

Abstract
Recombineering with Saccharomyces cerevisiae is a powerful methodology that can be used to clone multiple unmarked pieces of DNA to generate complex constructs with high efficiency. Here we introduce two new tools that utilize the native recombination enzymes of S. cerevisiae to facilitate the construction of a variety of useful tools for bacteria. First, yeast recombineering was used to make directed nested deletions in a bacterial-yeast shuttle plasmid using only one or two single stranded oligomers, thus obviating the need for a PCR step. Second, we have generated several new shuttle vectors for yeast recombineering capable of replication in a wide variety of bacterial genera. As a demonstration of utility, some of the approaches and vectors generated in this study were used to make a pigP deletion mutation in the opportunistic pathogen Serratia marcescens.
1. Introduction
Cloning by Saccharomyces cerevisiae homologous recombination is a powerful technique that has been harnessed for bacterial applications (Raymond, et al., 2002a, Shanks, et al., 2006). A “gap” or double stranded break (DSB) in a vector capable of replication in yeast can be repaired through homologous recombination with PCR-generated amplicons, because this repair event can be selected for with markers on the vector backbone such that the amplicon can be unmarked, i.e. have no selectable marker (DeMarini, et al., 2001, Jansen, et al., 2005, Mallet and Jacquet, 1996, Oldenburg, et al., 1997, Orr-Weaver and Szostak, 1983). The advantages of yeast recombination are A) that multiple unmarked pieces of DNA can be sewn together seamlessly without multiple rounds of amplification and without restriction sites on the ends, B) the site of recombination in a vector is not limited to a cut junction and can be hundreds of base pairs away, C) the method is efficient, robust and simple to perform. Yeast recombination can require longer overall time due to the growth rate of S. cerevisiae; however, the hands-on experimental time requirements are minimal.
Here we report two new tools that take advantage of yeast recombineering. The first is a method to make targeted deletions and short insertions in cloned genes using single stranded DNA oligomers. The second is a new set of bacterial-yeast shuttle vectors incorporating a number of replicons, promoters and selectable markers for a variety of uses in Gram-positive and Gram-negative organisms, which can be utilized for recombineering with budding yeast.
2. Materials and methods
2.1. Microbes and growth conditions
Bacterial and yeast strains used in this study are listed in Table 1. All bacteria were cultured using LB medium (Bertani, 1951). S. cerevisiae was cultured with YPD broth (1% Bacto-yeast extract, 2% Bacto-peptone and 2% dextrose), and selections were done using uracil drop-out medium (SC-ura) (Burke D., 2000). For β-galactosidase (β-gal) activity determination, SC-ura medium was modified by the addition of 80 μg/ml X-gal, was phosphate buffered (0.7% dibasic sodium phosphate and 0.3% monobasic sodium phosphate), and 2% galactose was substituted for dextrose (Burke D., 2000). Antibiotics were used at the following concentrations: for E. coli-ampicillin (100 μg/ml), gentamicin (10 μg/ml), and chloramphenicol (10 μg/ml) kanamycin (50 μg/ml), tetracycline (10 μg/ml) and streptomycin (25–100 μg/ml), for S. marcescens- kanamycin (100 μg/ml), gentamicin (20 μg/ml), for P. aeruginosa PA14- ampicillin (1500 μg/ml) and gentamicin (50 μg/ml), for S. aureus chloramphenicol (10 μg/ml) and kanamycin (25 μg/ml).
Table 1.
Plasmids and strains used in this study.
| Strain or Plasmid | Description | Reference or Source |
|---|---|---|
| E. coli EC100D | pir-116 | Epicentre |
| E. coli S17-1 λ-pir | conjugation donor strain | (Miller and Mekalanos, 1988) |
| S. marcescens D1 | wild type | Presque Isle Cultures |
| S. marcescens RSS1 | StrR, rpsL | This study |
| S. cerevisiae InvSc1 | diploid ura3-52/ura3-52 | Invitrogen |
| pYC2-CT-lacZ | oriColE1, PGAL1-lacZ, bla, CEN6, ARSH4, URA3 | Invitrogen |
| pSC189 | oriR6K-γ, nptII, bla, mariner-C9 | (Chiang and Rubin, 2002) |
| pRS416 | CEN6, ARSH4, bla, lacZa, oriColE1 | (Sikorski and Hieter, 1989) |
| pSK236 | bla, cat194, oripC194, oriColE1 | (Gaskill and Khan, 1988) |
| pGBNKBOR | Tn10, oriR6K-γ, replicon-pSC101ts, nptII | (Rossignol, et al., 2001) |
| pDG783 | aphA-3, oriColE1 | (Guerout-Fleury, et al., 1995) |
| pEX18-Gm | aacC1, oriT, sacB, lacZα, oriColE1 | (Hoang, et al., 1998) |
| pTV32-OK | Tn917-lacZ, oripWV01, aphA3 | (Cvitkovitch, et al., 2000) |
| pBBR1MCS-5 | aacC1, mob, rep, lacZα, oripBBR1 | (Kovach, et al., 1995) |
| pMMB66EH | bla, RSF1010 replicon, lacIQ, Ptac, oriT | (Fürste, et al., 1986) |
| pMQ30 | oriColE1, CEN6, ARSH4, aacC1, lacZα, oriT, sacB | (Shanks, et al., 2006) |
| pMQ75 | orip15a, CEN6, ARSH4, aacC1, lacZα, oriT, sacB | (Shanks, et al., 2006) |
| pMQ87 | CEN6, ARSH4, aacC1, lacZa, oriT, oriColE1 | (Shanks, et al., 2006) |
| pMQ91 | oriColE1, CEN6, ARSH4, bla, lacZα, oriT, oripRO1600 | (Shanks, et al., 2006) |
| pBK233 | oripUB110, tetL, oriColE1, bla, I-SceI | Joseph Horzempa |
2.2. Oligonucleotide-mediated recombination
Oligomers were ordered with no special cleaning (Integrated DNA Technologies) and were suspended in TE to 100μM. The pYC2-CT-lacZ plasmid (Table 1) was digested with AatII (10–20 fold over-digestion) and adjusted to 5 ng/μl. Full single colonies of S. cerevisae (ura3-52) were used to inoculate YPD broth (5 ml in a 20 ml test tube with aeration) and were grown over-night (~18 hours) with aeration. Aliquots (0.5 ml) of S. cerevisiae were centrifuged, washed with TE (0.5 ml) and covered with 0.5 ml “Lazy Bones” transformation solution (40% PEG 3350, 0.1M lithium acetate, 10mM Tris-Cl -pH 7.5, 1mM EDTA). To this mixture was added 25 μl of boiled salmon sperm carrier DNA (2 mg/ml), 10 μl of AatII-linearized or BamHI-linearized vector DNA (total of 50 ng), and the experimental oligomer(s) (25 μl for the single oligonucleotide experiment with AatII-linearized DNA, 0.1–10 μl (Table 3) with the BamHI-linearized DNA experiment (Table 4), and 10 μl of each oligonucleotide for the two-oligomer experiment (Table 5)), or an equal volume of TE for the no oligonucleotide control. This represents a 17,000 (10μl) or 42,500 (25μl):1 molar ratio of oligomer molecules to plasmid molecules. The mixture was vortexed for 60 s, incubated at room temperature for 20 hours, then heat shocked for 12 minutes at 42 C, centrifuged to pellet cells, the transformation mixture was removed, the cells were suspended in 150μl of TE, and plated onto SC-ura selection plates. These were incubated at 30 C for 3–4 days. A subset of colonies was patched onto SC-ura X-gal plates to determine β-gal activity, and these were incubated for 3 days at 30 C for unambiguous color development (Tables 3–4). Three independent experiments were conducted for each data set.
Table 3.
Oligmonucleotide-mediated recombination with one primer
| Oligomera | Number Ura+ CFUb | % Ura+ β-gal− c | P-valued |
|---|---|---|---|
| None | 324 | 20.2 | 1 |
| Δ13 | 1984 | 88.1 | >0.0001 |
| Δ96 | 470 | 63.3 | >0.0001 |
| Δ996 | 704 | 32.5 | 0.015 |
| Δ996-40 bp | 236 | 10.2 | 0.085 |
All oligomers were 60 bp except where noted, name indicates the number of deleted base pairs (bp), vector was digested with AatII.
Total number of transformants from three separate experiments.
Frequency of transformants with defective lacZ genes, as determined by absence of color on X-gal plates (n≥114 colonies per oligomer group).
Two-tailed Chi-Square test with Yates correction, comparing the frequency of lacZ mutations compared to the vector alone class.
Table 4.
Oligonucleotide-mediated recombination with one primer
| Oligomera | Number Ura+ CFUb | % Ura+ β-gal− c | P-valued |
|---|---|---|---|
| None | 280 | 7.4 | 1 |
| 0.1μl | 440 | 9.9 | >0.55 |
| 1.0μl | 390 | 29.6 | >0.0001 |
| 10.0μl | 606 | 44.3 | >0.0001 |
Oligomer lacZ-upstream-delta-100 was used at 100 μM, and targets a 100 base pair deletion. Vector was digested with BamH1.
Total number of transformants from three independent experiments.
Frequency of transformants with defective lacZ genes, as determined by absence of blue color on X-gal plates (n>150 colonies per oligomer class).
Two-tailed Chi-Square test with Yates correction, comparing the frequency of lacZ mutations compared to the no oligomer class.
Table 5.
Oligonucleotide-mediated recombination with two primers
| Oligomesa | Number Ura+ CFUb | % Ura+ β-gal− c | P-valued |
|---|---|---|---|
| None | 882 | 3.1 | 1 |
| Δ9/5 | 6512 | 49.0 | >0.0001 |
| Δ9/46 | 4182 | 38.6 | >0.0001 |
| Δ9/496 | 1998 | 23.4 | >0.0001 |
| Δ50/46 | 2792 | 13.1 | 0.0018 |
| Δ500/496 | 1252 | 15.2 | 0.0051 |
The oligomer names indicate the distance from the AatII mediated DSB.
Total number of transformants from three separate experiments.
Frequency of transformants with defective lacZ genes, as determined by absence of color on X-gal plates (n≥112 colonies per oligomer group).
Two-tailed Chi-Square test with Yates correction, comparing the frequency of lacZ mutations compared to the vector alone class.
2.3. Plasmid construction
Plasmids were made using yeast homologous recombination as previously described (Shanks, et al., 2006). PCR-generated DNA amplicons were made with primers that target recombination onto the previously described vectors that had been linearized with a restriction enzyme. Both the linearized vector and the amplicon were then used to cotransform a uracil auxotrophic (ura3-52) S. cerevisiae strain, InvSc1, as above. Uracil protrophs were pooled, plasmid DNA was isolated and was introduced into E. coli. Candidates were screened using restriction digest, diagnostic PCR and sequencing.
The pMQ117 plasmid was created by amplifying a temperature-sensitive version of the pSC101 replicon from pGBNKBOR with primers that target recombination into pMQ87 that had been cut with ApaL1, thus replacing the ColE1 origin (F-sc101, R-sc101, primers listed in Table 2).
Table 2.
Oligonucleotides used in this study for in S. cerevisae recombination.
| Name | Sequence |
|---|---|
| lacZ-delta-13 | CAGGATATGTGGCGGATGAGCGGCATTTTCTGCTGCATAAACCGACTACACAAATCAGCGa |
| lacZ-delta-96 | GGTGATGGTGCTGCGTTGGAGTGACGGCAGCCACTCGCTTTAATGATGATTTCAGCCGCG |
| lacZ-delta-996 | CTGGCGTAATAGCGAAGAGGCCCGCACCGATGATGAAGCAGAACAACTTTAACGCCGTGC |
| lacZ-delta-996-40 | AGCGAAGAGGCCCGCACCGATGATGAAGCAGAACAACTTT |
| F-lacZ-delta9 | aggatatgtggcggatgagcggcattttccAGTTACGCTAGGGATAACAGGGTAATATAG |
| R-lacZ-delta5 | cgctgatttgtgtagtcggtttatgcagcaCTATATTACCCTGTTATCCCTAGCGTAACT |
| F-lacZ-delta50 | ggtgatggtgctgcgttggagtgacggcagAGTTACGCTAGGGATAACAGGGTAATATAG |
| R-lacZ-delta50 | cgcggctgaaatcatcattaaagcgagtggCTATATTACCCTGTTATCCCTAGCGTAACT |
| F-lacZ-delta496 | ctggcgtaatagcgaagaggcccgcaccgaAGTTACGCTAGGGATAACAGGGTAATATAG |
| R-lacZ-delta500 | gcacggcgttaaagttgttctgcttcatcaCTATATTACCCTGTTATCCCTAGCGTAACT |
| F-lacZ-D2 | GATAGATCCCGTCGTTTTACAACGTCGb |
| R-lacZ-D3 | CGGAACTGGAAAAACTGCTGCTGGTGb |
| F-sc101 | ctttccagtcgggaaacctgtcgtgccagctgcattaatgCAGCGATTTGCCCGAGC |
| R-sc101 | ttgtcagaccaagtttactcatatatactttagattgattGCACCAAAAACTCGTAAAAG |
| F-wv01 | gtttgcgtattgggcgctcttccgcttcctcgctcacCCGATTTTTTATTAAAACGTCTC |
| R-wv01 | ccgctcatgagacaataaccctgataaatgcttcaataatctcgaGATTGCCTTGAATATATTGAC |
| F-pBBR1-y | ccgctttccagtcgggaaacctgtcgtgccagctgcattaatgCGCCCTACGGGCTTGC |
| R-p113-bbr | ctagataggggtcccgagcgcctacgaggaatttgtatcgGGCTTCCATTCAGGTCGAGG |
| R-p117-bbr | catatatactttagattgattgcaccaaaaactcgtaaaaGGCTTCCATTCAGGTCGAGG |
| R-ec-rpsL | cgctcatgagacaataaccctgataaatgcttcaataatCAGGATTGTCCAAAACTCTAC |
| F-Ec-rpsL | ctagaagcttctgcagacgGGTTTGACTGGTCAAATTTCG |
| R-kan-ALV | gaaatttgaccagtcaaacccgtctgcagaagcttctagaGGAACTTCGGAATAGGAAC |
| F-kan-ALV | ggaacacttaacggctgacagACGCTGCCGCAAGCACTCAG |
| R-r6k-ALV | ctgagtgcttgcggcagcgtCTGTCAGCCGTTAAGTGTTCC |
| F-r6k-ALV | ttgcgttgcgctcactgcccgctttccagtcgggaaaccGCTCTAGACCCCTATAGTGAG |
| F-Cen-D | GGTCCTTTTCATCACGTGC |
| R-oriT-D | gctgaaactctggctcaccc |
| F-t1t2 | cagaccgcttctgcgttctgc |
| R-r6k-mq | ctttagattgatttaaaacttcatttttaatttaaaaggCTGTCAGCCGTTAAGTGTTCC |
| F-oriR6k-p125 | gctgacccggcggggacgaggcaagctaaacagatcGACCCCTATAGTGAGTCGTATTAC |
| R-oriR6k-p125 | gggtggcgggcaggacgcccgccataaactgccaggcatCTGTCAGCCGTTAAGTGTTCC |
| F-kan p15gent | acaggcacattatgcatcgattaagctgtcaaacatgagcACGCTGCCGCAAGCACTCAG |
| R-kan- p15gent | ttcaataatattgaaaaaggaagagtatgagtattcaacCCGTAAGTCTGACGAAATCAG |
| R-pTac+ | gagtgagctgataccgctcgccgcagccgaacgaccgagCCAGTGAGACGGGCAACAGC |
| F-pTac+ | gctcgaattcgctagcccaaaaaaacgggtatggagaatctagaggatccAATTCTGTTTCCTGTGTGAAATTGTTATCC |
| R-amp-to-gmd | tttcggggaaatgtgcgcggaacccctatttgtttatttttGAAGAGTATGAGTATTCAAC |
| lacZ-upstream-delta-100 | AAAAAATTGTTAATATACCTCTATACTTTAgatcccgtcgttttacaacgtcgtgactgg |
| F-I-SceI-site | gacgagttcttctgagcgggactctggggtAGTTACGCTAGGGATAACAGGGTAATATAG |
| R-I-SceI-site | agaggaacttcggaataggaacttcgcggcCTATATTACCCTGTTATCCCTAGCGTAACT |
| F-pigP-delta.1 | caaggcgattaagttgggtaacgccagggttttcccaGGAGTGTTTGACGCTCGAAAAGG |
| R-pigP-delta.2 | ctgattcggttccatttataacctttctccaacgatgTGGCTGTCCTTACAAACATTACG |
| R-pigP-delta.3 | agcctgcgtaatgtttgtaaggacagccaCATCGTTGGAGAAAGGTTATAAATGGAAC |
| R-pigP-delta.4 | gagcggataacaatttcacacaggaaacagctatgaccGTAGGTCTCGCTGTACTCCCAC |
| F-I-SceI-117 | cgttgtaaaacgacggccagtgccaagcttgcatgcCCTTATTATTTCAGGAAAGTTTCG |
| R-I-SceI-117 | tgttgtgtggaattgtgagcggataacaatttcacacaggAGGAGAACGCATATGCATCA |
Lower case letters target homologous recombination, upper case nucleotides target amplification
Diagnostic primers
Primers that amplify recombination targeting sequence and amplify the DNA element to be cloned
Previously described (Shanks, et al., 2006)
The pMQ113 vector was created by amplifying a temperature-sensitive version of the pWV01 replicon and a kanamycin resistance gene originally from Enterococcus faecalis (aphA-3) from pTV32-OK (Cvitkovitch, et al., 2000) using primers (F-wv01, R-wv01) that direct recombination onto pMQ87 replacing the ColE1 origin and gentamicin resistance gene. The primer sequences are listed in Table 2. pMQ87 was first linearized with ApaL1 and BglII. E. coli candidates resistant to kanamycin and sensitive to gentimicin were chosen and validated as described above.
To make pMQ2, the pC194-containing HinDIII fragment from pSK256 (Gaskill and Khan, 1988) was ligated into the unique HinDIII site in the S. cerevisiae –E. coli shuttle vector pRS416 (Sikorski and Hieter, 1989).
The pMQ131 and pMQ132 shuttle vectors were created by replacing the pWV01 and pSC101 replicons of pMQ113 and pMQ117 with that of pBBR1MCS-2 using yeast in vivo recombination, using primers F-pBBR1-y and either R-p113-bbr or R-p117-bbr respectively. Plasmid pMQ113 had been digested with Nde1 and pMQ117 with MslI and SpeI.
Suicide vector pMQ118 was made by linearizing pMQ87 with BglII and ApaL1. The the rpsL gene and promoter from E. coli strain DH5a was amplified using primers F-ec-rpsL and R-ec-rpsL. The nptII gene and oriR6K-γ were amplified from pSC189 using primer pairs F-kan-ALV/R-kan-ALV and F-r6k-ALV/R-r6k-ALV respectively. The three amplicons with were simultaneously recombined in yeast replacing both the ColE1 replicon and the aacC1 gene of pMQ87.
The allelic replacement vector, pMQ150 was made by digesting pMQ118 with Xho1 and recombining the sacB gene amplified from pMQ30, using primers F-Cen-D and R-oriT-D that amplify DNA common to pMQ118 and pMQ30 flanking the sacB gene. The resulting plasmid was verified as conferring sucrose sensitivity to E. coli host strains.
Vector pMQ236 was made by digesting pMQ118 with MluI and transforming S. cerevisiae with this cut vector and two oligonucleotides (F-I-SceI-site, R-I-SceI-site) designed to direct the addition of the I-SceI meganuclease site and circularize the plasmid, as described above in the “oligonucleotide-mediated recombination” section. The addition of the I-SceI site was verified by sequencing.
Plasmid pMQ236 + pigP-Δ was made using primers pigP-delta.1 and pigP-delta.2 to amplify a ~600 bp region upstream, and pigP-delta.3 and pigP-delta.4 to amplify a ~700 bp region downstream of the pigP homolog of S. marcescens. These amplicons were used with EcoRI linearized pMQ236 to transform S. cerevisiae, resulting in creation of the pMQ236 + pigP-Δ construct as verified by sequencing. The last 4 codons of pigP are retained in this allele in order to maintain the start codon and ribosome binding site of the downstream gene.
The I-SceI delivery plasmid was generated by amplification of the I-SceI meganuclease gene from pBK233 with primers F-I-SceI-117 and R-I-SceI-117. These primers amplify the I-SceI gene and direct recombination with pMQ117, to replace the I-SceI gene under control of the E. coli Plac promoter. The resulting plasmid, pMQ240 was made with yeast recombineering and verified by sequencing.
The pMQ156 vector was made by digestion of pMQ125 with BspE1 and replacing the p15a replicon, the araC gene and the PBAD promoter with the oriR6K-γ replicon and Plac regions from pMQ118. The oriR6K- Plac region was amplified with primers F-t1t2 and R-r6k-mq.
The pMQ175 vector was made by digestion of pMQ125 with Msc1 and the pRO1600 replicon was replaced by the oriR6K-γ replicon from pMQ118 using yeast recombination. The oriR6K amplicon was generated using primers F-oriR6K-p125 and R-oriR6K-p125.
The pMQ200 vector was made by digesting pMQ175 with BglII and replacing both the aacC1 gene and the p15a gene with the nptII gene from pMQ118 using yeast in vivo recombination. Primers F-kan- p15gent and R-kan- p15gent were used to amplify the nptII-containing amplicon.
To generate pMQ123i, the araC- PBAD transcriptional system from pMQ97 was replaced by lacIQ-Ptac from pMMB66EH (Fürste, et al., 1986) using in vivo recombination. The promoter-repressor amplicon was made with primers F-pTac+ and R-pTac+II.
Expression vectors pMQ124 and pMQ125 were created by amplifying an aacC1 (gentamicin resistance) and p15a-origin containing amplicon from pMQ75 using primers R-amp-to-gm and F-pRMQS-D. This amplicon was recombined with pMQ91, a pUC-based, PBAD -bearing plasmid that has an ampicilin marker (bla) (Shanks, et al., 2006). The pMQ91 plasmid was digested in the bla gene using Adh1 and added along with the aacC1-p15a amplicon to a yeast transformation mixture. Because of two stretches of identity on the amplicon pMQ91 with the amplicon, two different recombination events could result in circulization of the plasmid. In the first possible recombination event, only the bla gene is replaced with the aacC1 gene leaving the pUC origin, yielding pMQ124 (Table 6). In the second possible recombination scenario, the bla gene and pUC origin of pMQ91 were both replaced with the aacC1 gene and p15a origin, yielding pMQ125.
Table 6.
Recombineering vectors and their relevant features.
| Plasmid Name | Resistance Marker1 | Replicon2 | Promoter Feature | Counter Selection | oriT (RP4) | Host Range3 | Yeast Replicon4 | Genbank# |
|---|---|---|---|---|---|---|---|---|
| pMQ117 | aacC1 | pSC101ts | Plac- lacZa | no | + | NHR, N | CEN6 | EU546817 |
| pMQ113 | aphA-3 | pWVO1ts | Plac- lacZa | no | + | BHR, N,P | CEN6 | EU546816 |
| pMQ2 | cat194, bla | ColE1, pC194 | Plac | no | − | BHR, N,P | 2μ | EU546815 |
| pMQ131 | aphA-3 | pBBR1 | PBAD-lacZa | no | + | BHR, N | CEN6 | EU546819 |
| pMQ132 | aacC1 | pBBR1 | PBAD-lacZa | no | + | BHR, N | CEN6 | EU546820 |
| pMQ118 | nptII | R6K-γ | Plac- lacZa | rpsL | + | NHR, N | CEN6 | EU546818 |
| pMQ150 | nptII | R6K-γ | Plac-lacZa | rpsL, sacB | + | NHR, N | CEN6 | EU546823 |
| pMQ236 | nptII | R6K-γ | Plac- lacZa | rpsL, I-SceI | + | NHR, N | CEN6 | |
| pMQ156 | aacC1 | R6K-γ/pRO1600 | Plac-lacZa | no | + | BHR, N | CEN6 | FJ380061 |
| pMQ175 | aacC1 | R6K-γ/p15a | PBAD-lacZa | no | + | NHR, N | CEN6 | FJ380062 |
| pMQ200 | nptII | R6K-γ | PBAD-lacZa | no | + | NHR, N | CEN6 | FJ380063 |
| pMQ123i | aacC1 | ColE1/RK2 | Ptac-gfpmut3 | no | + | BHR, N | 2μ | EU546814 |
| pMQ124 | aacC1 | ColE1/pRO1600 | PBAD-lacZa | no | + | BHR, N | CEN6 | EU546821 |
| pMQ125 | aacC1 | p15a/pRO1600 | PBAD-lacZa | no | + | BHR, N | CEN6 | EU546822 |
aacC1 is a gentamicin resistance determinant from Tn1696; aphA-3, from E. faecalis confers kanamycin resistance to Gram-positive and Gram-negative organisms; nptII from Tn903 confers kanamycin resistance to Gram-negative bacteria, cat194, from pC194 confers chloramphenicol resistance to Gram -positive and -negative organisms; bla from pBR322, confers ampicillin and carbenicillin resistance to Gram-negative organisms.
Replicon information is listed in text.
NHR= narrow host range, BHR= broad host range. N= Gram-negative, P=Gram-positive
CEN6 and 2μ are low- and high-copy replication determinants in S. cerevisiae, respectively.
2.5. Genetic manipulations and selection for S. marcescens rpsL mutation
All genetic manipulations were done as performed previously (Shanks, et al., 2006). A recessive streptomycin-conferring mutation in the rpsL gene of S. marcescens was acquired using selection. Overnight cultures of S. marcescens were grown to saturation in LB broth. Aliquots containing approximately 109 cells were plated on LB agar plates containing 25–50 μg/ml steptomycin. Resistant colonies were purified on plates containing 250 μg/ml streptomycin. A derivative of pMQ118 bearing a 500 bp piece of S. marcescens DNA (prgL), was introduced into one streptomycin resistant isolate of S. marcescens by conjugation. Kanamycin resistant colonies were found to be streptomycin sensitive indicating that the streptomyin resistance of the isolate was from an allele of rpsL, as pMQ118 has a wild-type copy of rpsL. The rpsL gene from S. marcescens and the streptomycin isolate was amplified with Taq DNA polymerase (New England Biolabs) using primers R-rpsL-CL and R-ec-rpsL, cloned using a TA-topo cloning kit (Invitrogen) and sequenced.
Plasmid maps and request information can be found at: http://www.dartmouth.edu/~google/vectors.html.
3. Results
3.1. Directed deletions and insertions without PCR
A previous report elegantly showed that single stranded DNA (ssDNA) oligomers could be used to target recombination between a double stranded DNA (dsDNA) amplicon and a dsDNA episome using S. cerevisiae recombination (Raymond, et al., 2002b). We hypothesized that ssDNA oligomers could be used to make targeted deletions in cloned genes using S. cerevisiae recombination. This would be very convenient as purchased oligomers could be used immediately, with no PCR steps, to target specific deletions or small insertions (Figure 1A). We also predicted that two oligomers could be used to generate a deletion and an insertion using two complementary ssDNA oligomers (Figure 1B). We also tested whether the distance from a DSB would make a difference in the frequency of desired recombination events. Once the mutation is made in a gene using budding yeast, it could then be moved into different organisms as determined by the host range of the vector used.
Figure 1. Oligonucleotide-mediated mutations using S. cerevisiae to modify bacterial genes.
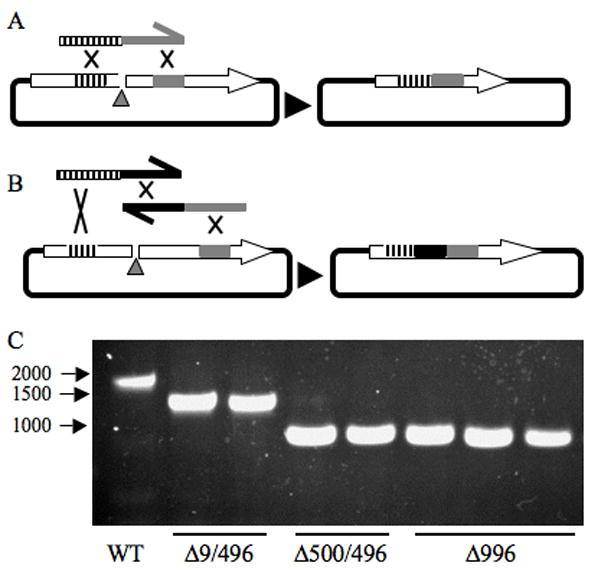
A and B) Model for oligonucleotide-mediated mutagenesis. A linearized plasmid vector (left) is repaired by targeted recombination by S. cerevisiae yielding either a deletion (A) or a deletion and an insertion (B). In A) one oligonuclotide is used to target recombination and in B) two oligomers are used. C) Agarose gel of diagnostic PCR amplicons from a lacZ gene that is intact (WT) or has been mutated using a single oligonuclotide (Δ997) or two oligonucleotides (Δ9/496 and Δ500/496). The size of the targeted deletion is indicated, but with two primers is the sum of the numbers with a 30 bp insertion, i.e. a 966 bp change in amplicon size for Δ500/496.
To determine the ability of a single ssDNA oligomer to mediate a deletion event we used pYC2-CT-lacZ. This S. cerevisiae-E. coli shuttle vector has the uracil biosynthetic gene, URA3, as a selectable marker, and the lacZ gene under control of a galactose inducible promoter. Therefore, we could measure the frequency of gap repair and of lacZ mutation events in S. cerevisiae by evaluation of lacZ gene product (β-galactosidase) activity on medium amended with X-gal and galactose. A double strand break was introduced into the lacZ gene with the AatII restriction nuclease, which linearized pYC2-CT-lacZ. When the linearized plasmid alone was used to transform a yeast strain with a ura3-52 mutation (plated on medium without uracil, SC-ura), we observed ~100 Ura+ colonies per experiment (Table 3), representing a frequency of 3.6×10−6 Ura+ CFU/total yeast cells per reaction. When the linearized plasmid was added with a single oligomer (60 bp in length) that targeted a 13bp deletion (Δ13), the number of Ura+ colonies increased to >600 colonies per transformation event (Table 3) with a frequency of 2.6×10−5 Ura+ CFU/reaction. Increasing the deletion size, i.e. a larger distance from the DSB to the targeting homology on the oligomer, reduced the frequency of Ura+ transformants (Table 3). Whereas, a 996 bp deletion could be made with a 60 bp oligomer, the use of a 40 bp oliogomer was not sufficient to distinguish from background levels of Ura+ CFU formation (Table 3), 2.96×10−5 Ura+ CFU/reaction.
The resulting Ura+ colonies could result from recombination events or from transformation with rare plasmids that did not experience a DSB at the AatII site. To differentiate between these classes, >100 colonies were patched onto SC-ura medium supplemented with X-gal and galactose. Blue colonies were indicative of an intact lacZ gene, whereas a white colony indicates a lack of β-gal activity and suggests mutation of the lacZ gene. A significant increase in the ratio of colonies without β-gal activity was observed when 60-bp oligomers were used to mediate recombination (Table 3). Diagnostic PCR was used to assess the 996 bp deletions, which could be distinguished from the WT lacZ gene on an agarose gel, and 5 out of 5 exhibited the correct size amplicon (Figure 1C, compare WT to 3 lanes on the far right, which shows the resulting PCR products from three such plasmids).
To test whether background colonies were a function of inefficient DSB formation due to potential limitations of the AatII enzyme, we designed a new oligonucleotide to target a deletion of 100 base pairs of the pYC2-lacZ plasmid spanning from the 3′ end of the GAL1 promoter through the first two codons of the lacZ gene. A successful deletion using this oligonucleotide should make a plasmid unable to confer β-galactosidase activity. Furthermore, we performed a dilution series of the oligonucleotide to determine the required amount of oligonucleotide necessary for efficient deletion formation. We observed absolutely no Ura+ colonies in the absence of vector (data not shown), and a similar amount of background Ura+ colonies with BamH1 digested vector compared to AatII digested vector (Table 3–4). The addition of 1–10μl of oligonucleotide (100μM) to the reaction produced a significant, four to six fold, enrichment in Ura+β-gal− colonies compared to the no oligonucleotide control (Table 4).
The same strategy was used to determine if a deletion could be made with two oligonucleotides with an over-lapping homology region. In this case we designed the oligomers with a common 30 bp sequence site. The oligomers were designed such that a successful recombination adds the 30 bp of homology as an insertion at the deletion site. The data in Table 5 indicate that two oligomers can be used to mediate recombination-based mutations. Furthermore, a significant increase in the frequency of β-galactosidase defective colonies was isolated with any group of primers compared to the vector alone (Table 5). Diagnostic PCR was used to assess the 9/496 bp deletions and the 500/496 bp deletions and 3 out of 3 for each deletion type exhibited the correct size amplicon (Figure 1C, two such examples for each are shown, compare WT to Δ9/496 and WT to Δ500/496). Random ®–gal deficient candidates were tested for the inserted 30-mer using PCR. The inserted 30 bp sequence was used as a primer-binding site with another primer in the lacZ sequence. Eight out of nine candidates exhibited a positive band of the correct size indicating a successful insertion, whereas a ®–gal positive candidate exhibited no band showing (data not shown). These data show that one or two oligomers can be used with budding yeast recombineering to target deletions and insertions.
3.2. Generation of Novel Recombineering Vectors
Most cloning vectors used for budding yeast are limited to replication in S. cerevisiae and high-copy replication in E. coli. In order to use the yeast recombineering system for a wider variety of purposes and bacterial genera, we created novel vectors that combine the ability to replicate in S. cerevisiae with a variety of bacterial replicons, selectable markers and promoters (Table 5). The use of S. cerevisiae recombination facilitated the production of the vectors to be described below, in that multiple pieces of DNA could be simultaneously cloned without leaving behind restriction sites that limit their future use. Also, unwanted parts of the previous plasmid are completely eliminated as targeted by the oligomer, whereas many commonly used vectors contain large areas of unwanted DNA left behind because of the location of restriction sites. The generation of the following vectors and their ability to be used for oligonucleotide-mediated deletions is a clear demonstration of their utility.
A pSC101-based shuttle vector
pSC101 is a 9.43 kBP R-plasmid originally isolated from Salmonella panama and is capable of replication in members of the Enterobacteriaceae (Kues and Stahl, 1989). It has its own replication machinery, and a copy number that has been estimated at 4–10 per E. coli chromosome (Hashimoto-Gotoh and Sekiguchi, 1977) (Hasunuma and Sekiguchi, 1977). pSC101-based plasmids are useful because 1) they are compatible with ColE1-based vectors, such as pUC19 and pBR322, (Churchward, et al., 1984), 2) they are maintained at a low-copy number, circumventing toxicity issues associated with high-copy plasmids, 3) they can support large inserts making it useful for making chromosomal libraries (Jiang, et al., 1987), and 4) temperature-sensitive versions of pSC101 have been made for use as suicide vectors, transposon delivery to organisms with low transformation efficiencies, and for targeted mutagenesis (Rossignol, et al., 2001).
We replaced a ColE1 origin from a yeast-Esherichia coli shuttle vector, pMQ87 (Shanks, et al., 2006), with a temperature-sensitive version of the pSC101 replicon using yeast in vivo recombineering, generating pMQ117 (Table 6). This temperature-sensitive replicon originated from the pSC101 derivative pHS1 (Hashimoto-Gotoh and Sekiguchi, 1977). pHS1 replicates as pSC101 at 30 C, but exhibits an almost complete loss of replication at 43 C (Hashimoto and Sekiguchi, 1976).
Gram-negative – Gram-positive – S. cerevisiae shuttle vectors
Two replicons were used to generate broad host-range vectors capable of use in both Gram-negative and positive bacteria. The replicons used are pWV01, a low-copy cryptic plasmid from Lactococcus lactis (Kok, et al., 1984), and pC194, a small R-plasmid from Staphylococcus aureus (Iordanescu, et al., 1978).
The pWV01 replicon is capable of replication in both Gram-positive and Gram-negative bacteria including such organisms as Actinobacillus actinomycetemcomitans, Bacillus sp., Clostridium sp., Enterococcus faecium, Lactobacillus acidophilus, Lactococcus sp., Listeria monocytogenes, Streptococcus sp., S. aureus, and E. coli. We chose a version of pWV01 that is thermosensitive for replication in both Gram-negative and –positive bacteria (Maguin, et al., 1992).
The kanamycin resistance determinant aphA-3, originally from Enterococcus faecalis, confers resistance to kanamycin in many Gram-positive and Gram-negative species, and the pWV01-temperature-sensitive replicon were combined using yeast in vivo cloning, generating pMQ113 (Table 6).
The pC194 plasmid has been described as having a copy number of 15 in S. aureus (Novick, 1991). The host range of pC194 has been described to include Gram-positive bacteria: S. aureus (Iordanescu, et al., 1978), Bacillus thuringiensis (Martin, et al., 1980), B. subtilis (Novick, 1991), and Gram-negative bacteria: E. coli (Goze and Ehrlich, 1980) and Francisella tularensis (Pomerantsev, et al., 1991). As replication efficiency is suboptimal outside of its native host, S. aureus, antibiotic selection is required for pC194 maintenance in other bacteria. The pC194 replicon is particularly inefficient in E. coli where it is maintained at 1–3 copies per cell (Goze and Ehrlich, 1980); therefore, the addition of a ColE1-based replicon is beneficial for shuttling between E. coli and other hosts. A plasmid, pMQ2 (Table 6), was made with has an ampicillin resistance marker for E. coli, and a chloramphenicol marker, cat194, that confers resistance in S. aureus, E. coli, Bacillus sp., and F. tularensis. The pMQ2 plasmid has been used to complement genes in S. aureus (Shanks and O’Toole, unpublished data) and replicates in F. tularensis (Horzempa, et al., 2008).
Broad-host range vectors with the pBBR1 replicon
The broad-host-range replicon from pBBR1MCS-5 was incorporated into this series of vectors (Kovach, et al., 1995). pBBR1 is a small cryptic plasmid isolated from Bordetella bronchiseptica that is capable of replicating in a wide range of Gram-negative bacteria and is compatible with IncP, IncQ, IncW, ColE1 and p15a plasmids (Antoine and Locht, 1992). pBBR1 is maintained at approximately 30–40 copies per cell in both E. coli and B. bronchiseptica making it easier to work with than many other broad host-range plasmids that are maintained at very low-copy number. The pBBR1 plasmid can replicate in at least 24 Gram-negative genera including Agrobacterium, Brucella, Burkholderia, Caulobacter, Pseudomonas, Rhizobium, and Vibrio (Lynch and Gill, 2006). A comprehensive series of vectors containing the pBBR1-replicon and multiple resistance elements has recently been reported, and the pBBR1-replicon was found to be highly appropriate for genomic library construction (Lynch and Gill, 2006). We made vectors with the pBBR1 replicon and kanamycin (pMQ131) or gentamicin (pMQ132) resistance markers, capable of use for yeast recombination. These vectors have been used successfully for complementation of mutations in S. marcescens (Shanks, et al., 2007) (Kalivoda, et al., 2008), in Rhodobacter capsulatus (Caiazza, et al., 2007) and pMQ132 has been used in P. aeruginosa strain PA14 (Shanks, unpublished).
We tested the ability of the pMQ132 vector to replicate in host-independent variants of Bdellovibrio bacteriovorus using conjugation (Medina, et al., 2008). Plasmid DNA was prepared from B. bacteriovorus gentamicin resistant transformants. This plasmid DNA was used to transform E. coli and the pMQ132 plasmid was verified by PCR and restriction analysis (data not shown). This expands the limited number of replicons reported for use in B. bacteriovorus.
An oriR6K-based vector for allelic exchange
R6K is a large R-plasmid (38 kb) from E. coli. R6K replication requires the plasmid-encoded pi protein (Crosa, 1980, Kontomichalou, et al., 1970). The pi protein can be provided in trans for replication of oriR6K -based plasmids, such that, vectors can be made that are able to replicate in E. coli strains that express the pi protein, but cannot replicate in strains without the pi protein (Kolter, et al., 1978) (Miller and Mekalanos, 1988) (Metcalf, et al., 1994). This plasmid is usually maintained at 10–20 copies per E. coli chromosome, but mutations in the pir gene can lead to elevated copy number (Crosa, 1980) (Filutowicz, et al., 1987, Greener, et al., 1990, Inuzuka, et al., 1980). Some commercially available E. coli strains harbour the pir-116 allele which confers a ~25-fold increase in copy number of oriR6K-based vectors. Hosts with the pir-116 mutation are advantageous for generating large amounts of plasmid DNA for a variety of applications, whereas hosts with wild-type pir are advantageous when working with genes with copy-number associated toxicicity.
The pMQ87 plasmid (Shanks, et al., 2006) was modified to incorporate the R6K-gamma origin as well as a different selectable marker and a counter-selectable marker. The ColE1 origin and aacC1 gene from pMQ87 was replaced by a kanamycin resistance determinant (originally from Tn903), the oriR6K-γ amma origin from pSC189 (Chiang and Rubin, 2002), and the wild-type rpsL gene from E. coli strain DH5a in a single step using yeast homologous recombination, generating pMQ118 (Table 6). The rpsL gene from E. coli confers streptomycin sensitivity to strains with specific rpsL mutations; therefore, it can be used as a counter selectable marker for allelic replacement applications or the selective loss of a plasmid (Russell and Dahlquist, 1989).
An rpsL mutant strain (RSS1) was made in order to use the pMQ118 plasmid for allelic replacement in S. marcescens. An rpsL mutant strain of S. marcescens was generated by selection on LB agar supplemented with 25 μg/ml of streptomycin. The rpsL region of one strain was cloned, sequenced and found to have an A to T transversion resulting in an asparagine 43 to isoleucine change (Genbank accession number DQ642044). A similar change, K43R, has been reported in the E. coli streptomycin-resistance associated rpsL150 mutation.
As many bacteria do not bear rpsL mutations enabling the use of rpsL as a counter-selectable marker, a plasmid was constructed that includes oriR6K replicon and both rpsL and sacB (pMQ150, Table 6). The sacB gene, which codes for a levansucrase from Bacillus subtilis, is commonly used as a counter-selectable marker in Gram-negative organisms as it confers sucrose-sensitivity (Ried and Collmer, 1987, Stavropoulos and Strathdee, 2001). Tandem counter-selection systems have been reported and shown to be highly effective (Stavropoulos and Strathdee, 2001). Both pMQ118 and pMQ150 have been used to make directed mutations in S. marcescens (Shanks, et al., 2007) (Kalivoda, et al., 2008).
The I-SceI meganuclease creates a double-stranded break in the integrated plasmid forcing a recombination event making it useful for allelic replacement strategies (Posfai, et al., 1999). To show the utility of the oligonucleotide recombination method, a third allelic replacment vector, we generated an I-SceI-based allelic replacement system. An I-SceI recognition site was added to allelic replacement vector pMQ118 by yeast in vivo cloning. To make pMQ236 (Table 6), pMQ118 was cut at a MluI site. The linearized pMQ118 was cotransformed with two oligonucleotides, which contain the I-SceI target sequence and pMQ118 sequence to target recombination with the vector, as described above. The resulting plasmid, pMQ236, had the I-SceI site as verified by sequencing. To deliver the I-SceI meganuclease into S. marcescens, we moved the I-SceI-coding gene into pMQ117, yielding, pMQ240.
To test the utility of some of these plasmids for allelic replacement, we used pMQ236 to make a chromosomal deletion of the S. marcescens pigP homolog. The Salmond group isolated the pigP gene in Serratia sp. ATCC 39006 in a mutant screen for regulators of prodigiosin (red pigment) production, and described PigP as the “master regulator” of pigment production (Fineran, et al., 2005). The closest homolog of this gene is annotated as SMA3564 in the Sanger Center sequenced S. marcescens strain. To test the importance of SMA3564 (pigP) in S. marcescens pigment production, and in-frame deletion construct was made using yeast recombineering (Figure 2A–C). Successful alleleic replacement of pigP with the pigP-Δ allele (Figure 2D, F) resulted in a marked decrease in pigment production (Figure 2E) indicating that this plasmid is useful for allelic replacement, and supporting the hypothesis that PigP has a conserved role in both Serratia species to promote red pigment production.
Figure 2. Use of oligonucleotide based recombineering, yeast recombineering and a novel allelic replacement vector to delete the pigP homolog from S. marcescens.
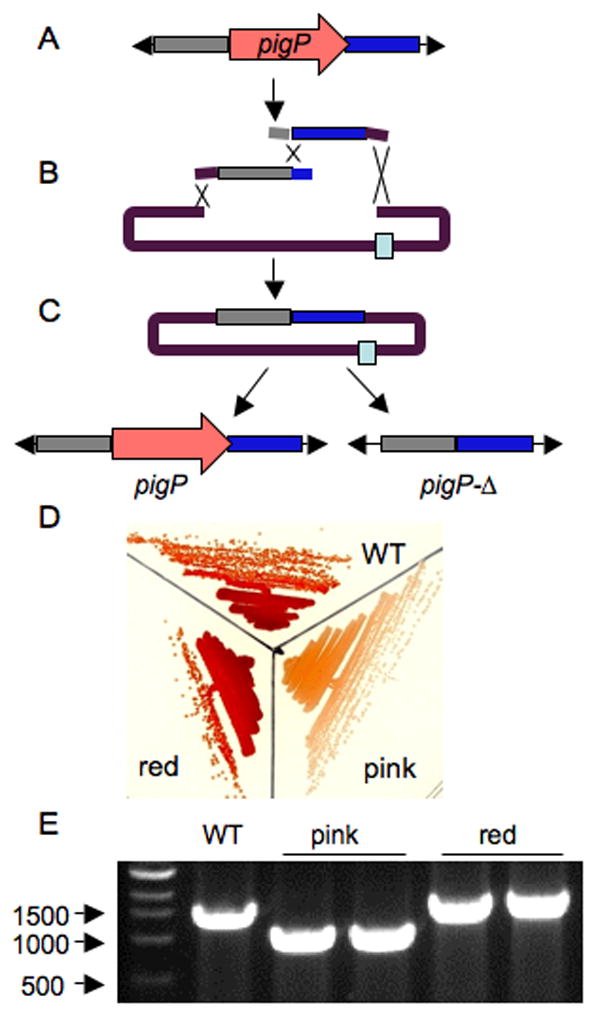
The I-SceI meganuclease sequence (light blue box) was introduced into pMQ118 by oligonuclotide directed recombination generating pMQ236 (as in Figure 1). A. DNA sequences flanking the pigP homolog from S. marcescens were amplified with primers that direct recombination with each other and with pMQ236. These amplicons were introduced into S. cerevisiae with linearized pMQ236, generating a seamless pigP-Δ mutagenesis construct (C). This pMQ236 + pig-Δ construct was introduced into wild-type (WT) S. marcescens to generate a merodiploid, and this strain was transformed with an I-SceI expressing plasmid. D. The result of this allelic replacement strategy is restoration of the WT gene (left), or deletion of the pigP gene (right). E. Two colony color-types resulted from this allelic replacement strategy, one type was pink and the other was indistinguishable from the WT parental strain. F. PCR analysis indicates that the pink strains have the pigP-Δ, whereas the red strains retained the full-length pigP gene. These results support the model that PigP is a positive regulator of red pigment production in both S. marcescens and Serratia sp. ATCC 39006.
An additional vector, pMQ156, was made with the aacC1 resistance marker, which has both the oriR6K and the pRO1600 replicons for variable copy-number maintenance in E. coli (due to oriR6K replicon), and replication in several other Gram-negative species (due to pRO1600 replicon) (Table 6). The pRO1600 replicon is a medium-copy, broad host-range replicon from Pseudomonas aeruginosa (Olsen, et al., 1982) and shuttle-vector derivatives have been wildly used (Schweizer, 2001).
Broad-host range expression vectors
Broad-range expression yeast shuttle vectors that feature the PBAD/araC expression system and RK2 based replicon have been described (Shanks, et al., 2006). We replaced the PBAD/araC expression system with Ptac/lacIQ from pMMB66EH (Fürste, et al., 1986). Yeast in vivo cloning was used to directly replace the PBAD/araC expression system on pMQ97 with Ptac/lacIQ from pMMB66EH generating pMQ123i (Table 6). This vector contains gentamicin resistance determinants and the RK2 and ColE1 replicons. The pMQ123i vector contains GFPmut3 as a reporter to determine relative expression levels (Cormack, et al., 1996). There are restriction sites on either side and within of the GFPmut3 gene so it can easily be used for transcriptional or translational fusions or replaced entirely using either traditional in vitro or recombineering techniques.
In addition, we have modified a previously described PBAD –containing vector, pMQ91 (Shanks, et al., 2006), by replacing either the ampicillin resistance marker with the aacC1 gentamicin resistance gene (pMQ124) or both the high-copy ColE1-based origin and bla gene with a p15a- replicon and aacC1 gene (pMQ125) (Table 6). Both retain the pRO1600 replicon of pMQ91. The p15a replicon is from a cryptic E. coli plasmid that is maintained at 10–14 copies per chromosome (Kretschmer, et al., 1975) (Hiszczynska-Sawicka and Kur, 1997). The low-copy vector, pMQ125, is valuable for controlled expression of genes in a broad range of Gram-negative bacteria. The pMQ125 vector has been used for controlled expression of the oxyR gene from S. marcescens (Shanks, et al., 2007).
Two more vectors combine the R6k replicon and the PBAD/araC expression system. The pMQ175 vector has both the oriR6K and p15a replicons and the aacC1 resistance gene, and pMQ200 has the oriR6K replicon and the nptII resistance gene (Table 3). These can be either used as episomes or as integrative vectors to create arabinose-inducible chromosomal constructs.
4. Discussion
This report describes both the use of S. cerevisiae-based recombineering to generate oligonucleotide-mediated deletions and insertions and an expanded vector tool-box. A previous group of yeast-recombineering vectors published by our group were useful for gene expression in a wide array of bacterial Gram-negative genera, but the allelic replacement vectors were focused for Pseudomonas species (Shanks, et al., 2006). This current vector set expands on the previous set, and includes shuttle vectors that can replicate in Gram-positive bacteria, and can be used for allelic replacement in genera such as Serratia and Escherichia. Furthermore, the ability of oligonuclotides to generate mutations in this format is very convenient, and takes very little hands-on time in the laboratory. Oligonucleotide-mediated generation of deletions and or insertions could be used for structure function analysis, or to add any number of short DNA sequences, such as sites for DNA-relevant sequences, e.g. loxP recombination, restriction nuclease or primer sites, or protein relevant sequences, such as protease recognition or phosphorylation sites into a gene. This technique is clearly limited by the presence or absence of restriction enzyme sites on a gene of interest.
From the nine separate experiments where ~100 base pair deletions were made (Tables 3–5), we observed a 3 to 6 fold ratio of mutants (Ura+, β-gal−) compared to false positives (Ura+, β-gal+), indicating the utility of this approach for making mutations in genes without an easily screened phenotype in yeast. However, we were surprised at the relatively high background (3–20%) in the experiments presented in Tables 3–5. These false positives (Ura+, β-gal+) could arise from four different events A) uncut vector, B) repair of vector through intramolecular recombination, or C) repair of the chromosomal ura3 allele and repaired vector, or D) vector integration into yeast chromosome. Classes A, and B are more likely because they require only one rare event to take place (the vector not being cut or repair of the cut vector), whereas class C requires two rare events, namely repair of the cut vector and recombination with the chromosome. Repair of the chromosomal ura3-52 allele through recombination with the URA3 gene on the vector alone is unlikely based on the fact that 80–97% of Ura+ transformants in the absence of oligonucleotides were β-gal+ (intact lacZ gene) and is not considered in class C. Class D requires integration of the vector into the chromosome retaining the URA3 and lacZ genes in a way that repairs lacZ (AatII digest scenario) or places lacZ under control of an active promoter (BamH1 digest scenario). Additionally, we would be less likely to isolate class D transformants because integration of pYC2-lacZ would introduce a second centromere into the chromosome in which it integrates, and two-centromere containing chromosomes are unstable in cell division. Between class A and B, the majority of background transformants are less likely to stem from uncut vector or a compromised enzyme preparation because of the 20-fold over-digestion performed and because different enzymes were used in different experiments. A minority of the background of Ura+ colonies was β-gal deficient (Tables 3–4), suggesting that the plasmid in these had been cut and repaired leading to a mutation in lacZ or through repair of the chromosomal ura3-52 allele.
The second reported tool is a set of vectors that represent a wealth of application possibilities for researchers who work with bacterial or eukaryotic genes, and will be made available to biomedical and basic science researchers.
Acknowledgments
We are grateful to Nicholas Stella and Christina Medaglia for technical assistance, and Eric Rubin, Frederic Boccard, Patrick Stragier, Dennis Cvitkovitch, Paul Kinchington, Eric Lambie, Karen Skorulpski, Victor Ambrose, Joseph Horzempa, and Ambrose Cheung labs for providing plasmids, strains, ideas and restriction enzymes. This work was supported by NIH R21-AI055774 grant to G.A.O., NIH fellowship F32 GM66658-01A1 and Research to Prevent Blindness Career Development Award to R.M.Q.S, and core grant NEI EY08098.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antoine R, Locht C. Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from Gram-positive organisms. Mol Microbiol. 1992;6:1785–1799. doi: 10.1111/j.1365-2958.1992.tb01351.x. [DOI] [PubMed] [Google Scholar]
- Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DDD, Stearns T. Methods In Yeast Genetics, A Cold Spring Harbor Laboratory Course Manual. Cold Harbor laboratory Press; Plainview, NY: 2000. Pages. [Google Scholar]
- Caiazza NC, Lies DP, Newman DK. Phototrophic Fe(II) oxidation promotes organic carbon acquisition by Rhodobacter capsulatus SB1003. Appl Environ Microbiol. 2007;73:6150–6158. doi: 10.1128/AEM.02830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SL, Rubin EJ. Construction of a mariner-based transposon for epitope-tagging and genomic targeting. Gene. 2002;296:179–185. doi: 10.1016/s0378-1119(02)00856-9. [DOI] [PubMed] [Google Scholar]
- Churchward G, Belin D, Nagamine Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene. 1984;31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Crosa JH. Three origins of replication are active in vivo in the R plasmid RSF1040. J Biol Chem. 1980;255:11075–11077. [PubMed] [Google Scholar]
- Cvitkovitch DG, Gutierrez JA, Behari J, Youngman PJ, Wetz JE, Crowley PJ, Hillman JD, Brady LJ, Bleiweis AS. Tn917-lac mutagenesis of Streptococcus mutans to identify environmentally regulated genes. FEMS Microbiol Lett. 2000;182:149–154. doi: 10.1111/j.1574-6968.2000.tb08889.x. [DOI] [PubMed] [Google Scholar]
- DeMarini DJ, Creasy CL, Lu Q, Mao J, Sheardown SA, Sathe GM, Livi GP. Oligonucleotide-mediated, PCR-independent cloning by homologous recombination. Biotechniques. 2001;30:520–523. doi: 10.2144/01303st02. [DOI] [PubMed] [Google Scholar]
- Filutowicz M, McEachern MJ, Mukhopadhyay P, Greener A, Yang SL, Helinski DR. DNA and protein interactions in the regulation of plasmid replication. J Cell Sci Suppl. 1987;7:15–31. doi: 10.1242/jcs.1987.supplement_7.2. [DOI] [PubMed] [Google Scholar]
- Fineran PC, Slater H, Everson L, Hughes K, Salmond GP. Biosynthesis of tripyrrole and beta-lactam secondary metabolites in Serratia: integration of quorum sensing with multiple new regulatory components in the control of prodigiosin and carbapenem antibiotic production. Mol Microbiol. 2005;56:1495–1517. doi: 10.1111/j.1365-2958.2005.04660.x. [DOI] [PubMed] [Google Scholar]
- Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Badgdasarian M, Lanka E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- Gaskill ME, Khan SA. Regulation of the enterotoxin B gene in Staphylococcus aureus. J Biol Chem. 1988;263:6276–6280. [PubMed] [Google Scholar]
- Goze A, Ehrlich SD. Replication of plasmids from Staphylococcus aureus in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1980;77:7333–7337. doi: 10.1073/pnas.77.12.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greener A, Filutowicz MS, McEachern MJ, Helinski DR. N-terminal truncated forms of the bifunctional pi initiation protein express negative activity on plasmid R6K replication. Mol Gen Genet. 1990;224:24–32. doi: 10.1007/BF00259447. [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. Antibiotic-resistance cassettes for Bacillus subtilis. Gene. 1995;167:335–336. doi: 10.1016/0378-1119(95)00652-4. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Sekiguchi M. Isolation of temperature-sensitive mutants of R plasmid by in vitro mutagenesis with hydroxylamine. J Bacteriol. 1976;127:1561–1563. doi: 10.1128/jb.127.3.1561-1563.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Gotoh T, Sekiguchi M. Mutations to Temperature Sensitivity in R Plasmid pSC101. Journal of Bacteriology. 1977;131:405–412. doi: 10.1128/jb.131.2.405-412.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasunuma K, Sekiguchi M. Replication of plasmid pSC101 in Escherichia coli K-12: requirement for dnaA function. Mol Gen Genet. 1977;154:225–230. doi: 10.1007/BF00571277. [DOI] [PubMed] [Google Scholar]
- Hiszczynska-Sawicka E, Kur J. Effect of Escherichia coli IHF mutations on plasmid p15A copy number. Plasmid. 1997;38:174–179. doi: 10.1006/plas.1997.1307. [DOI] [PubMed] [Google Scholar]
- Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosoally-located DNA sequences: applicaiton for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- Horzempa J, Carlson PE, Jr, O’Dee DM, Shanks RM, Nau GJ. Global transcriptional response to mammalian temperature provides new insight into Francisella tularensis pathogenesis. BMC Microbiol. 2008;8:172. doi: 10.1186/1471-2180-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka N, Inuzuka M, Helinski DR. Activity in vitro of three replication origins of the antibiotic resistance plasmid RSF1040. J Biol Chem. 1980;255:11071–11074. [PubMed] [Google Scholar]
- Iordanescu S, Surdeanu M, Della Latta P, Novick R. Incompatibility and molecular relationships between small Staphylococcal plasmids carrying the same resistance marker. Plasmid. 1978;1:468–479. doi: 10.1016/0147-619x(78)90005-7. [DOI] [PubMed] [Google Scholar]
- Jansen G, Wu C, Schade B, Thomas DY, Whiteway M. Drag & Drop cloning in yeast. Gene. 2005;344:43–51. doi: 10.1016/j.gene.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Jiang XM, Brahmbhatt HN, Quigley NB, Reeves PR. A low copy number cosmid. Plasmid. 1987;18:170–172. doi: 10.1016/0147-619x(87)90045-x. [DOI] [PubMed] [Google Scholar]
- Kalivoda EJ, Stella NA, O’Dee DM, Nau GJ, Shanks RM. The cAMP-dependent catabolite repression system of Serratia marcescens mediates biofilm formation through regulation of type 1 fimbriae. Appl Environ Microbiol. 2008 doi: 10.1128/AEM.02733-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok J, Van der Vossen JMBM, Venema G. Construction of plasmid cloning vectors for lactic streptococci which also replicate in Bacillus subtilis and Escherichia coli. Applied and Environmental Microbiology. 1984;48:726–731. doi: 10.1128/aem.48.4.726-731.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R, Inuzuka M, Helinski DR. Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell. 1978;15:1199–1208. doi: 10.1016/0092-8674(78)90046-6. [DOI] [PubMed] [Google Scholar]
- Kontomichalou P, Mitani M, Clowes RC. Circular R-factors molecules controlling penicillinase synthesis, replication in Escherichia coli under relaxed or stringent control. J Bacteriol. 1970;104:34–44. doi: 10.1128/jb.104.1.34-44.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris ME, Roop RM, Peterson KM. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- Kretschmer FJ, Chang AC, Cohen SN. Indirect selection of bacterial plasmids lacking identifiable phenotypic properties. J Bacteriol. 1975;124:225–231. doi: 10.1128/jb.124.1.225-231.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kues U, Stahl U. Replication of plasmids in gram-negative bacteria. Microbiol Rev. 1989;53:491–516. doi: 10.1128/mr.53.4.491-516.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Gill RT. Broad host range vectors for stable genomic library construction. Biotechnol Bioeng. 2006;94:151–158. doi: 10.1002/bit.20836. [DOI] [PubMed] [Google Scholar]
- Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. New thermosensitive plasmid for Gram-positive bacteria. Journal of Bacteriology. 1992;174:5633–5638. doi: 10.1128/jb.174.17.5633-5638.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet L, Jacquet M. Intergenic Flip Flop, a method for systematic gene disruption and cloning in yeast. Yeast. 1996;12:1351–1357. doi: 10.1002/(sici)1097-0061(199610)12:13<1351::aid-yea24>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Martin PAW, Lohr JR, Dean DH. Transformation of Bacillus thuringiensis protoplasts by plasmid deoxyribonucleic acid. J Bacteriol. 1980;145:980–983. doi: 10.1128/jb.145.2.980-983.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AA, Shanks RM, Kadouri DE. Development of a novel system for isolating genes involved in predator-prey interactions using host independent derivatives of Bdellovibrio bacteriovorus. BMC Microbiology. 2008:109J. doi: 10.1186/1471-2180-8-33. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, Wanner BL. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6K gamma origin plasmids at different copy numbers. Gene. 1994;138:1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]
- Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP. Genetic systems in staphylococci. Methods Enzymol. 1991;204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–452. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RH, DeBusscher G, McCombie WR. Development of broad-host-range vectors and gene banks: self-cloning of the Pseudomonas aeruginosa PAO chromosome. J Bacteriol. 1982;150:60–69. doi: 10.1128/jb.150.1.60-69.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW. Yeast recombination: the association between double-strand gap repair and crossing-over. Proc Natl Acad Sci U S A. 1983;80:4417–4421. doi: 10.1073/pnas.80.14.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantsev AP, Domaradskii IV, Doronin IP, Fursov VV. Study of cat-gene expression of pSa and pC194 in Escherichia coli, Francisella tuleransis, and Bacillus subtilis cells. Mol Gen Microbiol Virusol. 1991;9:21–24. [PubMed] [Google Scholar]
- Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999;27:4409–4415. doi: 10.1093/nar/27.22.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CK, Sims EH, Kas A, Spencer DH, Kutyavin TV, Ivey RG, Zhou Y, Karul R, Clendenning JB, Olson MV. Genetic variation at the O-antigen biosynthetic locus in Pseudomonas aeruginosa. J Bacteriol. 2002a;184:3614–3622. doi: 10.1128/JB.184.13.3614-3622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CK, Sims EH, Olson MV. Linker-mediated recombinatinal subcloning of large DNA fragments using yeast. Genome Res. 2002b;12:190–197. doi: 10.1101/gr.205201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried JL, Collmer A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 1987;57:239–246. doi: 10.1016/0378-1119(87)90127-2. [DOI] [PubMed] [Google Scholar]
- Rossignol M, Basset A, Espeli O, Boccard F. NKBOR, a mini-Tn10-based transposon for random insertion in the chromosome of Gram-negative bacteria and the rapid recovery of sequences flanking the insertion sites in Escherichia coli. Res Microbiol. 2001;152:481–485. doi: 10.1016/s0923-2508(01)01221-9. [DOI] [PubMed] [Google Scholar]
- Russell CB, Dahlquist FW. Exchange of chromosomal and plasmid alleles in Escherichia coli by selection for loss of a dominant antibiotic sensitivity marker. J Bacteriol. 1989;171:2614–2618. doi: 10.1128/jb.171.5.2614-2618.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer HP. Vectors to express foreign genes and techniques to monitor gene expression in Pseudomonads. Curr Opin Biotechnol. 2001;12:439–445. doi: 10.1016/s0958-1669(00)00242-1. [DOI] [PubMed] [Google Scholar]
- Shanks RM, Stella NA, Kalivoda EJ, Doe MR, O’Dee DM, Lathrop KL, Guo FL, Nau GJ. A Serratia marcescens OxyR homolog mediates surface attachment and biofilm formation. J Bacteriol. 2007:189. doi: 10.1128/JB.00859-07. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O’Toole GA. A Saccharomyces cerevisiae-Based Molecular Tool Kit for Manipulation of Gram-Negative Bacterial Genes. Appl Environ Microbiol. 2006;72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos TA, Strathdee CA. Synergy between tetA and rpsL provides high-stringency positive and negative selection in bacterial artificial chromosome vectors. Genomics. 2001;72:99–104. doi: 10.1006/geno.2000.6481. [DOI] [PubMed] [Google Scholar]