

Abstract
Muscle ring finger-1 (MuRF1) is a muscle-specific protein implicated in the regulation of cardiac myocyte size and contractility. MuRF2, a closely related family member, redundantly interacts with protein substrates, and hetero-dimerizes with MuRF1. Mice lacking either MuRF1 or MuRF2 are phenotypically normal whereas mice lacking both proteins develop a spontaneous cardiac and skeletal muscle hypertrophy indicating cooperative control of muscle mass by MuRF1 and MuRF2. In order to identify the unique role that MuRF1 plays in regulating cardiac hypertrophy in vivo, we created transgenic mice expressing increased amounts of cardiac MuRF1. Adult MuRF1 transgenic (Tg+) hearts exhibited a non-progressive thinning of the left ventricular wall and a concomitant decrease in cardiac function. Experimental induction of cardiac hypertrophy by trans-aortic constriction (TAC) induced rapid failure of MuRF1 Tg+ hearts. Microarray analysis identified that the levels of genes associated with metabolism (and in particular mitochondrial processes) were significantly altered in MuRF1 Tg+ hearts, both at baseline and during the development of cardiac hypertrophy. Surprisingly, ATP levels in MuRF1 Tg+ mice did not differ from wild type mice despite the depressed contractility following TAC. In comparing the level and activity of creatine kinase (CK) between wild type and MuRF1 Tg+ hearts we found that mCK and CK-M/B protein levels were unaffected in MuRF1 Tg+ hearts, however total CK activity was significantly inhibited. We conclude that increased expression of cardiac MuRF1 results in a broad disruption of primary metabolic functions, including alterations in CK activity that leads to increased susceptibility to heart failure following TAC. This study demonstrates for the first time a role for MuRF1 in the regulation of cardiac energetics in vivo.
Keywords: Muscle ring finger-1, MuRF1, ubiquitin ligase, cardiac hypertrophy, heart failure, creatine kinase
Introduction
The muscle ring finger (MuRF) proteins are striated muscle-specific proteins that have been implicated in various aspects of contractile regulation and myogenic responses 1. Muscle ring finger-1 (MuRF1) is a well-characterized RING-finger-dependent ubiquitin ligase that targets sarcomere proteins, such as cTnI, during the process of skeletal muscle atrophy 2, 3. MuRF1 has also been implicated in the regulation of cardiac myocyte size and contractility 3-5 and inhibits the development of cardiac hypertrophy 6, 7, a dynamic process commonly thought of as a precursor to heart failure 8. To date, MuRF1's regulation of cardiac muscle mass has centered around its involvement in the regulation of sarcomere protein degradation. While this is certainly an important function, in this report we propose that MuRF1 operates in a broader capacity that encompasses both protein turnover as well as control of cardiac metabolism.
Soon after the discovery of MuRF1, the related proteins MuRF2 and MuRF3 were identified as interacting proteins capable of forming hetero-dimers with MuRF1 9. Subsequently, functional redundancy between the MuRF1 and MuRF2 proteins was suggested by yeast 2-hybrid studies which identified that MuRF1 and MuRF2 interact with many of the same sarcomeric proteins 10. The idea of functional redundancy was strengthened by the finding that mice lacking either MuRF1 or MuRF2 appear relatively normal while mice lacking both MuRF1 and MuRF2 develop both cardiac and skeletal muscle hypertrophy 11. Although the studies involving these double knockouts clarified the importance of MuRF1 and MuRF2 in cardiac regulation, the individual roles that that these two proteins play in vivo have yet to be ascertained due to a lack of helpful transgenic models (i.e. single knockouts of these proteins have no cardiac phenotype at baseline while double knockouts are lethal). In order to overcome this issue, we created transgenic mice with increased cardiac expression of MuRF1 to dissect out the functions of MuRF1 in the heart. Using this animal model, we confirmed that MuRF1 regulates cardiac function during the development of cardiac hypertrophy in vivo. More importantly, we identified that MuRF1 alters cardiac energy metabolism in vivo, in part, by inhibiting creatine kinase activity, a rate-limiting step in the phospho-creatine ATP shuttle in the heart. These results shed new light on MuRF1's contributions to cardiac function in vivo via the regulation of key enzymes necessary to distribute ATP throughout the cell.
Materials and Methods
Experimental design
MuRF1 Tg+ and littermate wild type controls (50% male/50% female) at 10-12 weeks of age underwent trans-aortic constriction (TAC) to induce cardiac hypertrophy as previously described 7. No significant differences between genders were noted throughout the study. MuRF1 Tg+ and wild type controls underwent conscious echocardiographic analysis at baseline (prior to TAC) and then weekly for 4 weeks following the TAC procedure (10 mice/group). Additional mice (5/group) underwent conscious echocardiographic analysis at baseline and subsequently every 2 months for 18 months. M-mode and two-dimensional echocardiography was performed using the Vevo 660 ultrasound system as previously described 7. Hearts were dissected from the body and perfused with 4% paraformaldehyde. Paraffin sections were stained with hematoxylin and eosin, Masson's trichrome, or fluorescently labeled lectin as previously described 7. For cross-sectional area analysis of cardiomyocytes, TRITC conjugated lectin (Triticum vulgaris) staining was performed and measured as previously described and examined by fluorescence microscopy 7.
Additional materials and methods may be found in the online supplement.
Results
Mild baseline cardiac changes identified in MuRF1 Tg+ mice
In our first series of studies, MuRF1 Tg+ and wild type littermates were monitored for survival and cardiac function by echocardiography for up to 18 months. Equal numbers of male and female MuRF1 Tg+ and wild type offspring were identified at birth, indicating that the increased expression of cardiac MuRF1 did not affect developmental viability. No differences in overall cardiac mass were identified between adult MuRF1 Tg+ and wild type littermate controls (heart weight to body weight ratio 5.8±0.2 vs. 5.6±0.1 mg/g, respectively, Online Table I). However, by echocardiography, MuRF1 Tg+ mice displayed thinner anterior and posterior left ventricular wall thickness (19.3% and 19.8% less than wild type mice, respectively in diastole, Figure 1A, left and middle), an increase in left ventricular end diastolic dimension (9.9% greater than wild type mice, Figure 1A, right), and a decrease in function as measured by fractional shortening (FS% 39.5±1.3 vs. 54.5±0.5 in wild type mice, Figure 1B, left). Serial echocardiography revealed that this mild phenotype did not progress with age. Functional assessment by cardiac catheterization revealed no functional differences between MuRF1 Tg+ and wild type hearts (Online Table III), supporting the possibility that cardiac dysfunction did not precede decreases in CK activity in MuRF1 Tg+ hearts.
Figure 1. MuRF1 Tg+ hearts undergo an accelerated eccentric cardiac hypertrophy after trans-aortic constriction.
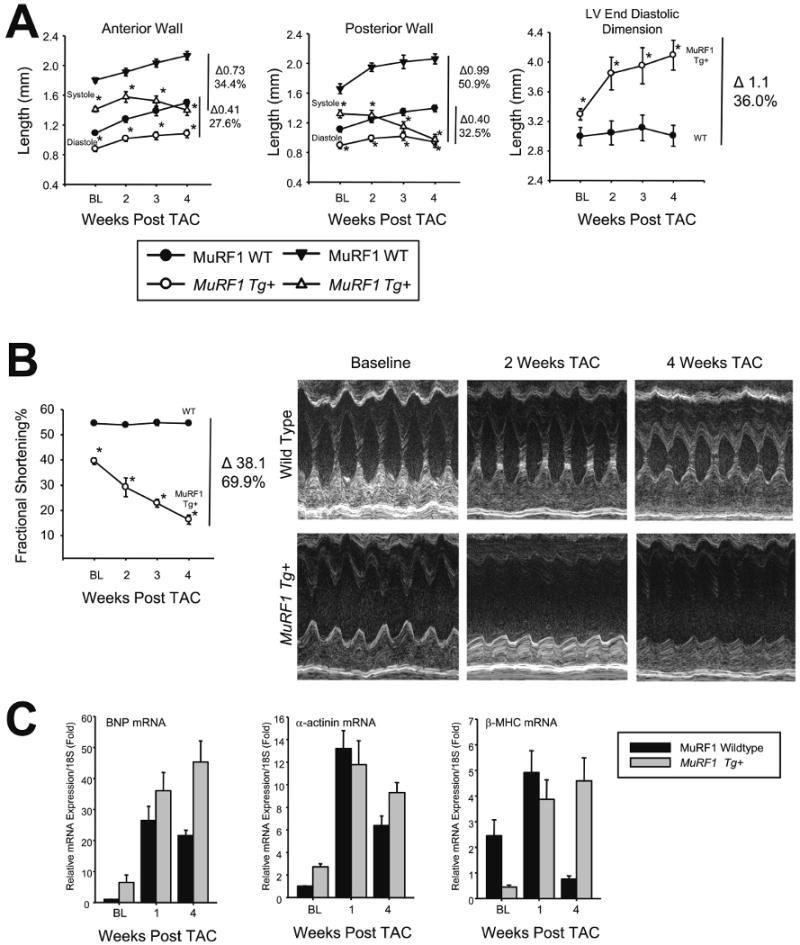
A. Echocardiographic assessment of anterior wall thickness, posterior wall thickness, and left ventricular dilation as measured by left ventricular dimension in diastole. TAC leads to a significant increase in left ventricle anterior and posterior wall thickness in wild type mice (left, middle), but not in MuRF1 Tg+ mice. MuRF1 Tg+ hearts, but not wild type, exhibited a concurrent increase in LV end diastolic dimension (right). N=10/group. B. Baseline decreases in FS% and wall thickness are exaggerated after TAC. N=10/group. C. MuRF1 Tg+ hearts have increased fetal gene gene expression compared to wild type mice after 4 weeks TAC. N=3/group. A student's T test was used to determine significance at each time point. *p<0.001.
Increased cardiac MuRF1 enhances susceptibility to heart failure in response to pressure overload
In our second series of studies, MuRF1 Tg+ and wild type littermates underwent surgery to induce pressure overload by trans-aortic constriction (TAC), and were monitored for survival daily, with cardiac function analyzed weekly by echocardiography. Following TAC, no differences in survival between MuRF1 Tg+ and wild type mice were identified for the duration of the 4-week study. MuRF1, but not MuRF2, mRNA levels significantly decreased to 40% of baseline in both wild type mice during the progression of cardiac hypertrophy (Online Figure 1C and 1D). Although MuRF1 Tg+ mice survival following TAC did not differ from littermate wild type controls, MuRF1 Tg+ mice rapidly progressed to heart failure during this time (Figure 1A and B), while maintaining 20 fold levels of endogenous cardiac MuRF1 (Online Figure 1B). The mild baseline anterior and posterior wall thickness deficits seen in MuRF1 Tg+ mice become exaggerated (27.6% and 32.5% less than wild type mice, respectively in diastole) as did left ventricular dilation, with interventricular diameter increasing by 36% (Figure 1A, right) after TAC. The anatomical changes seen in MuRF1 Tg+ mice following TAC were accompanied by a rapid functional decompensation, with the degree of FS decreasing by 70% in these mice.
MuRF1 Tg+ mice develop an eccentric cardiac hypertrophy after TAC
Typically, in response to pressure overload, a heart will develop “concentric” hypertrophy, characterized by an increase in cardiac mass due to a corresponding increase in cardiomyocyte cross sectional area and wall thickness. Importantly, maintenance of function is preserved in this form of hypertrophy. Conversely, in “eccentric” hypertrophy, pressure overload causes an increase in cardiac mass accompanied by cardiomyocyte elongation, rather than increasing cross sectional area. In the present study, following TAC both MuRF1 Tg+ and wild type mice developed cardiac hypertrophy as evidenced by increases in cardiac mass (heart weight to body weight 8.3±0.5 vs. 8.4±0.8 mg/g, respectively, Online Table I) and the up-regulation of “fetal” genes associated with cardiac hypertrophy (BNP, smooth muscle α-actin, and β-MHC) (Figure 1C). In wild type mice, TAC induced a typical concentric hypertrophy with increased wall thickness and cardiomyocyte area (Figure 2A & 2B). However, MuRF1 Tg+ mice did not develop increases in wall thickness (Figure 2A) or cardiomyocyte cross sectional area (Figure 2B) and had the characteristic loss of function associated with eccentric cardiac hypertrophy (FS% in Figure 1B, EF% in Online Table I). Apoptosis of cardiomyocytes often accompanies eccentric hypertrophy, adding to the overall deficit in contractility and heart function. However, analysis of TUNEL staining of sections of cardiac muscle taken from wild type and MuRF1 Tg+ determined no difference in the level of apoptosis between MuRF1 Tg+ and wild type mice (Figure 2C). Therefore, increased loss of cardiac cells was not an apparent contributing factor to the development of heart failure in the MuRF1 Tg+ mice.
Figure 2. Increased cardiac MuRF1 does not affect apoptosis, but does result in aberrant M-line formation.
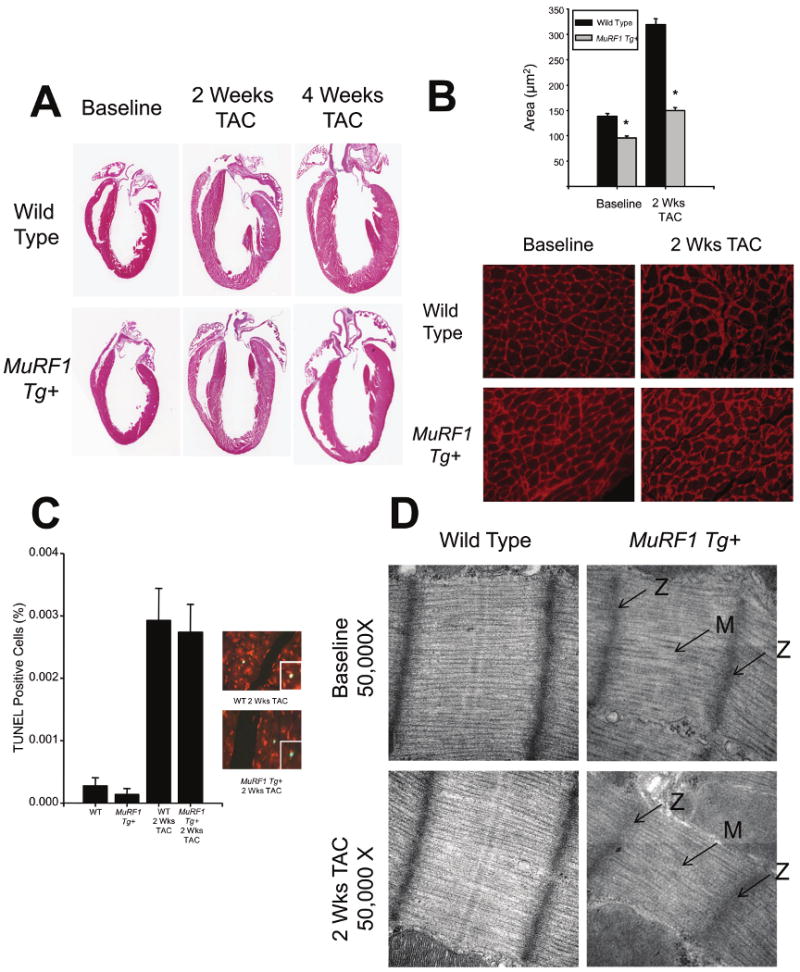
A. MuRF1 Tg+ hearts do not increase wall thickness in response to TAC. B. MuRF1 Tg+ cardiomyocytes have reduced cross sectional area at baseline, and after the induction of cardiac hypertrophy. N=3/group. C. TUNEL staining of whole heart sections from MuRF1 Tg+ and wild type mice indicate that no differences in apoptosis after the induction of cardiac hypertrophy. Inset: Higher power view of TUNEL positive cells. N=4/group. D. MuRF1 Tg+ hearts have sarcomere changes at the M-line, which is exaggerated after TAC. Arrows indicate disruption of the M line (M) and Z disk (Z). Photomicrographs represent at least 3 independent animals/group. A student's T test was used to determine significance at each time point. *p<0.001.
MuRF1 Tg+ mice have alterations in cardiac sarcomere M-line
MuRF1 has been localized to the M-line, and to a lesser extent the Z-disc of the sarcomere 9. Therefore, we sought to determine whether the loss of cardiac function observed in the MuRF1 Tg+ mice could be the result of defects in the structure of the cardiomyocyte contractile apparatus. Low power transmission electron microscopy revealed no gross defects in sarcomeric or mitochondrial structure in MuRF1 Tg+ cardiomyocytes (Online Figure 2B). However, under high power evaluation, MuRF1 Tg+ cardiomyocytes displayed a mild disruption in the M-line, which was exaggerated following TAC (Figure 2D). The dramatic deficits in cardiac function we observed in the MuRF1 Tg+ mice seem out of proportion to this relatively mild sarcomeric defect, leading us to consider additional causative factors leading to the heart failure in these mice.
Increased cardiac MuRF1 expression is not accompanied by increased ubiquitin-dependent degradation of sarcomeric proteins in vivo
We have previously demonstrated in vitro that MuRF1 recognizes and targets cTnI for degradation 3, and that the degree of degradation is directly proportional to the amount of MuRF1 present in the system. It is possible that elevated levels of MuRF1 could affect steady state levels of cardiac TnI in the MuRF1 Tg+ mice, thereby leading to disruption of sarcomeric function and subsequent heart failure. Upon investigation however, we found that steady state levels of cTnI did not differ between MuRF1 Tg+ and wild type hearts either at baseline or after TAC (Online Figure 3A). Similarly, steady state levels of other sarcomere proteins reported to interact with MuRF1 (including cTnT, cTnC, Telethonin, and MLC2 10) were also unaffected (Online Figure 3A and 3B). Furthermore, we detected no difference in cTnI mRNA levels between MuRF1 Tg+ and wild type mice (Online Figure 3C), ruling out transcriptional compensation as a means by which protein levels of cTnI were maintained in MuRF1 Tg+ mice. Although these findings contrast sharply with our previous in vitro studies 3, the apparent lack of effect of increased MuRF1 levels on these sarcomeric proteins is consistent with our findings in the hearts of MuRF1 -/- mice{Willis, 2007 #26}, where steady state levels of these proteins also did not differ from their strain-matched wildtype controls, either at baseline (Online Figure 4) or after TAC (data not shown). These results suggest that, in the hearts of MuRF1 Tg+ mice, MuRF1 is not obviously triggering ubiquitin-dependent degradation of sarcomeric proteins. This surprising finding is the first indication that MuRF1 may be playing a novel, and as yet unidentified role in regulating cardiac function, independent of its role as a ubiquitin ligase acting upon sarcomeric proteins.
Genes involved in cardiac energy metabolism are differentially expressed in MuRF1 Tg hearts
Our physiologic data indicate that MuRF1 Tg+ mice have only minor functional cardiac deficits at baseline (including a small decrease in both fractional shortening and ventricular wall thickness), but that upon the induction of cardiac hypertrophy via TAC, an eccentric cardiac hypertrophy rapidly develops and leads into heart failure. Examination of the levels of apoptosis and sarcomere protein degradation in the MuRF1 Tg+ mice did not reveal any mechanisms underlying the observed heart failure. Therefore, in order to gain a more global view of how increased MuRF1 expression affects cardiac physiology we used microarray analysis to identify changes in gene expression in MuRF1 Tg versus wild type mouse hearts, both at baseline and during the development of cardiac hypertrophy (1 and 4 weeks following TAC). We identified 3 major patterns of differentially expressed genes between MuRF1 Tg+ and wild type mice, designated Cluster 1, 2, and 3 (Online Figure 5).
Clusters 1 and 2 represent genes that were differentially expressed between the MuRF1 Tg and wild type mice ONLY at 4 weeks after TAC (Online Figure 5A, outline in blue). The genes comprising Cluster 1 exhibited decreased expression levels in both MuRF1 Tg and wild type mice (red) after 1 week of TAC. However, whereas the expression level of these genes returned to baseline in wild type mice by 4 weeks of TAC, in MuRF1 Tg mice these genes continued to exhibit decreased expression levels. Of the three clusters identified, it was only Cluster 1 that yielded a distinct pattern to the signaling pathways represented by the genes in this group. Cluster 1 genes were comprised mainly of members of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontologies (GO) associated with generation of energy from oxidative phosphorylation and mitochondrial genes (Mrlpl28, Slc25a10, Mrps28, Dci, Mrpl46, Ndufb8, Ndufs4, Cox4l1, Ndufb5, Ndufv2, Sdha, Dlat, Atp5j, and Bcl2l11) as outlined in more detail in Online Figure 5B. The pathways represented by these genes include the MSigDB categories of mitochondria, oxidative phosphorylation, and PGC; the KEGG categories of oxidative phosphorylation, TCA cycle; and the GO categories of precursor metabolites and energy, coenzyme metabolism, and cofactor metabolism (see Online Figure 5B). Interestingly, the PGC pathway, represents genes involved in PGC-1 signaling, which serve as inducible coregulators of nuclear receptors that control cellular energy metabolic processes 12.
The genes identified in Cluster 2 of our analysis were characterized by an increase in expression in both MuRF1 Tg and wild type mice (red) after 1 week of TAC, which at 4 weeks TAC decreased to baseline levels in the hearts of wild type mice but remained elevated in MuRF1 Tg hearts (Online Figure 5A). Only a few gene pathway signals were identified in Cluster 2 and these included genes in the MSigDB category PGC and lvad_heartfailure_up, consistent and overlapping with the metabolic pathways seen in cluster 1. Finally, Cluster 3 was comprised of those genes that displayed increased expression in MuRF1 Tg mouse hearts compared to wild type mouse hearts at any time point investigated (Online Figure 5A). Since these genes were consistently increased over time, they likely represent fundamental changes of the heart transcriptome brought about by increased MuRF1 expression, independent of the underlying cardiac hypertrophy seen in these mice. Analysis of Cluster 3 genes using MSigBD identified genes in the category of mitochondria, PGC, and lvad_heartfailure_up and the GO categories of precursor metabolites and energy and energy from oxidation of organic compounds. The results of pathway analysis of the genes found to be differentially expressed in wild type and MuRF1 Tg mice over the course of TAC strongly suggest that MuRF1 wields an effect on cardiac energy metabolism - both at baseline and after the development of cardiac hypertrophy. This realization led us to investigate next the ability of MuRF1 Tg hearts to forge metabolic adaptations after TAC.
Cardiac MuRF1 does not affect cardiac glucose and fatty acid utilization
Our microarray data supported the hypothesis that cardiac MuRF1 regulates key metabolic processes involved in oxidative phosphorylation, which is closely linked to ATP production. In addition, recent studies have identified that MuRF1 interacts with several proteins involved in glucose oxidation, including pyruvate kinase, Aldolase A, and pyruvate dehydrogenase kinase 10. This prompted us to investigate the possibility that MuRF1 was involved in the regulation of glucose oxidation, since maladaptive shifts in glucose oxidation during the development of cardiac hypertrophy could explain the functional deficits seen in MuRF1 Tg+ hearts. Using 14C-labeled glucose or 14C-labeled oleate (a long chain fatty acid) we were able to quantify the efficiency with which whole heart homogenates from MuRF1 Tg+ and wild type mice oxidized glucose and oleate at baseline and 4 weeks following TAC (Figure 3A and 3B). As expected, the hearts of wild type mice utilized more oleate (fatty acids) than glucose at baseline, and shifted toward glucose utilization after TAC (Figure 3C). Surprisingly though, MuRF1 Tg+ mice also utilized oleate and glucose to a similar extent (Figure 3A and B). This lack of differences in cardiac glucose and fatty acid utilization suggested that MuRF1 regulation of glucose oxidation is not a causative factor in the cardiac dysfunction seen in the MuRF1 Tg+ mice in response to pressure overload.
Figure 3. MuRF1 Tg+ hearts metabolize glucose and fatty acid to the same extent as littermate wild type controls at baseline and after TAC.
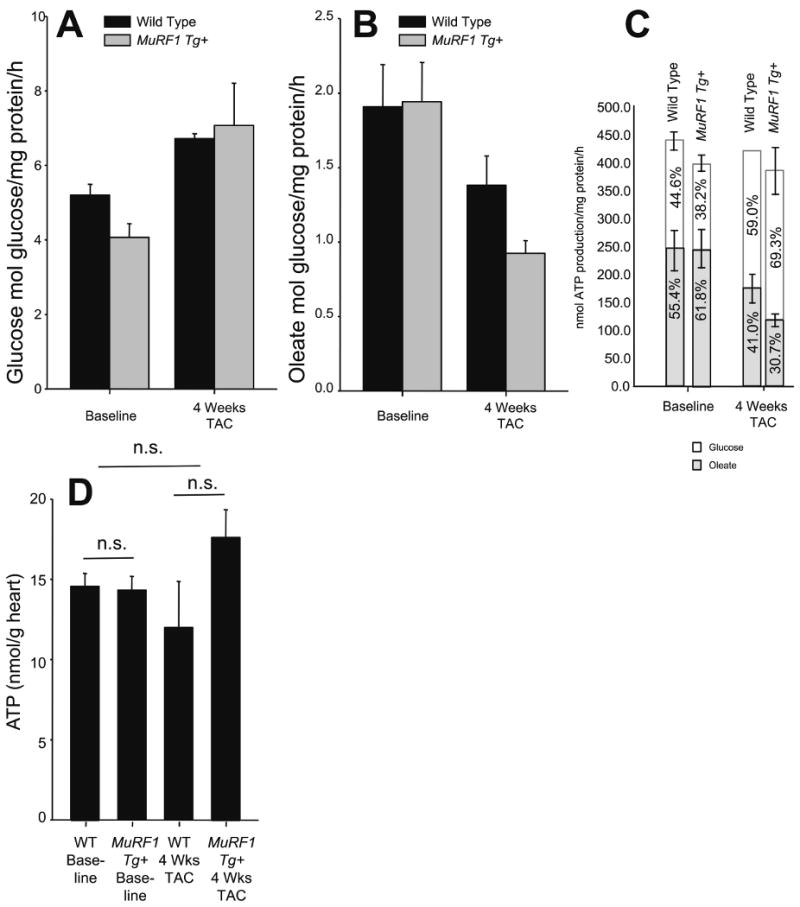
A. Glucose and B. Fatty acid (oleate) oxidation of wild type and MuRF1 Tg+ whole heart homogenates at baseline and after 4 weeks TAC. The relative contribution of glucose and oleate to ATP production shifts after the induction of hypertrophy (C), such that there is an enhanced shift in glucose contribution to ATP production, without significant changes in overall ATP production. N=3 animals/group run in triplicate. (D) Total ATP measured from whole heart homogenates. N=3 animals/group.
MuRF1 Tg+ hearts maintain total ATP concentrations
Data obtained from our microarray analysis indicated that genes found in mitochondria and involved in oxidative phosphorylation including Ndufb8, Ndufs4, Ndufb5, Ndufv2, Atp5j were significantly downregulated in the hearts of MuRF1 Tg+ mice. A decrease in ATP production could account for the deficient contractility exhibited by MuRF1 Tg+ hearts. However, analysis of cardiac ATP levels revealed that total ATP did not differ significantly between MuRF1 Tg+ and wild type mice at baseline or 4 weeks following TAC (Figure 3D), indicating that MuRF1 Tg+ hearts progressively failed during pressure overload despite having adequate levels of ATP, suggesting that the deficit may in fact lie in the ability of cardiac MuRF1 hearts to utilize ATP. This led us to investgate whether or not MuRF1 Tg+ hearts displayed any maladaptions in pathways necessary for effective ATP usage.
Increased cardiac MuRF1 inhibits creatine kinase activity in vivo
In the heart, the phospho-creatine-ATP shuttle transports ATP throughout the cell to maintain ATP-dependent processes such as ion (i.e. Ca2+, K+) transport through channels necessary for contractility. Vital to this ATP transport is the creatine kinase (CK) enzyme, which is responsible for the transport of ATP from the mitochondria to many areas of the cell through a phospho-creatine intermediate 13. A recent in vitro study suggested that CK is a MuRF1 substrate for ubiquitination 14, prompting us to investigate whether CK expression or activity was altered in the MuRF1 Tg+ mice. Indeed, we found that total CK activity was significantly depressed in MuRF1 Tg+ hearts compared to wild type hearts at baseline. In addition, the normal increase in enzyme activity seen in wild type mice following the induction of hypertrophy was considerably attenuated in the MuRF1 Tg+ hearts (Figure 4A).
Figure 4. MuRF1 Tg+ hearts have an attenuated total CK activity and increased mitochondrial number after TAC.
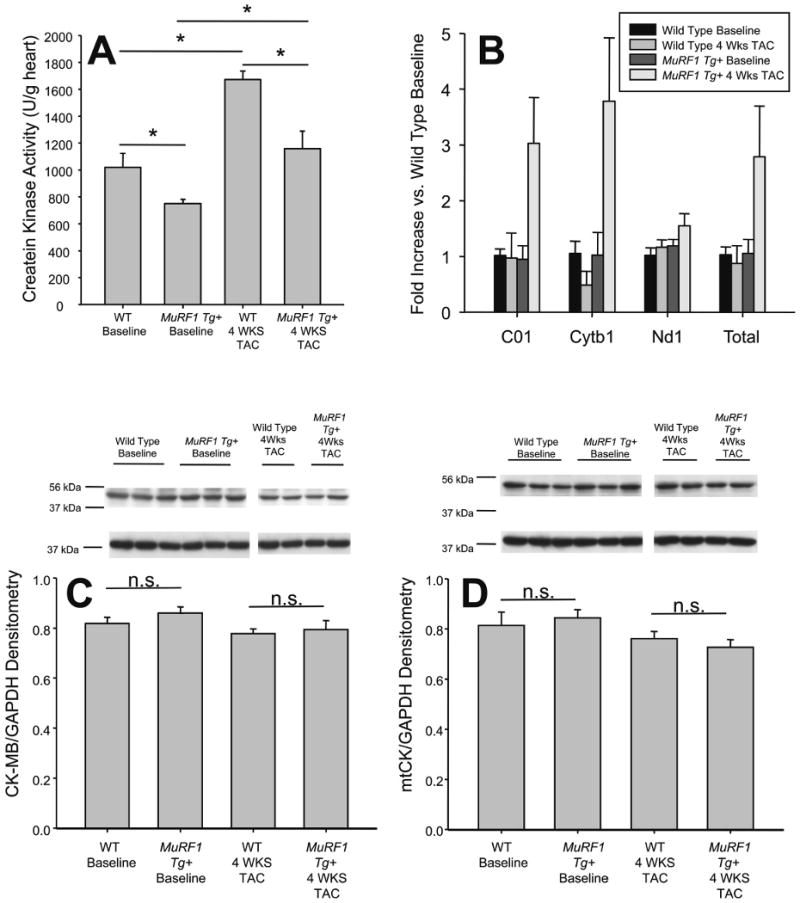
A. Creatine kinase (CK) activity of whole heart homogenates are attenuated in MuRF1 Tg+ mice at baseline, and after 4 weeks TAC. B. Real time analysis of cardiac mitochondrial genes C01, Cytb1, and Nd1 in MuRF1 Tg+ and wild type mice, referenced to genomic DNA at baseline and after TAC. C. Western immunoblot analysis of cardiac CK-M/B isoforms in wild type and MuRF1 Tg+ hearts at baseline and after TAC. B. Western immunoblot analysis of the mitochondrial CK isoform in wild type and MuRF1 Tg+ hearts at baseline and after TAC. A one way ANOVA was performed to determine significance, followed by a multiple comparison procedures (Holm-Sidak method) to determine significance between groups. *p<0.05. n.s.=not significant.
Several CK isoforms exist in the heart, which are able to form dimers, including CK-MM (∼70% of total cardiac CK) and CK-MB (∼25-30% of total cardiac CK). A mitochondrial isoform (mtCK) also exists which is essential for the transport of ATP out of the mitochondria. Using antibodies that recognize each of the CK isoforms, we determined that MuRF1 Tg+ heart CK-MM/CK-MB and mtCK steady state protein levels did not differ from wild type hearts (Figure 4C and D), although increased levels of CK-M/B modification in MuRF1 Tg+ hearts was evident (Online Figure 6C). The lack of decrease in CK isoform protein levels in MuRF1 Tg+ hearts was not due to compensatory incresases in CK mRNA, as measurements of CK-M, CK-B mRNA levels of were equal in both MuRF1 Tg+ hearts and wild type controls (Online Figure 6A). Interestingly though, quantitative analysis of 3 mitochondrial DNA components in MuRF1 Tg and wild type mice identified a nearly 2-3 fold increase in the number of mitochondria in the MuRF1 Tg mice (Figure 4B and Online Figure 7). This finding of increased mitochondrial numbers in the MuRF1 Tg+ hearts without a concomittent increase in mtCK levels might suggest that mtCK is preferentially degraded by MuRF1, although further studies would be needed in order to vailidate this. Regardless, the fact that CK protein levels did not differ between MuRF1 Tg+ and wild type hearts was a surprising finding given that we had detected a decrease in CK activity in these mice and previous studies had identified (in vitro) that CK is a target for MuRF1 ubiquitin ligase activity 3, 14. These results prompted us to investigate how CK activity is differentially regulated in MuRF1 Tg+ hearts compared to wild type hearts.
Our knowledge of the mechanisms by which creatine kinase activity is regulated is limited. Recent studies have identified that post-translational modification of CK by AMPK phosphorylation supports CK activity 15, 16. In an effort to determine if MuRF1 was possibly affecting CK activity by its affects on AMPK, we quantified the amount of phosphorylated AMPK in MuRF1 Tg and wild type hearts at baseline and after 4 weeks TAC (Online Figure 6B). We identified that MuRF1 Tg hearts had a small, but significant, decrease in phosphorylation AMPK compared to wild type mouse hearts. Although it remains to be determined whether or not this decrease in AMPK phosphorylation is due to imbalances in AMP/ATP levels secondary to heart failure, or due to other direct or indirect effects related to the increase in cardiac MuRF1 expression. These results suggest that MuRF1 regulates myocardial CK activity, possibly through its ubiquitin-mediated degradation and turnover of mtCK, or other non-degradatory pathways in which ubiquitin can regulate enzyme activity.
Discussion
In this report, we demonstrate that MuRF1 in vivo is able to manipulate cardiac function through its effects on the regulation of cardiac metabolism. We identify for the first time that increased cardiac MuRF1 pre-disposes the heart to failure, possibly due to its attenuation of CK activity, the enzyme responsible for the transfer of ATP from the mitochondria to cellular compartments throughout the cell.
CK has recently been identified as a target substrate of MuRF1 ubiquitination 14, 17. However, while these reports demonstrated MuRF1's ability to ubiquitinate CK, the in vitro platform on which these studies were performed did not allow investigations into whether or not this ubquitination was preemptive of actual degradation of CK. In our study, we observed a decrease in CK activity in MuRF1 Tg+ hearts in the absence of any change in steady state CK enzyme protein levels, or compensatory increases in CK mRNA. However, there was evidence of increased mitochondrial number, consistent with our microarray findings of regulation of the PGC-1 pathways in the MuRF1 Tg+ mice. Although these results do not prove a direct causal relationship between increased MuRF1 expression and decreased CK activity, it is possible that MuRF1 is degrading the possible increased mtCK, or that MuRF1 is regulating CK activity in a manner that is independent of a degradatory pathway. The ubiquitination of substrates by ubiquitin ligases, such as MuRF1, can result in outcomes other than degradation, such as protein relocalization, protein repair/refolding, and cellular signaling (reviewed by Willis et al., 200618). Our laboratory recently identified that the E3 ligase Atrogin-1 enhances the activity of the transcription factor FOXO by placing ubiquitin chains on it 19. Alternatively, the lack of degradation of CK proteins in the MuRF1 Tg+ mice could be due to a compensatory increase in de-ubiquitinating proteins (DUBs). In mammalian genomes there are more than 100 genes that encode putative DUBs, consistent with the diverse and specific functions they regulate 20-22. Our studies demonstrate that MuRF1 regulation of cardiac CK activity in vivo is more complex than the simple ubiquitin mediated degradation of a target. MuRF1 may play a role in regulating CK in a degradation-independent manner, or it is possible compensatory mechanisms are preventing the identification of MuRF1-mediated CK degradation.
Although further studies will be needed in order to identify the exact mechanisms involved, our results clearly demonstrate that MuRF1 plays a role in the largely unchartered area of the regulation of CK activity in vivo. Since CK is the key component of the ATP/Phospho-Creatine System 23, decreases in creatine kinase results in impaired transfer of ATP from the mitochondria to transporters, pumps, and enzymes in the cytosol and sarcomeres necessary to maintain cardiac function 23-25. In the present study, we identified that MuRF1 Tg+ hearts did have a decreased level of phospho (Thr172)-AMPK, which opens the possibility that MuRF1 may be influencing CK activity indirectly through AMPK. While a complete understanding of how CK phosphorylation is necessary for CK activity is not yet known, phosphorylation by AMPK may be necessary for optimal activity. Hence, the effects of MuRF1 on CK-MM/MB and/or mtCK activity could, in part, underlie the functional deficits seen in MuRF1 Tg+ mice that are amplified upon the induction of cardiac hypertrophy.
The decrease in cardiac CK activity, as seen in MuRF1 Tg+ mice in this study, has been associated with the development of heart failure. The effect of decreased CK activity in the heart has not been studied directly, however cardiac function following the complete loss of cardiac CK isoforms has been reported. CK deficient mice have a close to normal cardiac function at low or moderate workloads 26, and have been reported to develop a spontaneous hypertrophy on a mixed, but not pure background27. However, with increased workloads, fiber kinetics were found to be impaired 26. By echocardiography, mice deficient in CK have normal cardiac function at baseline, but their response to beta-adrenergic stimulation is blunted 28. These phenotypes parallel in many ways the near normal baseline cardiac function (determined by our catheter studies, Online Table III) we see in MuRF1 Tg+ hearts, which quickly decompensates upon stress brought on by TAC. MuRF1's attenuation of CK therefore may be largely responsible for the functional impairment of cardiac contractility of MuRF1 Tg+ mice. While it is possible that MuRF1 effects on CK activity are not a result of direct interaction, the interaction of MuRF1 and CK has been identified in multiple in vitro studies 10, 14, 17. It cannot be assumed that MuRF1 effects on CK activity results from the cardiac dysfunction identified, as abnormal phospho-creatine energetics have been shown to proceed, rather than are a consequence, of cardiac hypertrophy29.
Since we previously identified that MuRF1 interacts directly with troponin I, post-translationally modifies it with ubiquitin to target it for degradation by the proteasome in vitro 3, it was surprising to identify that cardiac MuRF1 levels in vivo did not affect steady state levels of cTnI in the present study. This finding might be explained by recent studies that have identified regulatory systems that prevent the UPS from interacting with sarcomeric proteins 30, 31. Briefly, activation of the calcium-activated proteins (calpains) have been implicated in the homeostatic turnover of sarcomere proteins 30, 31. Increased cardiac calpain-1 in mice resulted in increased substrate-specific proteolytic activity and hyper-ubiquitination of cardiac proteins with increased 26S proteasome activity and accelerated protein turnover 30. Similarly, forced over-expression of cardiac calpastatin (a naturally occurring calpain inhibitor) had opposite effects 30. This study demonstrated that adenovirus transfection of cardiomyocytes activated physiological calpain-1 30, which would explain why our adenoviral mediated expression of MuRF1 resulted in degraded cTnI in vitro 3. The ubiquitin ligase directed degradation of sarcomere proteins is part of a larger program of protein quality control just now being elucidated (recently reviewed by Willis et al., 200932).
Acknowledgments
The authors wish to thank Robert Bagnell and Vicky Madden in the Microscopy Services Laboratory in the UNC Department of Pathology & Laboratory Medicine for their assistance with the electron microscopy experiments. We also wish to thank Janice Weaver in the UNC Animal Histopathology Laboratory for assistance in preparing histological specimens.
Sources of Funding: This work was supported by NHLBI R01HL065619 (to CP) and support from the UNC Research Council, the UNC Foundation's R.J. Reynolds Faculty Development Award, and Children's Cardiomyopathy Foundation, and the AHA Scientist Development Grant (to MW).
Abbreviations
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- cTnC
cardiac troponin C
- CK
creatine kinase
- MLC2
myosin light chain 2
- MuRF1
muscle ring finger-1
- TAC
trans-aortic constriction
Footnotes
Disclosures: None.
References
- 1.Hoshijima M. Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am J Physiol Heart Circ Physiol. 2006;290:H1313–1325. doi: 10.1152/ajpheart.00816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 3.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregorio CC, Perry CN, McElhinny AS. Functional properties of the titin/connectin-associated proteins, the muscle-specific RING finger proteins (MURFs), in striated muscle. J Muscle Res Cell Motil. 2005;26:389–400. doi: 10.1007/s10974-005-9021-x. [DOI] [PubMed] [Google Scholar]
- 5.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, Patterson C. Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167:1147–1159. doi: 10.1083/jcb.200402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 9.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 10.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 11.Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008;27:350–360. doi: 10.1038/sj.emboj.7601952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 14.Zhao TJ, Yan YB, Liu Y, Zhou HM. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J Biol Chem. 2007;282:12022–12029. doi: 10.1074/jbc.M610363200. [DOI] [PubMed] [Google Scholar]
- 15.Palmer AK, Fraga D, Edmiston PL. Regulation of Creatine Kinase Activity by Phosphorylation of Serine-199 by AMP-Activated Kinase. FASEB J. 2008;22 1012.1010. [Google Scholar]
- 16.Ingwall JS. Is creatine kinase a target for AMP-activated protein kinase in the heart? J Mol Cell Cardiol. 2002;34:1111–1120. doi: 10.1006/jmcc.2002.2062. [DOI] [PubMed] [Google Scholar]
- 17.Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, Doi N, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, Franz T, Labeit S, Sorimachi H. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol. 2008;376:1224–1236. doi: 10.1016/j.jmb.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 18.Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41:567–579. doi: 10.1016/j.yjmcc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guterman A, Glickman MH. Deubiquitinating enzymes are IN/(trinsic to proteasome function) Curr Protein Pept Sci. 2004;5:201–211. doi: 10.2174/1389203043379756. [DOI] [PubMed] [Google Scholar]
- 21.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Swaminathan S, Amerik AY, Hochstrasser M. The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol Biol Cell. 1999;10:2583–2594. doi: 10.1091/mbc.10.8.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlattner U, Tokarska-Schlattner M, Wallimann T. Mitochondrial creatine kinase in human health and disease. Biochim Biophys Acta. 2006;1762:164–180. doi: 10.1016/j.bbadis.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Gross M, Lustig A, Wallimann T, Furter R. Multiple-state equilibrium unfolding of guanidino kinases. Biochemistry. 1995;34:10350–10357. doi: 10.1021/bi00033a005. [DOI] [PubMed] [Google Scholar]
- 25.Hornemann T, Stolz M, Wallimann T. Isoenzyme-specific interaction of muscle-type creatine kinase with the sarcomeric M-line is mediated by NH(2)-terminal lysine charge-clamps. J Cell Biol. 2000;149:1225–1234. doi: 10.1083/jcb.149.6.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boehm E, Ventura-Clapier R, Mateo P, Lechene P, Veksler V. Glycolysis supports calcium uptake by the sarcoplasmic reticulum in skinned ventricular fibres of mice deficient in mitochondrial and cytosolic creatine kinase. J Mol Cell Cardiol. 2000;32:891–902. doi: 10.1006/jmcc.2000.1130. [DOI] [PubMed] [Google Scholar]
- 27.Lygate CA, Hunyor I, Medway D, de Bono JP, Dawson D, Wallis J, Sebag-Montefiore L, Neubauer S. Cardiac phenotype of mitochondrial creatine kinase knockout mice is modified on a pure C57BL/6 genetic background. J Mol Cell Cardiol. 2009;46:93–99. doi: 10.1016/j.yjmcc.2008.09.710. [DOI] [PubMed] [Google Scholar]
- 28.Crozatier B, Badoual T, Boehm E, Ennezat PV, Guenoun T, Su J, Veksler V, Hittinger L, Ventura-Clapier R. Role of creatine kinase in cardiac excitation-contraction coupling: studies in creatine kinase-deficient mice. FASEB J. 2002;16:653–660. doi: 10.1096/fj.01-0652com. [DOI] [PubMed] [Google Scholar]
- 29.Maslov MY, Chacko VP, Stuber M, Moens AL, Kass DA, Champion HC, Weiss RG. Altered high-energy phosphate metabolism predicts contractile dysfunction and subsequent ventricular remodeling in pressure-overload hypertrophy mice. Am J Physiol Heart Circ Physiol. 2007;292:H387–391. doi: 10.1152/ajpheart.00737.2006. [DOI] [PubMed] [Google Scholar]
- 30.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW., 2nd Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res. 2007;100:1071–1078. doi: 10.1161/01.RES.0000261938.28365.11. [DOI] [PubMed] [Google Scholar]
- 31.Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet. 2005;14:2125–2134. doi: 10.1093/hmg/ddi217. [DOI] [PubMed] [Google Scholar]
- 32.Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up-Tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. 2009;81:439–448. doi: 10.1093/cvr/cvn289. [DOI] [PMC free article] [PubMed] [Google Scholar]