. 2002;68:761–775. doi: 10.1002/jnr.10246. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P. Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? Faseb Journal. 2004a;18:637–647. doi: 10.1096/fj.03-1112rev. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Moussa CEH, Vernier P. The role of alpha-synuclein in both neuroprotection and neurodegeneration. Protective Strategies For Neurodegenerative Diseases. 2004b;1035:250–270. doi: 10.1196/annals.1332.016. [DOI] [PubMed] [Google Scholar]
- Siman R, McIntosh TK, Soltesz KM, Chen ZM, Neumar RW, Roberts VL. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiology Of Disease. 2004;16:311–320. doi: 10.1016/j.nbd.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Levitt P. Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cereb Cortex. 2001;11:430–440. doi: 10.1093/cercor/11.5.430. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Parlaman JP, Levitt P. Anatomical abnormalities in dopaminoceptive regions of the cerebral cortex of dopamine D(1) receptor mutant mice. J Comp Neurol. 2005;487:270–282. doi: 10.1002/cne.20548. [DOI] [PubMed] [Google Scholar]
- Stredrick DL, Stokes AH, Worst TJ, Freeman WM, Johnson EA, Lash LH, Aschner M, Vrana KE. Manganese-induced cytotoxicity in dopamine-producing cells. Neurotoxicology. 2004;25:543–553. doi: 10.1016/j.neuro.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Mouri T, Suzuki Y, Nishiyama K, Fujii N. Study of subacute toxicity of manganese dioxide in monkeys. Tokushima J Exp Med. 1975;22:5–10. [PubMed] [Google Scholar]
- Takagi Y, Okada A, Sando K, Wasa M, Yoshida H, Hirabuki N. On-off study of manganese administration to adult patients undergoing home parenteral nutrition: new indices of in vivo manganese level. JPEN J Parenter Enteral Nutr. 2001;25:87–92. doi: 10.1177/014860710102500287. [DOI] [PubMed] [Google Scholar]
- Takeda A. Manganese action in brain function. Brain Research Reviews. 2003;41:79. doi: 10.1016/s0165-0173(02)00234-5. [DOI] [PubMed] [Google Scholar]
- Takeda A, Sotogaku N, Oku N. Manganese influences the levels of neurotransmitters in synapses in rat brain. Neuroscience. 2002;114:669–674. doi: 10.1016/s0306-4522(02)00353-6. [DOI] [PubMed] [Google Scholar]
- Takeda A, Sotogaku N, Oku N. Influence of manganese on the release of neurotransmitters in rat striatum. Brain Research. 2003;965:279–282. doi: 10.1016/s0006-8993(02)04157-4. [DOI] [PubMed] [Google Scholar]
- Tomás-Camardiel M, Herrera AJ, Venero JL, Cruz Sánchez-Hidalgo M, Cano J, Machado A. Differential regulation of glutamic acid decarboxylase mRNA and tyrosine hydroxylase mRNA expression in the aged manganese-treated rats. Molecular Brain Research. 2002;103:116. doi: 10.1016/s0169-328x(02)00192-4. [DOI] [PubMed] [Google Scholar]
- Wang XS, Ong WY, Connor JR. A light and electron microscopic study of divalent metal transporter-1 distribution in the rat hippocampus, after kainate-induced neuronal injury. Exp Neurol. 2002;177:193–201. doi: 10.1006/exnr.2002.7962. [DOI] [PubMed] [Google Scholar]
- Weber S, Dorman DC, Lash LH, Erikson K, Vrana KE, Aschner M. Effects of manganese (Mn) on the developing rat brain: Oxidative-stress related endpoints. Neurotoxicology. 2002;23:169–175. doi: 10.1016/s0161-813x(02)00014-1. [DOI] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit A, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. Journal of Neuroscience. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MTT. Cytochrome-Oxidase - An Endogenous Metabolic Marker For Neuronal-Activity. Trends In Neurosciences. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- Yin ZB, Aschner JL, dos Santos AP, Aschner M. Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Research. 2008;1203:1–11. doi: 10.1016/j.brainres.2008.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Derr-Yellin E, Nicklas WJ. Relative vulnerability of dopamine and GABA neurons in mesencephalic culture to inhibition of succinate dehydrogenase by malonate and 3- nitropropionic acid and protection by NMDA receptor blockade. J Pharmacol Exp Ther. 1995;275:1124–1130. [PubMed] [Google Scholar]
- Zhang DQ, Stone JF, Zhou TR, Ohta H, McMahon DG. Characterization of genetically labeled catecholamine neurons in the mouse retina. Neuroreport. 2004;15:1761–1765. doi: 10.1097/01.wnr.0000135699.75775.41. [DOI] [PubMed] [Google Scholar]
- Zhang FL, Xu ZF, Gao J, Xu B, Deng Y. In vitro effect of manganese chloride exposure on energy metabolism and oxidative damage of mitochondria isolated from rat brain. Environmental Toxicology and Pharmacology. 2008;26:232–236. doi: 10.1016/j.etap.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Zhang P, Hatter A, Liu B. Manganese chloride stimulates rat microglia to release hydrogen peroxide. Toxicology Letters. 2007;173:88–100. doi: 10.1016/j.toxlet.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]