

Abstract
In recent years, host cell caveolae/caveolins have emerged as potentially important targets for pathogenic microorganisms; therefore, we investigated the role of caveolin-1 (Cav-1) in T. cruzi infection using Cav-1 null mice. Cav-1 null and wild type mice were infected with the virulent Tulahuen strain. The mortality was 100% in both groups, but death was slightly delayed in the wild type mice. The parasitemia in the Cav-1 null mice was significantly reduced compared with wild type littermates. Histopathologic examination of the heart revealed numerous pseudocysts, myonecrosis, and marked inflammation, which was similar in both mouse groups. Real-time PCR confirmed these observations. Infection of cultured cardiac fibroblasts obtained from Cav-1 null and wild type mice revealed no differences in infectivity. Determination of serum levels of several inflammatory mediators revealed a striking reduction in IFN-γ, TNF-α and components of the nitric oxide pathway in infected Cav-1 null mice. Infection of wild type mice resulted in the expected enhancement of inflammatory mediators. The defective production of chemokines and cytokines observed in vivo is in part attributed to Cav-1 null macrophages. Despite these marked differences in the response to infection by inflammatory mediators between the two mouse strains, the final outcome was similar. These results suggest that Cav-1 may play an important role in the normal development of immune responses.
Keywords: Caveolin-1, null/knockout/deficient mice, Trypanosoma cruzi, Inflammation
1. Introduction
Chagas’ disease caused by the parasite Trypanosoma cruzi is an important cause of morbidity and mortality in endemic areas of Latin America. In addition, Chagas’ disease has also been described in individuals that have emigrated into the Unites States, Canada and Western Europe and is now appreciated as an opportunistic infection in the setting of HIV/AIDS [1, 2]. The parasite infects many organs including the heart and the reticuloendothelial system including liver, spleen and bone marrow. Although the role of caveolae in the pathogenesis of viral and bacterial diseases has been examined, there is little information regarding the role of caveolae and caveolin-1 in the pathogenesis of parasitic infections. Furthermore, the number of studies examining the role of caveolin-1 (Cav-1) in immunity is limited.
Caveolae are small, 50–100 nm, flask-shaped plasmalemmal organelles with a unique biochemical composition in comparison to the plasma membrane proper. Essentially invaginated lipid rafts, caveolae are membrane microenvironments enriched in cholesterol and sphingolipids, properties that impart a relative resistance to extraction in non-ionic detergents and a propensity to float in a sucrose density gradient. Such properties have allowed for the easy isolation of caveolae and subsequent analysis and characterization of their resident protein constituents. Although initially identified in endothelial and epithelial cells, caveolae are relatively ubiquitous structures found in most tissue and cell types, with particularly high abundance in adipocytes, fibroblasts, and endothelial cells [3–5].
The principal structural components of caveolae, the caveolin protein family members (caveolin-1, -2, and –3), have allowed for the detailed molecular analysis of these unique organelles [6–9]. Caveolin-1 and –2 (Cav-1, Cav-2) are expressed in the majority of cell types, with the exception of muscle, while Cav-3 expression is limited to muscular tissue [6, 7]. Caveolins serve as scaffolding proteins, responsible for the recruitment and regulation of numerous signaling molecules within caveolae [10, 11]. Thus, caveolae serve as concentrating platforms, whereby the activation of a cellular receptor can be localized to a specific microenvironment, subsequently regulating only those molecules within this structural domain [12].
In addition to their role in signal transduction, caveolae are also involved in non-clathrin mediated macromolecular endocytosis, thus providing an alternate route of cellular entry for many molecules [13–16]. Recent findings have demonstrated that viruses, bacteria, toxins, and parasites, have evolved to co-opt this function of caveolae in order to gain access to mammalian cells [17–22]. Unlike clathrin-mediated endocytosis, cellular entry via caveolae avoids lysosomal fusion, thereby providing an attractive mechanism by which pathogenic agents may invade host cells and subsequently evade the cytocidal mechanisms of the host cell [23].
We investigated the role of caveolae and Cav-1 in the pathogenesis of Chagas’ disease by infecting Cav-1 null mice with the Tulahuen strain of T. cruzi. The mechanisms involved in acute T. cruzi infection and host organism response remain the subject of intense study. In mouse models, several studies have shown that effective host defense against T. cruzi infection is associated with an upregulation of several cytokines and chemokines, as well as reactive nitrogen intermediates [24–26].
In the present report, we found that both Cav-1 null and wild-type T. cruzi infected mice had 100% mortality. However, in the case of the infected Cav-1 null mice the mortality occurred despite a reduction in parasitemia. There was no difference between parasite load in the hearts of wild-type or Cav-1 null mice, suggesting that loss of Cav-1 does not alter parasite entry and/or intracellular survival, as was confirmed by in vitro studies. Finally, we found a dramatic decrease in several serum chemokines/cytokines that have been associated with host resistance to infection. Cav-1 null macrophages were found to be partially responsible for this defect. Thus, Cav-1 null mice fail to elicit a normal immune response to T. cruzi infection.
2. Materials and Methods
2.1. Reagents
Polyclonal rabbit anti-T. cruzi IgG was obtained from Immunology Consultants Laboratories, Inc (Newberg, OR). Serum chemokine and cytokine analysis was performed using a simultaneous multianalyte Luminex-based bead array detection system according to manufacturer’s instructions (Linco Research, Inc., St. Charles, MO). Levels of serum IFN-γ and TNF-α from infected mice were tested using an ELISA kit (Endogen), following the manufacturer’s instructions. Serum nitric oxide production was assessed with a commercially available kit (Stressgen). All other reagents were of the highest purity grade and were obtained from the usual commercial sources.
2.2. Parasitology and Pathology
All animals were housed and maintained in a barrier facility at the Institute for Animal Studies, Albert Einstein College of Medicine. Wild-type and Cav-1 null mice and were generated and maintained by us, as previously described [27, 28], and are in the C57Bl/6 genetic background. The Tulahuen strain of T. cruzi was maintained in A/J mice. Female mice were infected i.p. with 103 trypomastigotes. Spleens and hearts from 14 day infected mice were fixed in 10% buffered formalin for routine histopathology and stained with H&E. Two microliters of blood, obtained by tail bleeding, were diluted in 18 μl of 0.89% ammonium chloride in phosphate-buffered saline (PBS) to lyse red blood cells. The number of trypomastigotes in the blood of each animal was determined microscopically using a Neubauer hemacytometer.
2.3. Quantitative real-time PCR
Heart tissue was collected from 17-day post-infected wild-type and Cav-1 null mice and stored at −80°C. Using the T. cruzi 195-bp repeat DNA specific primers TCZ-f (5′-GCTCTTGCCCACAAGGGTGC-3″) which amplify a 182 –bp product and genomic DNA purified from epimastigotes. Parallel reactions were carried out for each sample using 50ng murine genomic DNA and murine specific microglobulin primers β2F2 (5′-TGGGAAGCCGAACATACTG3′) AND β2R2 (5′-GCAGGCGTATGTATCAGTCTCA-3′), which amplify 190-bp product as an internal control. Light cycler-fast start master Syber green 1 (Roche) is used to amplify the templates using a Light cycler (Roche) instrument. The other reagents, parameters and data analysis used for the real time PCR as previously described in our laboratory.
2.4. Infection of primary cardiac fibroblast cell cultures
Isolated primary mouse cardiac fibroblasts were prepared and maintained as previously described [29]. Cells were infected with trypomastigotes at a multiplicity of infection of 1:1 parasite/host cell ratio and processed for immunoblotting and immunofluorescence.
2.5. Infection of primary mouse peritoneal macrophage cultures
Mice were i.p. injected with 1 ml of 3% Brewer's thioglycollate. Three days later, peritoneal cells were isolated by lavage with ice-cold PBS. After a 2hr. incubation at 37°C, non-adherent cells were removed and the remaining adherent macrophages were used for experiments.
2.6. Immunoblot analysis
Infected and uninfected wild-type and Cav-1 null cardiac fibroblasts were washed 5 times in sterile PBS and then lysed in buffer (10 mM Tris, pH 7.5, 50 mM NaCl, 1% Triton X-100, and 60 mM octyl glucoside), containing protease inhibitors (Boehringer Mannheim). Lysates were then centrifuged at 12,000 × g for 10 min to remove insoluble debris. Protein concentrations were analyzed using the BCA reagent (Pierce) and the volume required for 40 μg of protein was determined. Samples were then separated by SDS-PAGE and transferred to nitrocellulose. The nitrocellulose membranes were stained with Ponceau S (to visualize protein bands), followed by immunoblot analysis. All subsequent wash buffers contained 10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween-20, which was supplemented with 1% bovine serum albumin (BSA) and 2% nonfat dry milk (Carnation) for the blocking solution and 1% BSA for the antibody diluent. Primary antibodies were used at a 1:500 dilution. Horseradish peroxidase-conjugated secondary antibodies (1:5000 dilution, Pierce) were used to visualize bound primary antibodies with the Supersignal chemiluminescence substrate (Pierce).
2.7. Immunofluorescence
Isolated mouse cardiac fibroblasts were grown on glass coverslips, fixed in 2% paraformaldehyde, and permeabilized in 0.1% Triton X-100. Cells were then immunostained with polyclonal anti-T. cruzi antibodies. Bound primary antibodies were visualized with a rhodamine-conjugated anti-rabbit antibody (Jackson ImmunoResearch Inc.). As expected, omission of the primary antibodies prevented immunostaining.
2.8. Statistics
Statistical analysis of data was performed using the log-rank test for survival studies and the student’s unpaired t test from GraphPad Prism program (GraphPad, San Diego, CA). Differences between experimental groups were considered significant for P values of < 0.05.
3. Results
3.1. Survival and parasitemia of T. cruzi infected mice
Mice were infected with the Tulahuen strain of T. cruzi which causes an acute infection in wild type mice. Cav-1 null mice consistently displayed a moderate decrease in survival (median survival 26 days vs. 22 days, respectively; Fig. 1). However, this difference was not statistically significant. All infected mice died by 35 days post-infection. In both genotypes, trypomastigotes were first reproducibly quantifiable in the blood at 15 days post-infection. While Cav-1 null mice tend to exhibit less blood parasitemia at this stage, this difference was not statistically significant (Fig. 2). At day 20 post-infection, the parasitemia was significantly higher in wild-type than in Cav-1 null mice (Fig. 2), and this trend continued throughout days 25 and 30 post-infection.
Fig. 1.

Kaplan-Meier survival curve of wild-type and Cav-1 null mice following T. cruzi infection. Wild-type (WT) and Cav-1 null (KO) mice were infected with 1000 epimastigotes and their survival was followed over a period of 30 days. Note that Cav-1 null mice exhibit a decline in viability with a decrease in median time of survival from 26 to 22 days. n=8 mice for each group.
Fig. 2.

Parasitemia in wild-type and Cav-1 null mice following infection with T. cruzi. Daily parasitemia was assessed over a period of 30 days in wild-type (WT) and Cav-1 null (KO) animals. After 15 days of infection, parasitemia is first reproducibly quantifiable in both WT and KO mice. Note that Cav-1 null mice exhibit significantly less parasites in the blood between 20 and 30 days of infection. Results represent the mean ± SEM; *p<0.05; n=8 mice for each group.
For this study, we chose to analyze the heart because it is composed of nearly 50% fibroblasts and endothelial cells expressing caveolin-1, and it is a major target organ during T. cruzi infection. The histopathological examination of the heart revealed acute myocarditis with marked inflammation and numerous pseudocysts. Inflammation in the heart was mainly composed of scattered and small clusters of mononuclear cells. Unlike blood parasitism, there did not appear to be any differences in pathology or numbers of parasite nests between Cav-1 null and wild type mice at day 17 post-infection (data not shown). No alterations of the splenic architecture were observed.
In order to confirm the lack of difference in tissue parasite burden between wild-type and Cav-1 null mice, heart tissue was collected from infected mice and subjected to quantitative real-time PCR analysis. We found that there was no statistical difference between the amounts of T. cruzi DNA in the hearts of Cav-1 null mice compared to wild-type mice after 17 days of infection (Table 1). These results suggest that loss of Cav-1 does not alter the ability of T. cruzi to enter and infect tissue that normally expresses an abundance of Cav-1.
Table 1. Real-Time PCR Analysis.
Parasite burden in cardiac tissue from wild-type and Cav-1 null (KO) infected mice at 17 days post-infection. Wild-type (WT) and Cav-1 null (KO) mice were infected i.p. with 103 trypomastigotes. Data are a comparison of parasite burden in 50 ng of DNA isolated from infected hearts.
| Genotype | Tissue | DPI a | Parasites/Cell |
|---|---|---|---|
| Wild-type | Heart | 17 | 2.84 ± 0.18 |
| Cav-1 null | Heart | 17 | 2.78 ± 0.21 |
n ≥ 4 for each experimental group
DPI = days post-infection
3.2. Infection of cultured cardiac fibroblasts and peritoneal macrophages
Since T. cruzi exhibits cardiotropism, we next analyzed the ability of T. cruzi to enter cultured wild-type and Cav-1 null isolated mouse cardiac fibroblasts. In these experiments, we found that an equal number of intracellular parasites were detectable in Cav-1 null and wild-type cells by Western analysis after 48 and 72 hours of infection (Fig. 3A). Similarly, by visual inspection, a relatively equal amount of parasites were found in each of the cell lines, as assessed by phase contrast and immunofluorescence microscopy at 72 hours (Fig. 3B). Equivalent results were obtained after 48 hours of infection (data not shown).
Fig. 3. Infection of primary cardiac fibroblasts and peritoneal macrophages with T. cruzi.
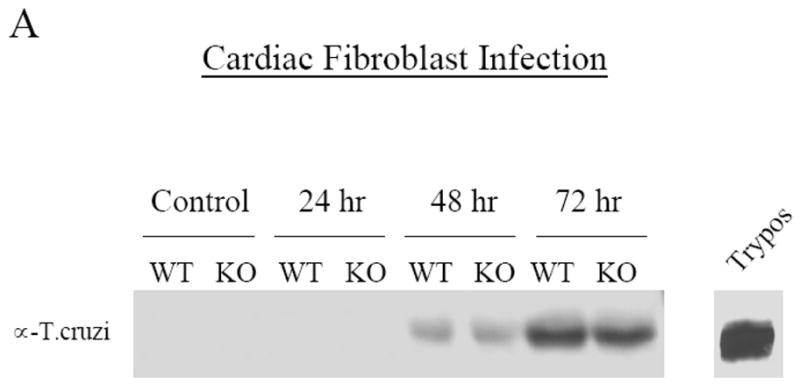


A) Analysis of infection by Western blot. Following a time-course of infection with T. cruzi, isolated primary cardiac fibroblasts from wild-type (WT) and Cav-1 null (KO) mice were subjected to Western blot analysis with antibodies that specifically recognize T. cruzi. At both time points where infection is evident by this methodology (48 and 72h), the amount of detectable T. cruzi does not vary between the genotypes.
B) Analysis of infection by immunofluorescence microscopy. Similar to the results presented in (A), infected primary cardiac fibroblasts were analyzed for intracellular parasite load at various time points. Following 72h of infection, abundant parasites are detectable in both genotypes by phase contrast (top panels) and by immunostaining with T. cruzi-specific antibodies (middle panels). In the merged image (bottom panels) parasites appear red. Similar numbers of intracellular parasites are evident in wild-type (WT) and Cav-1 null (KO) cells by this methodology.
C) Peritoneal macrophages were infected with culture derived trypomastigotes at a parasite/host cell ratio of 2:1. Cultures were maintained at 37°C for 24 hrs. Cells were stained with Giemsa and the number of intracellular parasites found in wild-type (WT) and Cav-1 null (KO) macrophages were counted under a light microscope (400X). A total of 500 cells were counted per well. Each bar represents the mean ± standard deviation of triplicate counts.
Infection of macrophages is important for the dissemination of trypomastigotes in the host for completion of its cell cycle. Therefore, we next examined the role of caveolin-1 in the ability of T. cruzi trypomastigotes to invade peritoneal macrophages. Macrophages from Cav-1 null mice exposed to trypomastigotes for 24 hrs. did not display any alterations in the numbers of amastigotes compared to wild-type mice (Fig 3C). Together these data suggest that T. cruzi does not utilize caveolae for entry either through active invasion or uptake.
3.3. Analysis of serum chemokines/cytokines and nitric oxide
Infection with T. cruzi causes increases expression of inflammatory cytokines and chemokines. These immune mediators have been implicated as playing a role in controlling parasitism. In order to gain further insight into the impaired resistance to T. cruzi infection in Cav-1 null mice, we analyzed the levels of several serum chemokines and cytokines. Our results demonstrated that, during the acute phase of T. cruzi infection (14 days post-infection), Cav-1 null mice produced significantly less IFN-γ, TNF-α, CCL3, CCL5, interleukin (IL)-6, and several other chemokines/cytokines when compared with infected control mice (Table 2, Fig. 4A and B). In addition, analysis of serum nitric oxide revealed similarly low levels in Cav-1 null animals (Fig. 5). Production of these factors is currently considered the hallmark of host resistance to T. cruzi infection. Taken together, these results suggest a regulatory role for caveolin-1 in the normal immune response against T. cruzi.
Table 2. Analysis of serum cytokines and chemokines at baseline and at 14 days post-infectiona.
Cav-1 null (KO) mice infected with T. cruzi trypomastigotes display reduced secretion of chemokines and cytokines. Wild-type (WT) and Cav-1 null (KO) mice were infected i.p. with 103 trypomastigotes and serum chemokine and cytokine levels were determined by LINCOplex. Results represent the mean ± SEM; *p<0.05, n ≥ 5 animals per group.
| Sample | MIP-1α | GM-CSF | MCP-1 | KC | Rantes | IFN-γ | G-CSF | TNF-α | IP-10 | IL-1α | IL-2 | IL-5 | IL-6 | IL-9 | IL-10 | IL-12 | IL-13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT baseline | 101.4 ±32 | 53.0 ±10 | 32.3 ± 5 | 29.4 ± 1 | 13.6 ± 2 | 12.4 ± 1 | 528.5 ± 34 | 8.2 ± 0.5 | 38.8 ± 6 | 49.8 ± 7 | <3.2 | 7.8 ± 0.3 | 8.6 ± 2 | 36.5 ± 6 | 48.9 ± 12 | 45.2 ± 10 | 110.9 ± 5 |
| KO baseline | 117.4 ±31 | 45.5 ±9 | 38.3 ± 7 | 13.5 ± 5 | 14.4 ± 3 | 10.4± 1.4 | 721.9 ± 22 | 10.6 ± 1.5 | 72.9 ± 13 | 48.1 ± 7 | <3.2 | 10.7 ± 1 | 18.4 ± 5 | 60.3 ± 20 | 35.1 ± 6 | 51.9 ± 3 | 108 ± 8 |
| p value | 0.383 | 0.362 | 0.317 | 0.037* | 0.434 | 0.130 | 0.060# | 0.144 | 0.052# | 0.450 | 0.041* | 0.096# | 0.195 | 0.218 | 0.340 | 0.399 | |
| WT Infected | 111.5 ±13 | <3.2 | 219 ± 29 | 188.8 ± 26 | 42.1 ± 6 | 508 ± 140 | 459 ± 46 | 110 ± 15 | 180 ± 21 | 80.6 ± 15 | 38.5 ± 20 | 30.8 ± 19 | 115 ± 13 | <3.2 | 41.2 ± 9 | 30.3 ± 5 | 132 ± 27 |
| KO Infected | 53.1 ± 4 | <3.2 | 103.6 ± 35 | 99.3 ± 28 | 16.1 ± 6 | 50.5 ± 13 | 975 ± 244 | 19.7 ± 7 | 179 ± 75 | 45.3 ± 8 | <3.2 | 17 ± 2 | 35.3 ± 5 | <3.2 | 42.2 ± 5 | 30.4 ± 6 | 73.5 ± 17 |
| p value | 0.041* | 0.085# | 0.073# | 0.037* | 0.050* | 0.076# | 0.005* | 0.390 | 0.095# | 0.292 | 0.003* | 0.474 | 0.338 | 0.126 |
Concentrations are [pg/ml]
p < 0.05
p < 0.10
Figure 4.


Serum chemokine and cytokine levels in wild-type and Cav-1 null mice during T. cruzi infection.
A) IFN-γ levels. Analysis of serum IFN-γ levels in wild-type (WT) and Cav-1 null (KO) animals before infection (day 0) and at 14 days post-infection reveals a normal increase in this cytokine following infection in WT animals, but a severely blunted response in Cav-1 null mice.
B) TNF-α levels. Serum levels of TNF-α are also significantly reduced in Cav-1 null mice at 14 days post-infection.
Results represent the mean ± SEM; *p<0.05, n ≥ 5 animals per group.
Figure 5.

Serum nitric oxide levels are significantly reduced in Cav-1 null (KO) animals during T. cruzi infection. Wild-type (WT) and Cav-1 null (KO) mice were infected i.p. with 103 trypomastigotes and serum nitric oxide levels were determined at 14 days post-infection. Results represent the mean ± SEM; *p<0.05, n ≥ 5 animals per group.
3.4. Chemokine and cytokine analysis from T. cruzi infected macrophages
Macrophages play an important role in the control and replication of T. cruzi. Trypomastigotes contain glycoinositolphospholipids (GIPLs) on their surfaces that have been shown to induce chemokine and cytokine production in macrophages. Previous studies and our unpublished observations have demonstrated that caveolin-1 is expressed in peritoneal macrophages [30, 31]. Therefore, we decided to evaluate whether the absence of caveolion-1 in macrophages was a contributing factor in the defective chemokine and cytokine production observed in vivo. After incubation of peritoneal macrophages with T. cruzi trypomastigotes for 24 hrs, culture supernatants were analyzed for the production of chemokines and cytokines. Cav-1 null macrophages showed decreased production of the chemokines CCL2, CCL5, CXCL1, CXCL10, and the cytokines G-CSF, TNF-α, IL-1α, and IL-6. However, there were no differences in the levels of CCL3 (Table 3). These results suggest that Cav-1 plays a role in regulating cytokine production in macrophages infected with T. cruzi.
Table 3. Analysis of cytokines and chemokines from wild-type and Cav-1 null macrophagesa.
Cav-1 null (KO) macrophages infected with T. cruzi trypomastigotes display reduced secretion of chemokines and cytokines. Peritoneal macrophages were infected with culture-derived trypomastigotes at a 2:1 parasite/host cell ratio. Cultures were maintained at 37°C for 24 hrs. Supernatants were collected and chemokine and cytokine levels from wild-type and Cav-1 null macrophages were determined. The bars represent mean ± standard deviation of triplicate counts.
| Sample | CCL3 | CCL2 | CXCL1 | CCL5 | G-CSF | TNF-α | CXCL10 | IL-1α | IL-6 |
|---|---|---|---|---|---|---|---|---|---|
| WT Infected | 73.75 ±12 | 776.1 ± 50 | 491 ± 20 | 14.5 ± 3 | 231 ± 16 | 26.5 ± 2 | 193.4 ± 7 | 30 ± 1 | 72.2 ± 1 |
| KO Infected | 65.25 ± 7 | 327.9 ± 56 | 77.6 ± 3 | 3.7 ± 0.2 | 25.1 ± 2 | 14.2 ± 0.5 | 83.1 ± 15 | 3.41 ± 0.1 | 7.5 ± 1 |
| p value | 0.426 | 0.014* | 0.001* | 0.028* | 0.003* | 0.011* | 0.011* | 0.002* | 0.003* |
Concentrations are [pg/ml]
p < 0.05
4. Discussion
Caveolae mediated entry of pathogens into cells has been reported with a number of viruses, bacteria, parasites and toxins. Examples of these are Ebola, Echovirus, influenza, FimH-expressing Escherichia coli, cholera toxin and malaria, which have been shown to co-localize or proposed to utilize caveolae to enter cells [32–35]. Caveolar entry is an additional mechanism utilized by pathogens to avoid their destruction by preventing their degradation via the endosomal or lysosomal compartments [34, 36]. However, cellular invasion by T. cruzi requires transient residence within an acidic lysosomal vacuole [37]. This allows activation of TxTox, which plays a role in vacuole disruption [38, 39]. In addition, lysosomal vacuoles also provide other necessary signals that permit the parasite to complete it’s intracellular life cycle [40]. Entry of T. cruzi into host cells is an active rather than passive process. Communication occurs between the host cell’s signaling pathways and the pathogen [41, 42]. Our in vitro invasion studies suggest that trypomastigotes of T. cruzi do not enter through caveolae and that Cav-1 does not play a role in host cell signaling required for its internalization.
Based on the findings presented in this study, it does not appear that caveolar endocytosis plays a major role in the entry of the parasite into cells. Endocytosis via caveolae occurs in a similar manner to that involving clathrin coated pits (CCPs), utilizing much of the same molecular machinery. For instance, the SNARE proteins, which are involved in the docking and fusion of vesicles, are employed by both caveolae and CCPs. In addition, the small GTPase dynamin, which mediated CCP internalization, has also been shown to localize to the necks of caveolae and be involved in their internalization as well [ 14, 15, 43]. While the exact processes mediating internalization via caveolae remain unknown, several studies have shown that ligand-mediated activation of a tyrosine kinase signaling pathway is an important step [16]. The list of ligands internalized by caveolae include several pathogenic agents which have been shown to utilize caveolae for entry into host cells [18, 20, 23, 44, 45].
Acute infection with the virulent T. cruzi Tulahuen strain resulted in mortality on both the Cav-1 and wild type mice, although death was slightly delayed in the wild-type mice. Interestingly, the mortality observed in infected Cav-1 mice was associated with a significant decrease in blood parasitemia. However, examination of heart tissue by real-time PCR and histopathology revealed no alterations in parasite load. Furthermore, in the absence of Cav-1 the ability of T. cruzi to enter cells was not compromised. These results suggest that the loss of Cav-1 does not alter infectivity and/or intracellular survival of the parasite.
There is limited information regarding the role of Cav-1 in the immune system. This is in part due to the previously contentious issue of its expression in cells of the immune system. We and others have observed that Cav-1 is a component of many immune cells and is expressed in a regulated manner. Expression of Cav-1 is low in unstimulated macrophages and increases with LPS stimulation [30, 31]. The induction of inflammatory mediators by macrophages during T. cruzi infection is mediated by glycoinositol phospholipids (GIPLs) [46]. Signaling is mediated through TLR-2 and possibly TLR-4 based on recent studies [47, 48]. It is of great importance that macrophages derived from Cav-1 null mice produced significantly lower amounts of chemokines and cytokines during T. cruzi infection. This suggests that Cav-1 may be involved in regulating their production under certain conditions.
Interestingly, our recent studies examined the role of Cav-1 in the pathogenesis of Salmonella typhimurium in the mouse model [49]. S. typhimurium infected Cav-1 null mice had a significant mortality and an increased bacterial tissue burden compared with wild type mice [49]. In contradistinction with the present data with T. cruzi infection, in Salmonella infected Cav-1 null mice there was an increased production of inflammatory mediators such as cytokines, chemokines, and nitric oxide. Nevertheless, infected Cav-1 null mice were unable to control this infection and the mice died [49]. In addition, macrophages obtained from Cav-1 null mice had an increased inflammatory responses and nitric oxide production in vitro in response to Salmonella LPS. These results demonstrate that Cav-1 plays a key role in regulating anti-inflammatory responses in macrophages in Salmonella infection. In addition, these observations suggest the increased production of toxic mediators from Cav-1 null mice may be responsible for the marked susceptibility of Cav-1 null mice to S. typhimurium [49]. The differences in the responses of Cav-1 null mice to infection with T. cruzi vs. S. typhimurium infection are very intriguing. They are both intracellular organisms that can reside in macrophages for extended periods of time. However, one is an intracellular parasite while the other an intracellular bacterium. In contradistinction to Salmonella infection, T. cruzi may interdict important signaling pathways in infected cells. These experiments are ongoing in our laboratory. It also underscores the fact that the response of Cav-1 null mice to infection is not the same for all microorganisms.
The production of chemokines and cytokines has been shown to play a role in controlling T. cruzi. A thorough examination of their levels in infected mice revealed that they were downregulated during infection in Cav-1 null mice compared to wild-type mice. The reason for the reduced peripheral parasitemia in infected Cav-1 null mice is not known since the reduction in inflammatory mediators in these mice might suggest that the parasitemia should be increased, There was no correlation between extent of parasite load in tissue and peripheral parasitemia. However, this is not always the case. Importantly, all mice wild type and null mice still died.
Our results showed that the decrease in chemokine and cytokine expression was not due to the limited number of parasites in Cav-1 null mice, but rather an intrinsic defect in Cav-1 null macrophages. However, we cannot exclude the contribution of other immune cells in the overall reduction in serum chemokine and cytokine production in infected Cav-1 null mice. In addition, although immune cells play a major role in the production of inflammatory mediators during T. cruzi infection, other cells may also be involved. Infection of endothelial cells, which express high levels of Cav-1, with T. cruzi causes direct induction of IL-1β and IL-6 [50].
Despite the impaired immune response in Cav-1 null mice during an acute infection with T. cruzi, they do not display an increased mortality rate. This suggests that the lack of chemokine and cytokine production in this mouse model is not a primary cause of death. It is possible that Cav-1 may play a role in cytokine/cytokine receptor systems. Surprisingly, despite the deficiency in serum chemokine production we did not observe any significant difference in recruitment of immune populations to the heart of Cav-1 null mice compared to wild-type mice.
To our knowledge this is the first report examining the role of Cav-1 in the host mediated response to infection with the protozoan parasite, T. cruzi. The results in this study show that Cav-1 null mice did not display an increased susceptibility to T. cruzi despite their reduced inflammatory profile. The role of cytokines and chemokines in the host defense against this parasite requires additional study.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH), and the American Heart Association (AHA). Cecilia J. de Almeida was supported by a Fogarty International Training Grant 1D43TW007129.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kirchhoff LV. American trypanosomiasis (Chagas' disease)--a tropical disease now in the United States. N Engl J Med. 1993;329:639–644. doi: 10.1056/NEJM199308263290909. [DOI] [PubMed] [Google Scholar]
- 2.Harms G, Feldmeier H. The impact of HIV infection on tropical diseases. Infect Dis Clin North Am. 2005;19:121–135. ix. doi: 10.1016/j.idc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Palade GE. Fine structure of blood capillaries. Journal of Applied Physiology. 1953;24:1424. [Google Scholar]
- 4.Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol. 1955;1:445–458. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 6.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci, USA. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang ZL, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 8.Glenney JR. The sequence of human caveolin reveals identity with VIP 21, a component of transport vesicles. FEBS Lett. 1992;314:45–48. doi: 10.1016/0014-5793(92)81458-x. [DOI] [PubMed] [Google Scholar]
- 9.Glenney JR. Tyrosine phosphorylation of a 22 kD protein is correlated with transformation with Rous sarcoma virus. J Biol Chem. 1989;264:20163–20166. [PubMed] [Google Scholar]
- 10.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, A family of scaffolding proteins for organizing "pre-assembled signaling complexes" at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 11.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 12.Lisanti MP, Scherer P, Tang ZL, Sargiacomo M. Caveolae, caveolin and caveolin-rich membrane domains: A signaling hypothesis. Trends In Cell Biology. 1994;4:231–235. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 13.Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141:905–915. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnitzer JE, Liu J, Oh P. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J Biol Chem. 1995;270:14399–14404. doi: 10.1074/jbc.270.24.14399. [DOI] [PubMed] [Google Scholar]
- 15.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol. 1998;141:101–114. doi: 10.1083/jcb.141.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311–320. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 17.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 18.Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 19.Norkin LC. Simian virus 40 infection via MHC class I molecules and caveolae. Immunol Rev. 1999;168:13–22. doi: 10.1111/j.1600-065x.1999.tb01279.x. [DOI] [PubMed] [Google Scholar]
- 20.Norkin LC, Anderson HA, Wolfrom SA, Oppenheim A. Caveolar Endocytosis of Simian Virus 40 Is Followed by Brefeldin A- Sensitive Transport to the Endoplasmic Reticulum, Where the Virus Disassembles. J Virol. 2002;76:5156–5166. doi: 10.1128/JVI.76.10.5156-5166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- 22.Shin JS, Abraham SN. Caveolae as portals of entry for microbes. Microbes Infect. 2001;3:755–761. doi: 10.1016/s1286-4579(01)01423-x. [DOI] [PubMed] [Google Scholar]
- 23.Norkin LC. Caveolae in the uptake and targeting of infectious agents and secreted toxins. Adv Drug Deliv Rev. 2001;49:301–315. doi: 10.1016/s0169-409x(01)00143-0. [DOI] [PubMed] [Google Scholar]
- 24.Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation. 2000;102:3003–3008. doi: 10.1161/01.cir.102.24.3003. [DOI] [PubMed] [Google Scholar]
- 25.Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, Ropert C, Gazzinelli RT. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. 2004;172:1711–1718. doi: 10.4049/jimmunol.172.3.1711. [DOI] [PubMed] [Google Scholar]
- 26.Holscher C, Mohrs M, Dai WJ, Kohler G, Ryffel B, Schaub GA, Mossmann H, Brombacher F. Tumor necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. 2000;68:4075–4083. doi: 10.1128/iai.68.7.4075-4083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 28.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, Lisanti MP. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, Pereira de Souza A, Kitsis RN, Russell RG, Weiss LM, Tang B, Jelicks LA, Factor SM, Shtutin V, Tanowitz HB, Lisanti MP. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:C457–474. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 30.Gargalovic P, Dory L. Caveolin-1 and caveolin-2 expression in mouse macrophages. High density lipoprotein 3-stimulated secretion and a lack of significant subcellular co-localization. J Biol Chem. 2001;276:26164–26170. doi: 10.1074/jbc.M011291200. [DOI] [PubMed] [Google Scholar]
- 31.Lei MG, Morrison DC. Differential expression of caveolin-1 in lipopolysaccharide-activated murine macrophages. Infect Immun. 2000;68:5084–5089. doi: 10.1128/iai.68.9.5084-5089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marjomaki V, Pietiainen V, Matilainen H, Upla P, Ivaska J, Nissinen L, Reunanen H, Huttunen P, Hyypia T, Heino J. Internalization of echovirus 1 in caveolae. J Virol. 2002;76:1856–1865. doi: 10.1128/JVI.76.4.1856-1865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duncan MJ, Shin JS, Abraham SN. Microbial entry through caveolae: variations on a theme. Cell Microbiol. 2002;4:783–791. doi: 10.1046/j.1462-5822.2002.00230.x. [DOI] [PubMed] [Google Scholar]
- 35.Montesano R, Roth J, Robert A, Orci L. Non-coated membrane invaginations are involved in binding and internalization of cholera and tetanus toxins. Nature (Lond) 1982;296:651–653. doi: 10.1038/296651a0. [DOI] [PubMed] [Google Scholar]
- 36.Amer AO, Swanson MS. A phagosome of one's own: a microbial guide to life in the macrophage. Curr Opin Microbiol. 2002;5:56–61. doi: 10.1016/s1369-5274(02)00286-2. [DOI] [PubMed] [Google Scholar]
- 37.Ley V, Robbins ES, Nussenzweig V, Andrews NW. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J Exp Med. 1990;171:401–413. doi: 10.1084/jem.171.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrews NW, Abrams CK, Slatin SL, Griffiths G. A T. cruzi-secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell. 1990;61:1277–1287. doi: 10.1016/0092-8674(90)90692-8. [DOI] [PubMed] [Google Scholar]
- 39.Andrews NW, Whitlow MB. Secretion by Trypanosoma cruzi of a hemolysin active at low pH. Mol Biochem Parasitol. 1989;33:249–256. doi: 10.1016/0166-6851(89)90086-8. [DOI] [PubMed] [Google Scholar]
- 40.Tomlinson S, Vandekerckhove F, Frevert U, Nussenzweig V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 1995;110( Pt 5):547–554. doi: 10.1017/s0031182000065264. [DOI] [PubMed] [Google Scholar]
- 41.Moreno SN, Docampo R. Calcium regulation in protozoan parasites. Curr Opin Microbiol. 2003;6:359–364. doi: 10.1016/s1369-5274(03)00091-2. [DOI] [PubMed] [Google Scholar]
- 42.Burleigh BA. Host cell signaling and Trypanosoma cruzi invasion: do all roads lead to lysosomes? Sci STKE. 2005;2005:pe36. doi: 10.1126/stke.2932005pe36. [DOI] [PubMed] [Google Scholar]
- 43.Henley JR, Krueger EW, Oswald BJ, McNiven MA. Dynamin-mediated internalization of caveolae. J Cell Biol. 1998;141:85–99. doi: 10.1083/jcb.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Norkin LC. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp Cell Res. 1999;246:83–90. doi: 10.1006/excr.1998.4301. [DOI] [PubMed] [Google Scholar]
- 46.Campos MA, Gazzinelli RT. Trypanosoma cruzi and its components as exogenous mediators of inflammation recognized through Toll-like receptors. Mediators Inflamm. 2004;13:139–143. doi: 10.1080/09511920410001713565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO, Travassos LR, Smith JA, Golenbock DT, Gazzinelli RT. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167:416–423. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- 48.Oliveira AC, Peixoto JR, de Arruda LB, Campos MA, Gazzinelli RT, Golenbock DT, Akira S, Previato JO, Mendonca-Previato L, Nobrega A, Bellio M. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. 2004;173:5688–5696. doi: 10.4049/jimmunol.173.9.5688. [DOI] [PubMed] [Google Scholar]
- 49.Medina FA, de Almeida CJ, Dew E, Li J, Bonnucelli G, Williams TM, Cohen AW, Pestell RG, Frank PG, Tanowitz HB, Lisanti MP. Salmonella Pathogenesis in Caveolin-1 (−/−) Deficient Mice: Role of Cav-1 in Innate Immunity and the Inflammatory Immune Response. Infect Immun. 2006 doi: 10.1128/IAI.00949-06. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanowitz HB, Gumprecht JP, Spurr D, Calderon TM, Ventura MC, Raventos-Suarez C, Kellie S, Factor SM, Hatcher VB, Wittner M, et al. Cytokine gene expression of endothelial cells infected with Trypanosoma cruzi. J Infect Dis. 1992;166:598–603. doi: 10.1093/infdis/166.3.598. [DOI] [PubMed] [Google Scholar]