

Abstract
Modularity is a highly sought after feature in engineering design. A modular catalyst is a multi-component system whose parts can be predictably interchanged for functional flexibility and variety. Nearly two decades after the discovery of the first modular polyketide synthase (PKS), we critically assess PKS modularity in the face of a growing body of atomic structural and in vitro biochemical investigations. Both the architectural modularity and the functional modularity of this family of enzymatic assembly lines are reviewed, and the fundamental challenges that lie ahead for the rational exploitation of their full biosynthetic potential are discussed.
Modularity is a highly sought after feature in engineering design. Large-scale integrated circuits, automobile assembly lines and multipurpose chemical plants are just some examples of the power of modular engineering. (In contrast, notwithstanding its exquisite engineering elegance, a Swiss watch is not known for its modularity.) A modular system may be defined as a multi-component system that can be divided into smaller subsystems, which interact with each other and can be predictably interchanged for functional flexibility and variety. The two highlighted words merit particular attention as one considers the modularity of catalysts.
Guided by the above definition, the functional flexibility of a catalyst would either refer to its range of chemical transformations or the scope of its substrate tolerance. In the chemical catalysis literature, the term “modular” is frequently used, although it most often refers to the preparative modularity of a catalyst. (Unlike proteins, the synthesis of man-made catalysts is not necessarily governed by modular principles.) A few man-made catalysts with modular reactive or molecular recognition features are known. In such cases, modular reactivity is achieved by swapping the metal center of an organometallic catalyst [1]. Alternatively, modular substrate range results from systematically altering a particular feature of the ligand structure [2]. In practice, this kind of predictable functional modularity invariably encounters serious limitations due to the intimate interplay between the metal and ligand. The ability to fully decouple catalysis from recognition as is, for example, the case for the ultimate catalytic machine, the ribosome, remains a lofty but elusive goal for man-made catalysts.
Against this general backdrop, it is worth reassessing modular polyketide synthases (PKSs), a family of multifunctional catalysts that has received much attention owing to their ability to synthesize a seemingly endless variety of complex natural products [3,4]. As implied in the above definition, it is the prospect of tapping into the functional modularity of these megasynthases (not merely their architectural modularity) that makes them attractive targets for engineering. Schemes 1 and 2 illustrate two different forms of functional modularity that one desires in a modular PKS. In both schemes E1, E2, E3 and E4 are sequentially acting catalysts, and B, C and D are intermediates in the polyketide biosynthetic pathway. In Scheme 1, E2 is replaced with a different catalyst E2′ to alter a targeted functional group or stereocenter without affecting the rest of the natural product or the PKS turnover rate. Examples include replacing a ketoreductase (KR) domain of a PKS module with (i) a KR having different stereospecificity [5], or (ii) a tridomain comprised of a ketoreductase, dehydratase (DH) and enoylreductase [6], or (iii) an aminotransferase (AMT) for reductive amination of the β-ketoacyl intermediate (not yet demonstrated).
Scheme 1.

Scheme 2.

Scheme 2 illustrates a distinct modular principle, in which E1 and E2 are replaced by E1′ and E2′, respectively, so as to accommodate the processing of an alternative functional group or stereocenter (introduced via the substrate A′) without affecting the PKS turnover rate. Examples include regiospecific introduction of unnatural primer [7–9] or extender units [10,11] into a polyketide backbone.
It should be noted that in both of the above schemes, wild-type E3 and E4 have adequately broad substrate tolerance so as to process C′ and D′ with chemical and kinetic fidelity. Hence, modularity is not required of these PKS components. Below we review our current knowledge of the architectural modularity of a modular PKS, followed by a critical assessment of its functional modularity as exemplified in Schemes 1 and 2.
ARCHITECTURAL MODULARITY
The depiction of modular PKS-catalyzed biosynthetic pathways through schemes as in Figure 1 has become common practice. The derivation of such schemes rests upon analysis of the primary sequence of the modular PKS, which reliably identifies individual catalytic centers, generally known as domains, within the megasynthase. The organization of these catalytic domains into modules is governed by the principle that, insofar as possible, active sites clustered along the covalent polypeptide backbone are responsible for catalyzing successive reactions in the polyketide biosynthetic pathway and vice versa. While this approach reliably maps individual enzymes from the “assembly line” onto corresponding transformations in the biosynthetic pathway, its oversimplification precludes any description of limitations to the architectural (or functional) modularity of the PKS. There are a significant number of examples, for instance, of apparently competent catalytic KR or DH domains that clearly are functionally silent, based on the structure of the resulting polyketide natural product. Unfortunately, there are as yet no clear guidelines for identifying such catalytically silent domains absent knowledge of product structure.
Figure 1. Modular organization of 6-deoxyerythronolide B synthase (DEBS).

Chain elongation occurs minimally through the combined action of the ketosynthase (KS), acyl transferase (AT), and acyl carrier protein (ACP) domains. The final oxidation state of the ®-carbon is controlled by the specific combination of ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) domains present in a given module. Once processed, the polyketide chain is either passed to the KS domain of the downstream module or cyclized and released by the thioesterase (TE) domain at the C-terminus of the polyketide synthase. The loading didomain (LDD) is responsible for the selection and subsequent loading of the appropriate priming unit. KR°, inactive ketoreductase domain.
Within the past few years, several high-resolution structures of prototypical components of the 6-deoxyerythronolide B synthase (DEBS) have been solved [12••, 13••,14••,15•,16]. Together, these structures have facilitated the assembly of an atomic structure model of a typical PKS module (Figure 2). Not surprisingly, this model bears a close resemblance to the experimentally determined atomic structure model of the mammalian fatty acid synthase [17]. At the onset, it must be recognized that such models present a snapshot of the PKS (or fatty acid synthase) as opposed to a motion picture of the megasynthase during its catalytic cycle. It is conceivable that conformational dynamics, both within and between domains, are critical to PKS function [18,19,20••,21•,22•,]. Notwithstanding this caveat, available atomic models provide a relatively clear picture of the architectural modularity of a PKS, in conjunction with other experimental information. In particular, using an engineering analogy, three architecturally relevant questions can now be addressed. First, can the assembly line be deconstructed into structurally intact subsystems? Second, to what extent are sub-systems from heterologous sources architecturally compatible with each other? And finally, how universal are the connectors linking subsystems that we wish to interchange in order to achieve functional flexibility?
Figure 2. Composite atomic model of DEBS module 5.

Orange, N-terminal coiled coil linker that facilitates docking between modules 4 and 5; blue, ketosynthase (KS) domain; green, acyltransferase (AT) domain; yellow, KS-AT linker; red, AT-KR linker; cyan, ketoreductase (KR) domain; magenta, acyl carrier protein (ACP) domain. (Only a single ACP domain is shown for clarity.) The sequences (colored by domain/linker of origin) of junctions at which the KS, AT, KR and ACP domains can be deconstructed into stand-alone proteins, or alternatively recombined, are indicated with black arrows. The latter half of the AT-KR linker (red, residues 902–911) and the KR-ACP linker (black, residues 1360–1377) are not shown, as there is no reliable data based on which they can be modeled. The relative orientation of the ACP and KS domains is predicted by in silico docking analysis, whereas the relative orientation of the KR domains and the remainder of the module is based on the X-ray structure of homologous porcine fatty acid synthase [17].
It has been known for some time now that individual PKS modules retain structural and functional integrity as stand-alone, yet still multifunctional proteins [23,24]. More recently, it has also become clear that the ketosynthase (KS), acyltransferase (AT), acyl carrier protein (ACP), KR and DH domains of a PKS module all retain structural and catalytic integrity as stand-alone proteins so long as the domain boundaries have been judiciously selected [16,25,26,27••,]. (Note that “catalytic integrity” here refers to the intrinsic reactivity of individual active sites. Domain disconnection markedly reduces the turnover number of the module (Figure 3), which is of course a property of the full system.) Therefore, assuming that an intact ER domain can also be disconnected into a stand-alone protein, the domains and modules of greatest interest to a PKS engineer can be regarded as architecturally autonomous subsystems.
Figure 3. Relative turnover rates of alternatively deconstructed versions of DEBS module 3.

Domains and linker regions are colored as in Figure 2. The relative turnover number of the intact module (first entry) is compared with that of a derivative in which: (i) a stand-alone ACP is used (second entry); (ii) the KS and AT are discrete proteins (third entry); or (iii) an analog of (ii) lacking the post-AT linker (fourth entry). In the third and fourth entries, only the KS is shown as a dimer.
In silico analysis of the architecture of a typical PKS module (e.g. Figure 2) also suggests that the KS, AT and KR domains do not directly contact each other. Direct interactions involving these domains are limited to contacts with inter-domain linkers and, of course, with the mobile ACP domain. We speculate that heterologous ACP domains are architecturally compatible with KS, AT and KR domains. Our assumption is based on the fact that ACP-KS, ACP-AT and ACP-KR interactions are weak (but functionally important; vide infra), as well as on the extensively documented structural and functional integrity of a hybrid module in which the ACP of DEBS module 5 has been replaced by its counterpart from module 6 [24,28]. If so, then the question of whether heterologous combinations of catalytically active domains are architecturally compatible is less important than whether the individual domains are compatible with their scaffolding linkers.
Considerably less attention has been focused thus far on understanding the universality of the connectors that link catalytically active domains. Indeed, the very existence of these linkers is essentially ignored in commonly used cartoon representations of modular PKSs such as that in Figure 1. As seen in Figure 2, however, these linkers comprise a significant fraction of the total protein in a PKS module and have well defined architectural features of their own. Some linkers, such as the N-terminal coiled coil linker of module 5, do not engage in significant protein-protein interactions with the remainder of the module and not only can be expressed as stand-alone proteins [29••] but also can be interchanged without loss of function [24,30]. These connectors appear to be truly universal. In contrast, connectors such as the KS-to-AT linker or the AT-to-KR linker may be universal or context dependent. On one hand, they engage in extensive interactions with adjacent catalytic domains; on the other, a majority of such non-covalent interactions involve residues that are highly conserved across different modules [13••]. This aspect of the architectural modularity of a PKS module remains to be explored. If the recombination of catalytic domains and linkers from heterologous sources results in perturbed protein-protein interactions, then the PKS turnover rate will likely suffer even if individual active sites are well folded and retain their intrinsic catalytic properties. A vivid example is illustrated in the third and fourth entries in Figure 3, which summarize the adverse consequence of deleting the AT-to-KR linker (shown in red) on the rate of chain elongation catalyzed by the KS and ACP domains [27••]. Experiments suggest that this impairment is not due to changes in the structure or reactivity of individual domains. Rather, it highlights the potentially critical role of the AT-to-KR linker in facilitating important protein-protein interactions between the ACP and the paired KS and AT domains.
In summary, whereas the complete modules of a multimodular PKS can be accurately viewed as architecturally modular sub-systems connected by universal linkers, the architectural modularity of individual modules remains to be fully explored. To do so, it will be essential to understand at an atomic level the structural implications of recombining heterologous catalytic domains and their scaffolding linkers at an atomic level.
FUNCTIONAL MODULARITY
The sub-systems of a digital signaling device may be architecturally modular, and the connectors that interface these sub-systems may also be universal. However, if the replacement of a sub-system with a functionally equivalent unit results in impedance mismatching, signal transfer across the modified interface is incomplete and some of the signal is reflected back. By analogy, architectural modularity of a modular PKS is a necessary but of itself insufficient condition for the design of kinetically competent hybrid PKSs. Developing methods for accurately predicting the scope and limitations to the functional modularity of PKS domains and modules, as illustrated in Schemes 1 and 2, is arguably the most far-reaching goal for this field of research.
Thus far, the predominant method for exploring functional modularity has been through the construction and in vivo (qualitative) product analysis of domain- or module-swapped PKSs. Notwithstanding the conceptual simplicity and power of this approach, it has limited ability to differentiate between kinetically robust versus attenuated hybrid PKSs and even lesser capacity to dissect the nature of the kinetic bottleneck in an unproductive hybrid PKS. More recently, it has been possible to analyze biochemically the properties of hybrid modules and multimodular PKSs [31,32••], which in turn has provided useful insights into the nature of the problems and how they may be surmounted. Perhaps not surprisingly, many of the early chimeric PKS modules were kinetically impaired due to the selection, largely based on analysis of multiple alignments of primary sequence, of what have turned out to be suboptimal fusion junctions between heterologous domains and linkers (for example, see [31]). With the emergence of high-resolution structural data, it is now possible to exploit the architectural modularity of PKSs based on direct structural knowledge of domain junctions in the design of structurally robust chimeric modules and multi-modules. Therefore, we will highlight the major outstanding challenges for understanding and predictably exploiting the functional modularity of PKSs through three illustrative examples below.
As shown in Figure 1, module 3 of DEBS lacks a functional KR domain. Thus, introducing KR activity into this module without compromising its overall turnover rate constitutes a good test of the functional modularity outlined in Scheme 1. Chen et al. attempted to do so by fusing the KS-AT didomain of module 3 to a heterologous KR-ACP didomain at a junction that maintains the module’s architectural integrity [32••]. To do so would require effective collaboration between a KS and an ACP derived from different modules. Therefore, they first evaluated the specificity of the KS domain of module 3 for ACP domains from modules 1, 2, 5 and 6 (all of which bear a functional KR). Surprisingly, a 1000-fold range in reaction rate was observed in the catalysis of C-C bond formation (Figure 4). Because ACP5 had the closest kcat and kcat/KM to those of ACP3, a chimeric module was designed in which KS-AT from module 3 was fused to KR-ACP from module 5. Not only did the resulting hybrid module have a kcat 70% that of the wild-type module, but it also catalyzed reduction of the corresponding β-ketoacyl biosynthetic intermediate. While this example illustrates the potential for functional domain interchange, it could be argued that it falls short of the above definition of modularity because the selection of the KR-ACP didomain from module 5 required trial-and-error evaluation of several ACP domains harboring functional KR neighbors. Predictable interchange of subsystems in this case would require the identification of a common “epitope” on all ACP domains of PKS modules that dictates KS-ACP specificity so as to enable selection of the appropriate KR-ACP didomain based on in silico sequence or structure analysis. Some progress towards the identification of such an epitope has been made in related (Type II) PKSs and fatty acid synthases [33,34]; however, a general solution to this problem has not yet been proposed in the context of modular PKSs. Importantly, it must be recognized that this epitope does more than simply facilitate specific KS-ACP recognition but profoundly influences the transition state of the C-C bond forming reaction, as exemplified by the dramatically different values of kcat for C-C bond formation in the presence of alternative ACPs [32••].
Figure 4. Steady-state kinetic analysis of chain elongation by the KS domain of DEBS module 3 in the presence of five different ACP domains from DEBS.

A 1000-fold variation in specificity is observed, with both kcat and kcat/KM values decreasing as the natural ACP domain (ACP3) is replaced by the ACP domains from modules 1, 2, 5 and 6 of DEBS. Data taken from [32••].
Another aspect of the functional modularity of a modular PKS that is still incompletely understood is the flexibility for regiospecific introduction of an unnatural extender unit. Because extender unit acyl transfer onto the ACP is not typically rate-limiting within the catalytic cycle of a given module, the challenge is two-fold. On one hand the need to maintain the architectural integrity of the module is paramount as one interchanges (or engineers) AT domains (see, for example, Figure 3). On the other hand, downstream active sites may also require exchange (or engineering) in order to accommodate the unnatural extender unit (Scheme 2). The architectural modularity of a PKS module is discussed above. We simply note that at least two alternatives to AT domain interchange have been proposed that minimize architectural perturbations in the PKS module. In one approach, the substrate binding pocket of the AT domain is mutated to alter its specificity [11,35]. In another, the AT domain is inactivated and complemented with a co-expressed acyl transferase with the desired specificity [36]. In contrast, no satisfactory approaches have yet emerged for exploiting PKS modularity in order to process the unnatural extender unit through downstream active sites without an appreciable kinetic penalty. For example, a two- to four-fold reduction in C-C bond formation rates was observed in the case of two DEBS KS domains when the natural nucleophilic substrate, methylmalonyl-ACP, was replaced by the unnatural analog malonyl-ACP [32••]. Similarly, a 6-fold reduction in kinetic specificity constant was observed in chain transfer between modules, when the incoming polyketide chain harbored an unnatural α-carbon substituent [37]. Developing predictable strategies for alleviating these kinetic penalties will require a deeper understanding of the structural basis for KS domains to discriminate between subtly different substrates, something that is beyond the scope of available structural models.
The challenges that lie ahead for understanding and exploiting the functional modularity of modular PKSs are perhaps best exemplified by the especially fascinating (but vexing) example of DEBS module 2. Table 1 shows the specificity of this module for accepting and elongating seven diketides and triketides [38••]. Remarkably, whereas certain substrates are transferred onto the KS active site with extremely high efficiency, the KS is still incapable of catalyzing their elongation. (A similar feature has also been observed with module 3 of the pikromycin synthase [39]. Thus it appears that the very same KS can exhibit entirely different specificity for the two successive chemical reactions that it catalyzes, self-acylation and decarboxylative condensation. How does one take this Janus-like behavior into account as one exchanges KS domains or even entire modules? Clearly it is not possible to divide the same active site into subsystems that can be separately interchanged to alleviate kinetic bottlenecks in chain transfer versus chain elongation. Presumably, the trick will lie in predicting which heterologous KS domains have the most desirable combination of recognition features for a given situation, although it remains to be seen how this will be accomplished.
Table 1.
Relative rates of chain transfer to the KS domain of DEBS module 2, and chain elongation by this module of alternative diketide and triketide analogs.
| Substrate | Relative rate of chain transfer | Relative rate of chain elongation |
|---|---|---|
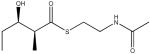 |
1 | 1 |
 |
0.21 | 0.01 |
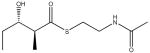 |
0.16 | 0 |
| 7.6 | 1.5 | |
| 0.48 | 0.2 | |
| 1.9 | 0 | |
| 9.8 | 0 |
CONCLUSION
In summary, the foundational knowledge for the assessment and rational exploitation of the functional modularity of modular PKSs is only just beginning to emerge, even for a prototypical PKS such as DEBS. Given the embarrassment of riches that awaits exploitation within nature’s vast repertoire of modular PKSs, one clearly cannot wait for a case-by-case dissection of individual PKS structures and properties. The challenge therefore lies in the accurate identification of those features of a PKS that are functionally modular, and in the development of predictive algorithms that exploit this modularity. The old serenity prayer, “give us grace to accept the things that cannot be changed, courage to change the things that can be changed, and the wisdom to distinguish one from the other” is perhaps more applicable to modular PKSs than any other family of biochemical machines discovered in recent times!
Acknowledgments
This work has been supported by grants from the NIH (CA 66736 to C.K. and GM 22172 to D.E.C.) and a Stanford Graduate Fellowship to S.K. A more detailed description and coordinates of the atomic model of module 5 of DEBS (Figure 2) are available upon request.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Yoon TP, Jacobsen EN. Privileged chiral catalysts. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 2.Burk MJ. Modular Phospholane Ligands in Asymmetric Catalysis. Acc Chem Res. 2000;33:363–372. doi: 10.1021/ar990085c. [DOI] [PubMed] [Google Scholar]
- 3.McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci U S A. 1999;96:1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol. 2005;23:1171–1176. doi: 10.1038/nbt1128. [DOI] [PubMed] [Google Scholar]
- 5.Kao CM, McPherson M, McDaniel R, Fu H, Cane DE, Khosla C. Alcohol stereochemistry in polyketide backbones is controlled by the beta-ketoreductase domains of modular polyketide synthases. J Am Chem Soc. 1998;120:2478–2479. [Google Scholar]
- 6.Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. Gain of Function Mutagenesis of the Erythromycin Polyketide Synthase. 2. Engineered Biosynthesis of an Eight-Membered Ring Tetraketide Lactone. J Am Chem Soc. 1997;119:11339–11340. [Google Scholar]
- 7.Kuhstoss S, Huber M, Turner JR, Paschal JW, Rao RN. Production of a novel polyketide through the construction of a hybrid polyketide synthase. Gene. 1996;183:231–236. doi: 10.1016/s0378-1119(96)00565-3. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsen JR, Hutchinson CR, Cane DE, Khosla C. Precursor-directed biosynthesis of erythromycin analogs by an engineered polyketide synthase. Science. 1997;277:367–369. doi: 10.1126/science.277.5324.367. [DOI] [PubMed] [Google Scholar]
- 9.Marsden AF, Wilkinson B, Cortes J, Dunster NJ, Staunton J, Leadlay PF. Engineering broader specificity into an antibiotic-producing polyketide synthase. Science. 1998;279:199–202. doi: 10.1126/science.279.5348.199. [DOI] [PubMed] [Google Scholar]
- 10.Oliynyk M, Brown MJ, Cortes J, Staunton J, Leadlay PF. A hybrid modular polyketide synthase obtained by domain swapping. Chem Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- 11.Reeves CD, Murli S, Ashley GW, Piagentini M, Hutchinson CR, McDaniel R. Alteration of the substrate specificity of a modular polyketide synthase acyltransferase domain through site-specific mutations. Biochemistry. 2001;40:15464–15470. doi: 10.1021/bi015864r. [DOI] [PubMed] [Google Scholar]
- 12••.Keatinge-Clay AT, Stroud RM. The structure of a ketoreductase determines the organization of the beta-carbon processing enzymes of modular polyketide synthases. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. This paper reports the crystal structure of the region of DEBS module 1 that spans the AT and ACP domains. It includes the KR domain and a previously unclassified “linker”; the latter turns out to be a structural subdomain of the KR. The paper facilitates redefinition of the boundaries of the KR domain. [DOI] [PubMed] [Google Scholar]
- 13••.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. The high resolution structure of the [KS][AT] didomain of DEBS module 5 revealed the architectural organization of the core domains of all PKS modules and offered an atomic level insight into the specific role played by the different classes of linkers in mediating interdomain interactions. The structure also demonstrated that the prevailing “swinging arm” model for interaction between ACP and partner domains is inadequate, thereby highlighting a need for large conformational changes during the PKS catalytic cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 2007;16:2093–2107. doi: 10.1110/ps.073011407. The paper reported the solution NMR structure of the ACP domain of DEBS module 2. Its close sequence identity to other ACP domains of DEBS enabled the authors to compare and contrast the electrostatic and steric differences within this set of ACP domains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Structural and Mechanistic Analysis of Protein Interactions in Module 3 of the 6-Deoxyerythronolide B Synthase. Chem Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. This paper reports the crystal structure of the [KS][AT] fragment of DEBS module 3 bound to a covalently bound inhibitor to enable clearer visualization of the KS active site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keatinge-Clay A. Crystal Structure of the Erythromycin Polyketide Synthase Dehydratase. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 18.Li Q, Khosla C, Puglisi JD, Liu CW. Solution structure and backbone dynamics of the holo form of the frenolicin acyl carrier protein. Biochemistry. 2003;42:4648–4657. doi: 10.1021/bi0274120. [DOI] [PubMed] [Google Scholar]
- 19.Findlow SC, Winsor C, Simpson TJ, Crosby J, Crump MP. Solution structure and dynamics of oxytetracycline polyketide synthase acyl carrier protein from Streptomyces rimosus. Biochemistry. 2003;42:8423–8433. doi: 10.1021/bi0342259. [DOI] [PubMed] [Google Scholar]
- 20••.Koglin A, Mofid MR, Lohr F, Schafer B, Rogov VV, Blum MM, Mittag T, Marahiel MA, Bernhard F, Dotsch V. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–276. doi: 10.1126/science.1122928. Showed that some protein-protein interactions between carrier protein domains and partner domains in non-ribosomal peptide synthetases (NRPSs, a related enzymatic assembly line) enabled the selection of one conformation of the carrier protein from a pre-existing equilibrium, highlighting the dynamic nature of these domains. [DOI] [PubMed] [Google Scholar]
- 21•.Frueh DP, Arthanari H, Koglin A, Vosburg DA, Bennett AE, Walsh CT, Wagner G. Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature. 2008;454:903–906. doi: 10.1038/nature07162. This paper reported the structures of the dynamic complexes between apo carrier protein domain and the thioesterase domain of the EntF NRPS subunit of enterobactin synthetase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Koglin A, Lohr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Guntert P, Wagner G, Walsh CT, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. doi: 10.1038/nature07161. This paper reported the structures of dynamic complexes between a carrier protein domain and a stand-alone type II thioesterase (from the third module of tyrocidine A synthetase and surfactin synthetase systems respectively), thereby highlighting the dynamic nature of these domains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aparicio JF, Caffrey P, Marsden AF, Staunton J, Leadlay PF. Limited proteolysis and active-site studies of the first multienzyme component of the erythromycin-producing polyketide synthase. J Biol Chem. 1994;269:8524–8528. [PubMed] [Google Scholar]
- 24.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Dissecting and exploiting intermodular communication in polyketide synthases. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 25.Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Reconstituting modular activity from separated domains of 6-deoxyerythronolide B synthase. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]
- 26.Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, et al. Molecular basis of Celmer’s rules: stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 27••.Chen AY, Cane DE, Khosla C. Structure-Based Dissociation of a Type I Polyketide Synthase Module. Chem Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. This paper reported the complete deconstruction of DEBS module 3, which facilitated examination of the relative importance of specific domain-domain and domain-linker interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kao CM, Luo G, Katz L, Cane DE, Khosla C. Manipulation of macrolide ring size by directed mutagenesis of a modular polyketide synthase. J Am Chem Soc. 1995;117:9105–9106. [Google Scholar]
- 29••.Weissman KJ. The structural basis for docking in modular polyketide biosynthesis. Chembiochem. 2006;7:485–494. doi: 10.1002/cbic.200500435. The paper reports the solution NMR structure of the docking domain that non-covalently connects modules 4 and 5 of DEBS. [DOI] [PubMed] [Google Scholar]
- 30.Tsuji SY, Cane DE, Khosla C. Selective protein-protein interactions direct channeling of intermediates between polyketide synthase modules. Biochemistry. 2001;40:2326–2331. doi: 10.1021/bi002463n. [DOI] [PubMed] [Google Scholar]
- 31.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 32••.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. This papers describes the remarkable range of specificity exhibited by KS and ACP domains in chain elongation. The insight gained thereby allowed the rational engineering of a kinetically competent hybrid module. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Y, Lee TS, Kobayashi S, Khosla C. Ketosynthases in the initiation and elongation modules of aromatic polyketide synthases have orthogonal acyl carrier protein specificity. Biochemistry. 2003;42:6588–6595. doi: 10.1021/bi0341962. [DOI] [PubMed] [Google Scholar]
- 34.Zhang YM, Wu B, Zheng J, Rock CO. Key residues responsible for acyl carrier protein and beta-ketoacyl-acyl carrier protein reductase (FabG) interaction. J Biol Chem. 2003;278:52935–52943. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]
- 35.Petkovic H, Sandmann A, Challis IR, Hecht HJ, Silakowski B, Low L, Beeston N, Kuscer E, Garcia-Bernardo J, Leadlay PF, et al. Substrate specificity of the acyl transferase domains of EpoC from the epothilone polyketide synthase. Org Biomol Chem. 2008;6:500–506. doi: 10.1039/b714804f. [DOI] [PubMed] [Google Scholar]
- 36.Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J Am Chem Soc. 2003;125:14307–14312. doi: 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- 37.Chuck JA, McPherson M, Huang H, Jacobsen JR, Khosla C, Cane DE. Molecular recognition of diketide substrates by a beta-ketoacyl-acyl carrier protein synthase domain within a bimodular polyketide synthase. Chem Biol. 1997;4:757–766. doi: 10.1016/s1074-5521(97)90314-8. [DOI] [PubMed] [Google Scholar]
- 38••.Wu J, Kinoshita K, Khosla C, Cane DE. Biochemical analysis of the substrate specificity of the beta-ketoacyl-acyl carrier protein synthase domain of module 2 of the erythromycin polyketide synthase. Biochemistry. 2004;43:16301–16310. doi: 10.1021/bi048147g. Utilizing a series of biochemical assays, the authors quantitatively examined the two consecutive reactions catalyzed by the KS domain – intermodular chain transfer by self-acylation and intramodular chain elongation by decarboxylative condensation – for a panel of di- and tri-ketide substrates. This analysis revealed clear but as yet unexplained differences in the specificity of the same KS domain for the two core reactions. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe K, Wang CC, Boddy CN, Cane DE, Khosla C. Understanding substrate specificity of polyketide synthase modules by generating hybrid multimodular synthases. J Biol Chem. 2003;278:42020–42026. doi: 10.1074/jbc.M305339200. [DOI] [PubMed] [Google Scholar]