

Abstract
The critical role of the beta cell in the pathogenesis of type 2 diabetes is now well established. When examined in individuals with type 2 diabetes and those at increased risk, reductions in beta cell mass and abnormalities of beta cell function can both be demonstrated. Whether one alone is sufficient or both are necessary for the development of hyperglycaemia has been debated. Based on human and animal studies, it appears that neither alone is sufficient. Rather, for glucose to rise to the level diagnostic of diabetes, defects in both beta cell mass and beta cell function are required.
Keywords: amyloid, animal models, beta cell function, beta cell mass, beta cell volume, genes, glucagon, insulin secretion, islets, pancreatectomy
Introduction
Science frequently resembles a pendulum. We learn, we forget, we relearn. Put another way, new tools frequently allow us to rediscover principles that were already known, sometimes quite well known. Type 2 diabetes represents the perfect example.
Consider the divide between insulin resistance and beta cell dysfunction. These two factors are both critically important in the pathogenesis of the hyperglycaemia of type 2 diabetes. However, the focus on each is such that one would never believe that both were critical players, but rather the onset of diabetes was simply due to one or the other. Certainly, only about 15 years ago, a visit to many diabetes research centres or clinics in the United States would have left you convinced that insulin resistance was the only important player [1]. The beta cell and its maladies were viewed as a late occurrence and of little import. Nowadays, on both sides of the Atlantic and in most regions of the world, the critical nature and early occurrence of beta cell abnormalities is no longer in question [2]
When one digs a little deeper into the saga of the beta cell, one discovers another debate. Is it mass or is it function that is important? Does reduced mass explain reduced function? Can an individual develop the disease with one and not the other? Again there is an element of polarisation. In this perspective we examine this issue in a little more detail and come to the conclusion that no beta cell biologist has to be wrong. Rather, the consensus should be that both, and indeed several aetiologies, may exist and are critical in the demise of the beta cell in type 2 diabetes.
Is Beta Cell Mass Reduced in Type 2 Diabetes?
It has been known for more than a century that there are morphological abnormalities in the islet in individuals with type 2 diabetes. In fact, in 1901 Opie published hand drawn colour images of haematoxylin and eosin staining clearly demonstrating decreased cellularity and the presence of hyaline material in the islets of Langerhans [3] (Figure 1). This was subsequently followed by papers from McLean and Ogilvie [4] as well as Klöppel et al [5] highlighting that the volume of beta cells was reduced in type 2 diabetes (Figure 2). Interestingly, alpha cell mass was not decreased, resulting in a relative increase in the alpha to beta cell ratio [4]. More recently, Butler et al [6] confirmed these findings and extended them by showing that beta cell volume is decreased in subjects with impaired fasting glucose (IFG) to a degree intermediate between normal and diabetes (Figure 2). The exact degree of beta cell reduction remains somewhat unclear and possibly even controversial. Studies by a number of investigators suggest that it is between zero and 63% [5–11]; however, recent work by Rahier and colleagues [12] we believe represents the best assessment. The reason for believing so is that it includes sampling in over 100 individuals from both the body and tail of the pancreas, two regions which vary with respect to the proportion of the islet comprised of beta cells; with the tail known to contain greater amounts of these insulin-secreting cells. This comprehensive study places the loss of beta cells at under 40%, occurring similarly in islets in the body and tail of the pancreas [12] (Figure 3). Further, this reduction occurred similarly in subjects who were normal weight (BMI <25 kg/m2) or overweight/obese (BMI 26–40 kg/m2), and was only 24% in subjects who had the diagnosis for up to 5 years compared to 54% in those known to have the disease for greater than 15 years.
Figure 1.
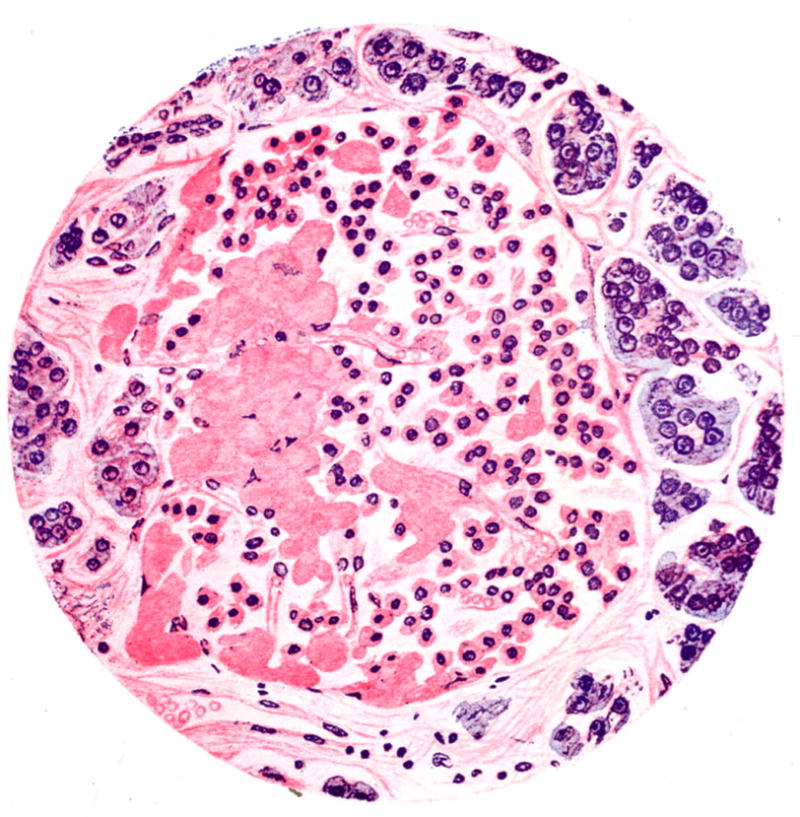
Hand drawn micrograph of an islet from a patient with type 2 diabetes stained with haematoxylin and eosin. Cellularity of the islet is decreased and it contains eosinophilic material termed “hyalinosis”, which we now know as amyloid. Reprinted with permission from [3].
Figure 2.

Relationship of obesity and glucose tolerance with islet beta and alpha cell volume and mass based on autopsy assessments. (a) Beta cell mass (blue) is reduced in subjects with type 2 diabetes (n=25) compared to controls (n=25), while alpha cell mass (orange) is not different. Subjects with type 1 diabetes and those with amyloidosis were excluded. Data are mean ± se and from [4] with permission. (b) Beta cell volume is increased in obese (pink; n=4 controls and 6 with type 2 diabetes) compared to normal weight (white; n=7 controls and 8 with type 2 diabetes) subjects. Diabetes is associated with reduced beta cell volume in both obese and normal weight subjects. Data are mean ± sd and from [5] with permission. (c) Beta cell volume is reduced in subjects with type 2 diabetes (DM, green; n=16 normal weight and 41 obese) compared to those who do not have diabetes (ND, yellow; n=17 normal weight and 31 obese), whether subjects are normal weight of obese. Subjects with impaired fasting glucose (IFG, pink; n=19 obese) have reduced beta cell volume that is intermediate between no diabetes and diabetes. Data are mean ± se and adapted with permission from [6].
Figure 3.
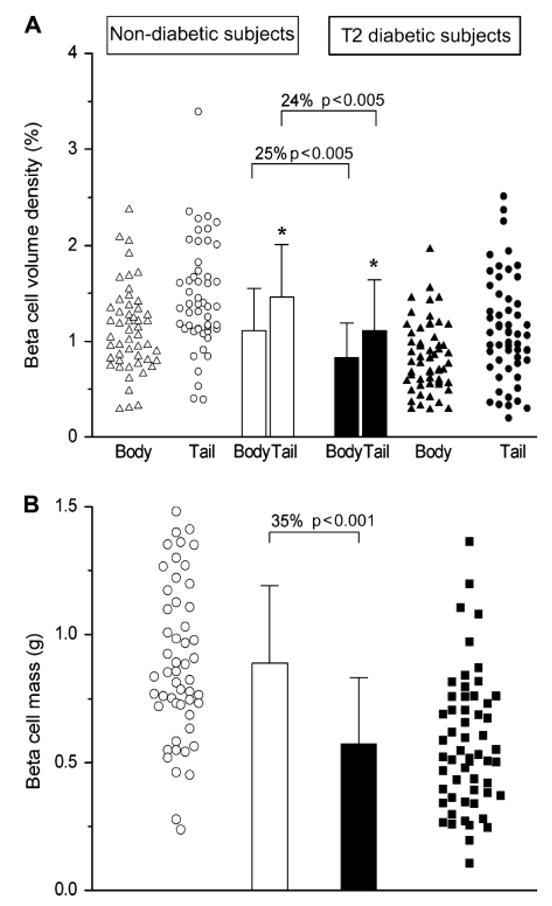
Differences in beta cell volume and mass in 52 subjects without (open symbols and bars) and 57 with type 2 diabetes (closed symbols and bars). (a) The volume of beta cells varies markedly in non-diabetic and diabetic subjects with a lot of overlap between the two groups. Despite this large variability, beta cell volume is greater in the pancreatic tail than body in both non-diabetic and diabetic subjects. In subjects with diabetes compared to those without, beta cell volume is reduced in both the body and tail of the pancreas. (b) Beta cell mass determined based on volume density and pancreatic weight is reduced by 35% in subjects with type 2 diabetes. Data are mean ± sd. Reproduced with permission from [12].
If beta cell mass is reduced, can this be the sole explanation for the development of hyperglycaemia in type 2 diabetes? To answer this, we can turn to studies in both humans and animals to gain a better understanding. Hemi-pancreatectomy in human donors fails to produce frank hyperglycaemia. In first-degree relatives of the recipient, no significant change in fasting glucose and a small change in intravenous glucose tolerance were observed [13], while in patients who for medical reasons underwent a pancreatectomy of a similar magnitude, glucose tolerance either improved or deteriorated mildly [14]. The findings in these human studies are supported by work in rodents, dogs and monkeys. Studies in rodents undertaken in large part by the Boston group have highlighted the need to remove close to 90% of pancreatic tissue in healthy animals before hyperglycaemia can be demonstrated [15]. Similarly, two canine studies clearly demonstrated that resection of 50% [16] and 65% [17] of the pancreas did not result in the development of diabetes. It is quite possible that the lack of a significant increase in fasting glucose in the studies of reduced beta cell mass produced by resection of pancreatic tissue may be due to the simultaneous reduction of alpha cells, new beta cell formation or functional adaptation of the remaining beta cells. However, this has not been clearly delineated.
Textbox 1
50% pancreatectomy in humans does not result in diabetes or compensatory regeneration of beta cells.
Surgical reduction of islet mass reduces both alpha and beta cell number, and thus would be expected to reduce both glucagon and insulin release. Prospective human data on this are rather limited, but available information suggests that while fasting levels of glucagon and insulin are not different, the glucagon and insulin responses to arginine are probably decreased [13]. Thus, the glucagon to insulin ratio is not markedly altered, which is not the case in two non-human primate models where predominantly beta cells are lost. These two models of reduced beta cell mass, one chemical [18] and the other secondary to islet amyloid deposition [19], suggest it is unlikely that a reduction in the number of glucagon-producing cells explains the lack of recurrence of hyperglycaemia. In the study where beta cells were destroyed with streptozotocin, normoglycaemia was maintained despite the fact that the number of insulin secreting cells was clearly diminished [18]. In the other, only in the presence of both beta and alpha cell secretory dysfunction and after islet amyloid deposition had reduced beta cell mass by over 50%, was an elevation in the fasting glucose observed [19]. It seems therefore one has to look beyond changes in the number of alpha cells as the basis for the rather mild impairments in glucose tolerance and fasting glucose observed with simple beta cell mass reduction.
Could the lack of diabetes following beta cell mass reduction be explained by new beta cell formation? While one should always be cautious extrapolating the rodent literature to humans, when considering this question, one has to be even more careful. That being said, there are still some similarities. There is clear evidence that the greatest turnover of beta cells occurs in the neonatal period in rodents and a similar event occurs in humans [20]. With obesity-associated insulin resistance, the volume of beta cells is increased [5, 6, 12], which appears to be in part the result of an increase in secretory demand on the beta cell. Pregnancy is also associated with insulin resistance and in rodents beta cell expansion occurs followed post-partum by involution [21], a process likely also occurring in humans [22]. However, later in life such expansion is less likely as aging is clearly associated with a decrease in the ability of the beta cell to regenerate [23]. While in young rodents regeneration of beta cells may occur by replication of existing beta cells or neogenesis from exocrine ducts [24], the evidence for this occurring in humans and even in non-rodent animal models is minimal. In fact, a recent study underscored the difficulty of using the human pancreas to produce new beta cells [25]. In this work, examination of tissue obtained at consecutive partial pancreatectomies showed that the loss of beta cells did not result in compensatory regeneration, as there were no differences in the fractional beta cell area, proportion of proliferating beta cells or duct cells expressing insulin [25]. A similar observation has been made in dogs following surgical reduction of beta cell mass where, several months following the procedure, the pancreatic remnant did not differ from the earlier resected pancreas morphologically, including the number and size of islets and the number of beta cells [16]. Thus, a compensatory response involving new beta cell formation seems to be a less likely mechanism for compensation for reduced beta cell volume in non-rodent models of reduced beta cell mass and in humans where beta cell mass is actually reduced.
Where does that leave us? A likely conclusion has to be that a simple reduction in beta cell mass, often greater than that typically observed in humans with type 2 diabetes, fails to result in a clinically meaningful increase in glucose levels. From this it seems likely that when the beta cell is normal, the maintenance of glucose homeostasis following a reduction of beta cell mass is the consequence of an adaptive increase in beta cell function. That this may be the case is discussed in a subsequent section.
Is Beta cell Function Reduced in Type 2 Diabetes?
Here again, the answer has been known for a substantial period of time and there is absolutely no doubt that it is diminished. In fact, Yalow and Berson’s development of the insulin radioimmunoassay half a century ago provided the first clear evidence of a defect in beta cell function [26] (Figure 4). In their study they showed that even in individuals with “early maturity onset diabetes”, there was a defect in early insulin release following glucose ingestion. Subsequently it has been shown that this early insulin secretory defect following oral glucose is already well established in individuals with impaired glucose tolerance (IGT), irrespective of their ethnicity [27, 28] (Figure 5).
Figure 4.
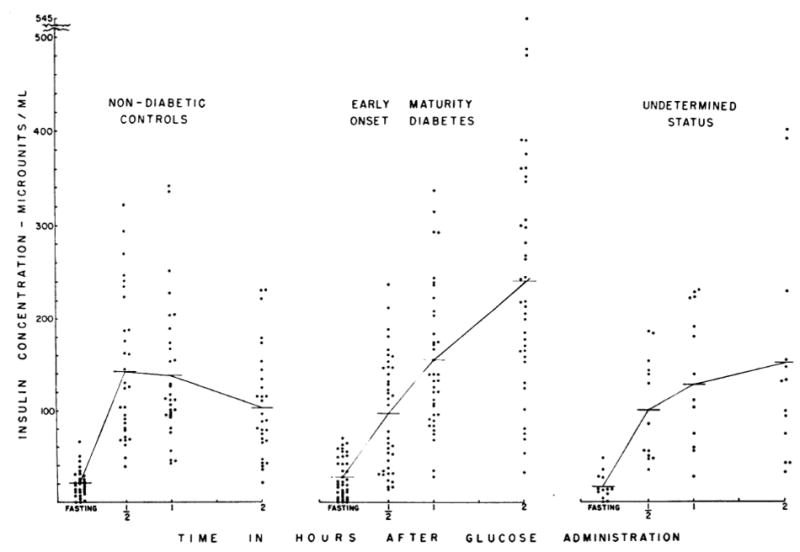
Plasma insulin concentrations before and following a 100-gram glucose load in healthy control subjects and two groups of subjects, one known to have early (“maturity onset”) type 2 diabetes and a second group in whom the duration of the diabetes was undetermined. The insulin response at 30 minutes was clearly diminished in individuals with recent onset of hyperglycaemia. Reproduced with permission from [26].
Figure 5.
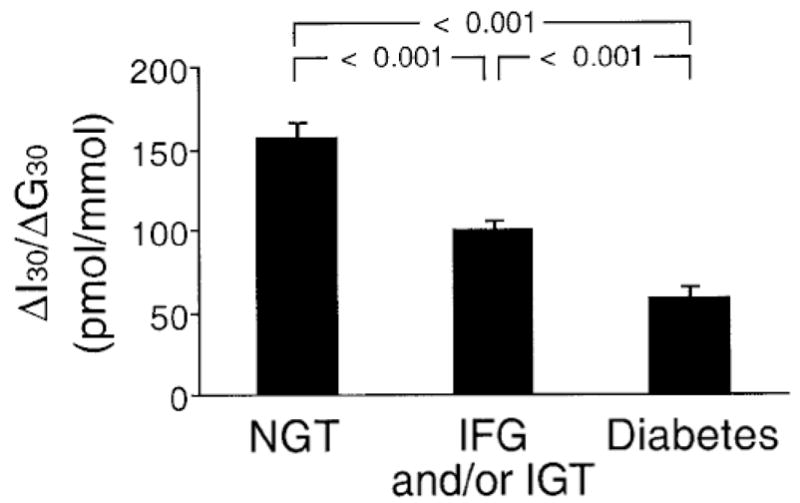
Incremental early insulin response (insulinogenic index) measured 30 minutes after glucose ingestion (ΔI30/ΔG30) in 240 subjects with normal glucose tolerance (NGT), 191 with impaired fasting glucose/impaired glucose tolerance (IFG/IGT) and 100 with diabetes. As glucose tolerance declined, beta cell function deteriorated. Reproduced with permission from [27].
Textbox 2
In humans with type 2 diabetes, first-phase insulin release is absent when beta cell volume is reduced by only 30–60%.
While this early insulin response to oral glucose is reduced in subjects with diminished glucose tolerance and is further decreased in those with type 2 diabetes, insulin is still released (Figures 4 and 5). In contrast, the acute insulin response following intravenous glucose injection is completely lacking in type 2 diabetes [29]. In fact, it has clearly been shown to decline with increasing fasting plasma glucose levels, commencing at a level of 5.0 mmol/l and frequently being absent when the concentration is below the diagnostic cut point for diabetes [30, 31] (Figure 6). Thus, there is a clear dissimilarity in the responses to oral and intravenous glucose, with these distinctions highlighting that the beta cell functional defect differentially affects the pathways that couple stimulus and secretion. Importantly, in contrast to the pathway responsible for the rapid release of insulin following intravenous glucose stimulation, coupling of signalling and secretion through pathways responsive to non-glucose secretagogues is not totally absent even at glucose concentrations that are well above 7.0 mmol/l, the level that defines human type 2 diabetes [32]. Interestingly, this is not simply a function of the in vivo situation as analogous observations have been made in vitro using donor islets from healthy and type 2 diabetic subjects [33–35]. Perifusion or static incubation of islets from subjects with type 2 diabetes demonstrated the total loss of the response to glucose with lesser reductions in the responses to potassium, arginine, glibenclamide and glucagon-like peptide-1 (GLP-1). Further, the impact of this secretory defect was highlighted in studies in which transplantation of isolated islets from healthy humans into diabetic immunodeficient mice resulted in the reversal of hyperglycaemia, while transplantation of equivalent numbers of islets from type 2 diabetic cadaveric donors did not [33]. However, it must be recognized that a limitation of this report is that while similar numbers of islets were transplanted, the quantity of beta cells transplanted was not reported.
Figure 6.
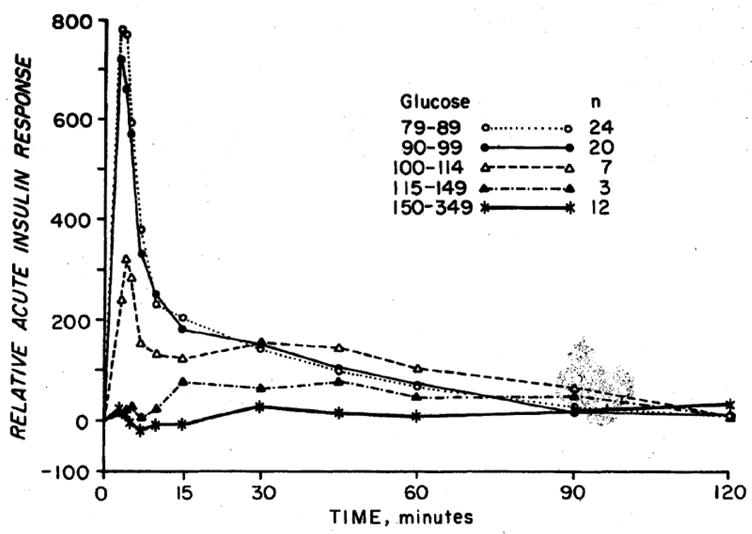
Acute insulin response to intravenous glucose in subjects grouped based on their fasting glucose levels. As the fasting glucose increases, the insulin response decreases. The response is absent when fasting glucose exceeds 6.4 mmol/l. Reproduced with permission from [30].
A number of abnormalities of continuous insulin release have also been described in individuals with type 2 diabetes and those at risk. The beta cell has an intrinsic pacemaker that generates pulses of insulin release approximately every 15 minutes [36, 37]. These rapid pulses occur on a background of slower, ultradian oscillations tightly coupled to natural oscillations in plasma glucose that have a periodicity of 80–150 minutes [38]. In type 2 diabetes and in high-risk states, these secretory patterns are altered. Thus, in individuals with a family history of type 2 diabetes, the rapid pulses are lost [37], while in subjects with type 2 diabetes and those with IGT, the tight temporal oscillatory pattern between glucose and insulin is disturbed so that the oscillatory changes in insulin secretion lack normal periodicity [38]. Further, the normal beta cell can be entrained to oscillate in concert with exogenously produced glucose oscillations, with the amplitude of these oscillations increasing in proportion to the magnitude of change in plasma glucose [38]. In contrast, beta cells in individuals with IGT and type 2 diabetes are incapable of entraining and fail to increase insulin release appropriately in response to changes in plasma glucose [38].
In addition to these secretory changes, the biosynthesis of insulin has been shown to be abnormal in type 2 diabetes. Insulin production involves its proteolytic processing from a larger precursor proinsulin to the final products insulin and C-peptide. This series of steps occurring within the beta cell is not 100% efficient, so that exocytosis of secretory granule content results in the release of small amounts of proinsulin and partially processed intermediates, along with C-peptide and insulin [39]. In type 2 diabetes, peptide processing is less efficient so that a greater proportion of proinsulin is released resulting in disproportionate proinsulinaemia [39]. Furthermore, disproportionate proinsulinaemia has been shown to be a marker for the subsequent development of diabetes in individuals at increased risk [40, 41].
Can these functional changes be recapitulated in individuals without beta cell mass reduction and thus lend credence to the concept that a simple functional lesion is all that is necessary for the development of type 2 diabetes? Based on observations with a number of different interventions, the answer to this question is “unlikely”. When beta cell granule exocytosis is reduced by administration of somatostatin, insulin release is clearly impaired, but at the same time this inhibitory peptide is not beta cell selective so that glucagon release from the alpha cell is also diminished. The result is only a relatively small rise in the fasting glucose concentration, even when glucagon is replaced [42]. When activity of the beta cell’s potassium channel is inhibited with the anti-hypertensive diazoxide in order to impair glucose-stimulated insulin secretion, the release of beta cell products is impaired but again only a relatively mild change in glycaemia occurs [43]. Interestingly, while glucocorticoids are well recognized to induce insulin resistance [44], they also have islet effects that result in a disproportionate increase in proinsulin release and increased glucagon output as observed in type 2 diabetes [45–48]; yet, in otherwise healthy individuals they do not induce major reductions in glucose tolerance. Finally, administration of catecholamines to mimic increased alpha adrenergic activity as a mechanism underlying beta cell dysfunction results in reduced insulin secretory responses, but again does not produce the abnormalities typically associated with the hyperglycaemia of type 2 diabetes [49]. Thus, it is not possible to intervene in humans with one of these approaches and reproduce the severity of the functional beta cell lesion of type 2 diabetes. However, it is unclear whether simultaneous abnormalities of, for example, sympathetic tone and potassium channel activity could result in the development of frank hyperglycaemia.
Where does that then leave us? It is clear that the defect in beta cell function in type 2 diabetes is global: reductions in insulin secretory responses to orally ingested nutrients as well as to intravenous glucose and non-glucose secretagogues, dampened pulsatile insulin release, and impaired insulin biosynthesis. However, the magnitude of the functional abnormality differs based on the measure being examined. And, as there is no easy way to chemically reproduce the beta cell abnormalities of type 2 diabetes, the lesion is likely at a minimum comprised of a number of changes in cellular function.
Textbox 3
| Islet Function | |
| In Type 2 Diabetes | Post Partial Pancreatectomy |
| Absent first-phase insulin response to glucose | Zero or minimal change in first-phase insulin response to glucose |
| Reduced insulin responses to non-glucose secretagogues | Reduced insulin responses to non-glucose secretagogues |
| Loss of periodicity of pulsatile insulin release | Maintenance of periodicity of pulsatile insulin release |
| Insulin release not entrainable | Insulin release entrainable |
| Inappropriately increased glucagon responses | Reduced glucagon responses |
| Results in hyperglycaemia | Zero or minimal change in glycaemia |
Does Either Reduced Beta Cell Mass or Beta Cell Dysfunction Beget the Other?
It appears therefore that reductions in both beta cell mass and beta cell function are present in individuals with type 2 diabetes. A critical question then arises. Does one follow the other, with one possibly even causing the other? As we cannot assess beta cell mass non-invasively, information in humans is non-existent and it is difficult to know exactly what happens in the progression from normal glucose tolerance through impaired glucose tolerance to type 2 diabetes. Thus, we have no choice but to turn to either animal models or in vitro studies, both of which clearly have limitations, in extrapolating findings to the human disease process. When considering this important question, one has to include as part of this discourse consideration of additional aspects many of the studies discussed earlier.
First, let’s consider the possibility of simple reductions in beta cell mass of the order observed in type 2 diabetes begetting beta cell dysfunction. The available evidence in animal models would suggest this simple mechanism is not likely to be operative. A 50% percent surgical reduction of pancreatic mass in dogs failed to produce changes in either fasting or arginine-stimulated insulin levels, fasting glucagon concentrations, the peak insulin response to intravenous glucose, or insulin sensitivity [16]. In the face of this lack of change, it is not surprising that the animals did not develop hyperglycaemia. This contrasts with both type 2 diabetes and IGT where to a variable extent reduced insulin responses, excessive glucagon release and insulin resistance exist [1, 2, 48]. In order to produce a state of hyperglycaemia in these dogs with a reduced number of islets, it was necessary to infuse glucose continuously for two weeks to maintain the plasma glucose level above 13.9 mmol/l [16]. Similarly, in rats following a 60% pancreatectomy, glycaemia was not altered six weeks following surgery unless sucrose was introduced into the drinking water, and then it took three weeks with the increase in non-fasting glucose still being <1 mmol/l [50]. In Gottingen minipigs treated with both streptozotocin and nicotinamide, a model of beta cell mass reduction in which alpha cells would not be anticipated to be reduced, mild hyperglycaemia developed but derangements in the periodicity or entrainability of insulin release could not be reproduced and all that was observed was a simple reduction in the amplitude of the pulses [51]. These findings contrast with the derangements of continuous insulin release observed both in type 2 diabetic subjects and those at increased risk [37, 52]. Consequently, as simple mass reduction does not seem to beget the functional abnormalities, one has to conclude that beta cell mass has been replenished and/or beta cell function has improved.
That it is unlikely that replenishment of beta cell mass is occurring is suggested by the fact that there does not appear to even be a period of transient hyperglycaemia in the immediate days or weeks post-partial pancreatectomy, even when up to two-thirds of the pancreas has been removed [16, 17]. More importantly, as discussed and in keeping with this observation, six months following removal of 50% of the pancreas the size and number of islets as well as the number of beta cells was not different to that in the portion of pancreas obtained at the time of resection [16]. Interestingly, this observation in dogs is compatible with the recent report of a lack of new beta cell formation following 50% pancreatectomy in humans [25]. Thus, it would seem more likely that the normal beta cell is adapting functionally to the loss of a large proportion of its compatriots.
That the normal beta cell has an innate capacity to obviate the development of hyperglycaemia by enhancing its function is suggested by studies in healthy animals that have undergone a surgical reduction in islet mass. Following a 65% pancreatectomy in dogs, which reduces beta cell mass more than typically found in human type 2 diabetes, the beta cell’s sensitivity to glucose is enhanced [17]. This adaptive change means that at physiological glucose concentrations each beta cell is releasing insulin more efficiently and thereby maintaining euglycaemia. Observations compatible with such an enhancement of beta cell sensitivity to glucose have also been made in rodent models of reduced beta cell mass [53]. Thus, it would appear that a dysfunctional beta cell might be unable to adapt to a reduction in beta cell mass, resulting ultimately in the development of hyperglycaemia.
More recent work using genetically modified rodents strongly support the thesis that reduced beta cell mass alone is insufficient and that beta cell dysfunction is a sine qua non if mass reduction is to result in the development of hyperglycaemia. In a mouse lacking the critical transcription factor FoxM1, proliferation of existing beta cells cannot occur; yet, despite this inability to form new beta cells, a 60% pancreatectomy does not result in a change in glucose tolerance [54]. On the other hand, a 70% pancreatectomy in GLP-1 receptor null mice that fail to release insulin in response to GLP-1 [55] results in the development of marked hyperglycaemia [56]. Thus, one is left to conclude that when the remaining beta cells are functionally normal, a simple reduction in mass alone does not produce beta cell dysfunction, while when the remaining beta cells are dysfunctional, the additional insult of a reduction in beta cell mass results in the development of hyperglycaemia.
Could beta cell dysfunction then be the basis for a reduction in beta cell mass? Here again the inability to monitor possible changes in beta cell mass in situ has required the use of in vitro systems or examination of animal models in which pancreatic material is typically only accessible post mortem. It is clear from in vitro work that elevations in glucose and fatty acids, either alone or together, are associated with the induction of beta cell dysfunction and the loss of s cells by apoptosis [57]. However, these changes are not easily recapitulated in vivo.
Glucose-induced changes in beta cell morphology that could be considered indicative of reduced beta cell mass have been observed. In dogs, following two months of sustained hyperglycaemia only attainable after two weeks of continuous glucose administration on a background of reduced beta cell mass, a profound reduction in the number and size of islets was found with marked depletion of insulin stores [16]. In contrast, in rats 96 hours of hyperglycaemia produced by continuous infusion of glucose is associated with an increase in beta cell mass as a result of hypertrophy and replication, likely averting further hyperglycaemia [58]. These findings contrast with those in hyperglycaemic humans where a compensatory generation of new beta cells does not occur [5, 6, 12]. While these glucose-infused animal models are of interest, their relevance as a primary mechanism to explain human type 2 diabetes is questionable since the human condition does not typically transition rapidly from normal glucose tolerance through IGT to marked hyperglycaemia, but instead progresses over years.
While classically thought of in terms of peptide release, beta cell dysfunction could lead to beta cell loss through a mechanism(s) unrelated solely to exocytosis. Insulin is not the only unique peptide synthesized by the beta cell; the cell produces and co-releases islet amyloid polypeptide (IAPP or amylin) in parallel with insulin [59]. Whereas the physiology of IAPP is poorly understood, its association with the loss of beta cell mass in type 2 diabetes is clearer. Normally this amyloidogenic peptide does not aggregate to form amyloid fibrils, whereas in type 2 diabetes it does so and during this process beta cell death and mass loss likely ensue [60]. It is not clear in human type 2 diabetes what beta cell environment is permissive for IAPP aggregation to occur, but it has been suggested that impaired processing of the larger precursor proIAPP to native IAPP may be an important contributor [61, 62]. Further work is needed to determine the basis for IAPP aggregation and may throw light on whether this represents an instance where beta cell dysfunction may commence the process of beta cell loss.
It would appear therefore that any ability of reduced beta cell mass to beget beta cell dysfunction or beta cell dysfunction to beget reduced beta cell mass is complex. Of the two processes, current data would suggest the latter is more likely. However, the evidence is far from definitive and more work is clearly required.
So is the Beta Cell Lesion of Type 2 Diabetes Simply “Decreased Functional Beta Cell Mass” or is it “Decreased Beta Cell Mass and Function”?
The former and rather interesting description of the beta cell lesion of type 2 diabetes has come about in recent years. Its basis and meaning appear to us somewhat mysterious and perhaps even misleading.
Textbox 4
The meaning of the term “functional beta cell mass” appears somewhat mysterious and even misleading.
If one accepts the premise that beta cell mass is decreased in type 2 diabetes, does this term then mean that the reason hyperglycaemia develops is because normally functioning beta cells have been lost and all that remains are dysfunctional cells? Alternatively, are the changes simply one of beta cell mass reduction resulting in a critical reduction in the volume of normally functioning beta cells? On the other hand, could this represent the loss of beta cells that then leads through “stress” to the loss of beta cell function? Based on the evidence we have presented, it is truly unclear. In addition, the fact that in the presence of a sizeable number of beta cells that contain plenty of insulin, the first-phase response to intravenous glucose can be totally absent when others such as the response to intravenous GLP-1 or oral glucose are still present, truly underscores the inadequacy of this term. Therefore, we feel it is a misnomer and consideration should be given to striking it from discussion.
What would we then propose as a better descriptor? Our opinion is that the beta cell lesion of type 2 diabetes is multifactorial. It is likely genetically determined and influenced by the environment, both intra-uterine and from birth throughout life. The genetic underpinning may be two-fold, with genetically determined reductions in both beta cell secretory function and beta cell mass co-existing (Figure 7a). Alternatively, though we believe less likely, it may result from a genetic susceptibility that simultaneously affects both mass and function (Figure 7b). Finally, we cannot exclude that on the background of a genetic abnormality, a dysfunctional beta cell may lie upstream of a mass lesion and coexistence of a “toxic” environment further elaborates this beta cell abnormality leading to the loss of beta cells (Figure 7c). Therefore, we are of the conviction that amalgamating the processes of the loss of beta cell mass and beta cell function together as a single entity is unwise. To us they represent separate and yet critical interdependent processes and should be seen that way. We propose that as the complex beta cell lesion of type 2 diabetes is the result of “decreased beta cell mass and function”, it should be considered and termed that way. Understanding the basis for the failure of these two components holds part of the key to the pathogenesis of the islet lesion of type 2 diabetes and ultimately to better approaches to its therapy.
Figure 7.


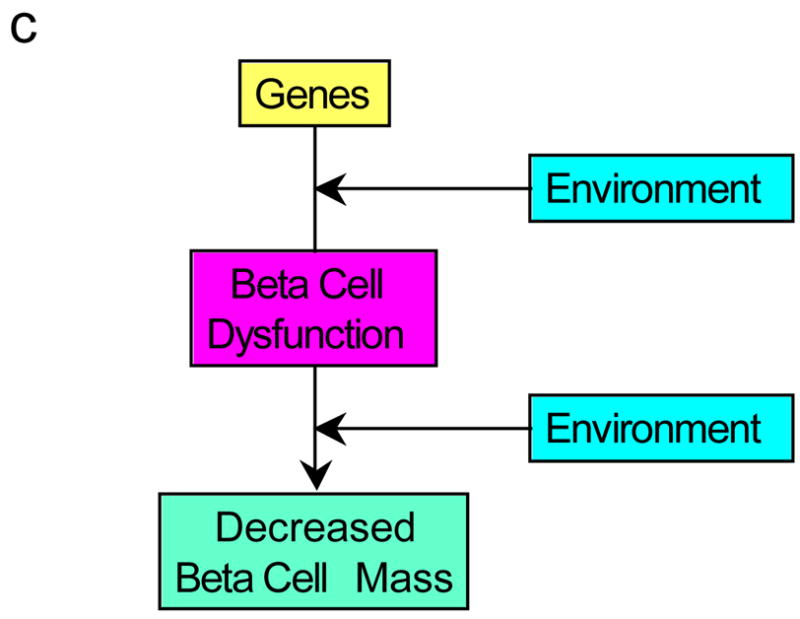
Possible pathways for the development of the reduced beta cell mass and beta cell dysfunction observed in type 2 diabetes. (a) Genetic defects combined with environmental changes determine reductions in both beta cell function and mass. (b) A genetic defect in combination with environmental changes simultaneously affects both mass and function. (c) The interaction of genetic susceptibility with environmental factors results in beta cell dysfunction that in turn leads to a loss of beta cells. The effect of this combination of changes is associated with the development of hyperglycaemia.
Acknowledgments
This work was supported in part by the Department of Veterans Affairs, NIH grants DK-075998, DK-074404 and a Juvenile Diabetes Research Foundation Fellowship to S.Z.
Footnotes
Duality of Interest
The authors declare that they have no duality of interest.
References
- 1.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 3.Opie E. The relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islets of Langerhans. J Exp Med. 1901;5:527–540. doi: 10.1084/jem.5.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maclean N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes. 1955;4:367–376. doi: 10.2337/diab.4.5.367. [DOI] [PubMed] [Google Scholar]
- 5.Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. s-cell deficit and increased s-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 7.Clark A, Wells CA, Buley ID, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed] [Google Scholar]
- 8.Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia. 1983;24:366–371. doi: 10.1007/BF00251826. [DOI] [PubMed] [Google Scholar]
- 9.Saito K, Yaginuma N, Takahashi T. Differential volumetry of A, B, and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J Exp Med. 1979;129:273–283. doi: 10.1620/tjem.129.273. [DOI] [PubMed] [Google Scholar]
- 10.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 11.Yoon KH, Ko SH, Cho JH, et al. Selective s-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 12.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic s-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10 (Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 13.Seaquist ER, Robertson RP. Effects of hemipancreatectomy on pancreatic alpha and beta cell function in healthy human donors. J Clin Invest. 1992;89:1761–1766. doi: 10.1172/JCI115779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menge BA, Schrader H, Breuer TG, et al. Metabolic consequences of a 50% partial pancreatectomy in humans. Diabetologia. 2009;52:306–317. doi: 10.1007/s00125-008-1219-1. [DOI] [PubMed] [Google Scholar]
- 15.Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest. 1983;71:1544–1553. doi: 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imamura T, Koffler M, Helderman JH, et al. Severe diabetes induced in subtotally depancreatized dogs by sustained hyperglycemia. Diabetes. 1988;37:600–609. doi: 10.2337/diab.37.5.600. [DOI] [PubMed] [Google Scholar]
- 17.Ward WK, Wallum BJ, Beard JC, Taborsky GJ, Jr, Porte D., Jr Reduction of glycemic potentiation: a sensitive indicator of s-cell loss in partially pancreatectomized dogs. Diabetes. 1988;37:723–729. doi: 10.2337/diab.37.6.723. [DOI] [PubMed] [Google Scholar]
- 18.McCulloch DK, Raghu PK, Johnston C, et al. Defects in s-cell function and insulin sensitivity in normoglycemic streptozocin-treated baboons: a model of preclinical insulin-dependent diabetes. J Clin Endocrinol Metab. 1988;67:785–792. doi: 10.1210/jcem-67-4-785. [DOI] [PubMed] [Google Scholar]
- 19.Howard CF., Jr Longitudinal studies on the development of diabetes in individual Macaca nigra. Diabetologia. 1986;29:301–306. doi: 10.1007/BF00452067. [DOI] [PubMed] [Google Scholar]
- 20.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. s-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49:1325–1333. doi: 10.2337/diabetes.49.8.1325. [DOI] [PubMed] [Google Scholar]
- 21.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 22.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 23.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 24.Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1220. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 25.Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke s-cell regeneration. Diabetes. 2008;57:142–149. doi: 10.2337/db07-1294. [DOI] [PubMed] [Google Scholar]
- 26.Yalow RS, Berson SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39:1157–1175. doi: 10.1172/JCI104130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE American Diabetes Association GENNID Study Group. s-cell function is the major determinant of oral glucose tolerance in four ethnic groups in the United States. Diabetes. 2002;51:2170–2178. doi: 10.2337/diabetes.51.7.2170. [DOI] [PubMed] [Google Scholar]
- 28.Mitrakou A, Kelley D, Mokan M, et al. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med. 1992;326:22–29. doi: 10.1056/NEJM199201023260104. [DOI] [PubMed] [Google Scholar]
- 29.Pfeifer MA, Halter JB, Porte D., Jr Insulin secretion in diabetes mellitus. Am J Med. 1981;70:579–588. doi: 10.1016/0002-9343(81)90579-9. [DOI] [PubMed] [Google Scholar]
- 30.Brunzell JD, Robertson RP, Lerner RL, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42:222–229. doi: 10.1210/jcem-42-2-222. [DOI] [PubMed] [Google Scholar]
- 31.Godsland IF, Jeffs JA, Johnston DG. Loss of beta cell function as fasting glucose increases in the non-diabetic range. Diabetologia. 2004;47:1157–1166. doi: 10.1007/s00125-004-1454-z. [DOI] [PubMed] [Google Scholar]
- 32.Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D., Jr Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328. doi: 10.1172/JCI111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng S, Vatamaniuk M, Huang X, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004;53:624–632. doi: 10.2337/diabetes.53.3.624. [DOI] [PubMed] [Google Scholar]
- 34.Del Guerra S, Lupi R, Marselli L, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–735. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 35.Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes. 2006;55:435–440. doi: 10.2337/diabetes.55.02.06.db04-1575. [DOI] [PubMed] [Google Scholar]
- 36.Bergstrom RW, Fujimoto WY, Teller DC, De Haen C. Oscillatory insulin secretion in perifused isolated rat islets. Am J Physiol. 1989;257:E479–E485. doi: 10.1152/ajpendo.1989.257.4.E479. [DOI] [PubMed] [Google Scholar]
- 37.O’Rahilly S, Turner RC, Matthews DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med. 1988;318:1225–1230. doi: 10.1056/NEJM198805123181902. [DOI] [PubMed] [Google Scholar]
- 38.O’Meara NM, Sturis J, Van Cauter E, Polonsky KS. Lack of control by glucose of ultradian insulin secretory oscillations in impaired glucose tolerance and in non-insulin-dependent diabetes mellitus. J Clin Invest. 1993;92:262–271. doi: 10.1172/JCI116560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46:1725–1732. doi: 10.2337/diab.46.11.1725. [DOI] [PubMed] [Google Scholar]
- 40.Kahn SE, Leonetti DL, Prigeon RL, Bergstrom RW, Fujimoto WY. Proinsulin as a marker for the development of NIDDM in Japanese-American men. Diabetes. 1995;44:173–179. doi: 10.2337/diab.44.2.173. [DOI] [PubMed] [Google Scholar]
- 41.Mykkanen L, Haffner SM, Kuusisto J, Pyorala K, Hales CN, Laakso M. Serum proinsulin levels are disproportionately increased in elderly prediabetic subjects. Diabetologia. 1995;38:1176–1182. doi: 10.1007/BF00422366. [DOI] [PubMed] [Google Scholar]
- 42.Ward WK, Halter JB, Best JD, Beard JC, Porte D., Jr Hyperglycemia and s-cell adaptation during prolonged somatostatin infusion with glucagon replacement in man. Diabetes. 1983;32:943–947. doi: 10.2337/diab.32.10.943. [DOI] [PubMed] [Google Scholar]
- 43.Raju B, Cryer PE. Mechanism, temporal patterns, and magnitudes of the metabolic responses to the KATP channel agonist diazoxide. Am J Physiol Endocrinol Metab. 2005;288:E80–E85. doi: 10.1152/ajpendo.00188.2004. [DOI] [PubMed] [Google Scholar]
- 44.Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab. 1982;54:131–138. doi: 10.1210/jcem-54-1-131. [DOI] [PubMed] [Google Scholar]
- 45.Ward WK, LaCava EC, Paquette TL, Beard JC, Wallum BJ, Porte D., Jr Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and experimental insulin resistance. Diabetologia. 1987;30:698–702. doi: 10.1007/BF00296991. [DOI] [PubMed] [Google Scholar]
- 46.Kalhan SC, Adam PAJ. Inhibitory effect of prednisone on insulin secretion in man: model for duplication of blood glucose concentration. J Clin Endocrinol Metab. 1975;41:600–610. doi: 10.1210/jcem-41-3-600. [DOI] [PubMed] [Google Scholar]
- 47.Marco J, Calle C, Roman D, Diaz-Fierros M, Villaneuva ML. Hyperglucagonism induced by glucocorticoid treatment in man. N Engl J Med. 1973;288:128–131. doi: 10.1056/NEJM197301182880305. [DOI] [PubMed] [Google Scholar]
- 48.Raskin P, Aydin I, Yamamoto T, Unger RH. Abnormal alpha cell function in human diabetes: the response to oral protein. Am J Med. 1978;64:988–997. doi: 10.1016/0002-9343(78)90454-0. [DOI] [PubMed] [Google Scholar]
- 49.Robertson RP, Halter JB, Porte D., Jr A role for alpha-adrenergic receptors in abnormal insulin secretion in diabetes mellitus. J Clin Invest. 1976;57:791–795. doi: 10.1172/JCI108338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leahy JL, Bonner-Weir S, Weir GC. Minimal chronic hyperglycemia is a critical determinant of impaired insulin secretion after an incomplete pancreatectomy. J Clin Invest. 1988;81:1407–1414. doi: 10.1172/JCI113470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen MO, Gotfredsen CF, Wilken M, Carr RD, Porksen N, Rolin B. Loss of beta-cell mass leads to a reduction of pulse mass with normal periodicity, regularity and entrainment of pulsatile insulin secretion in Gottingen minipigs. Diabetologia. 2003;46:195–202. doi: 10.1007/s00125-002-1011-6. [DOI] [PubMed] [Google Scholar]
- 52.Porksen N. Early changes in beta-cell function and insulin pulsatility as predictors for type 2 diabetes. Diabetes Nutr Metab. 2002;15 (Suppl):9–14. [PubMed] [Google Scholar]
- 53.Leahy JL, Bonner-Weir S, Weir GC. Abnormal glucose regulation of insulin secretion in models of reduced B-cell mass. Diabetes. 1984;33:667–673. doi: 10.2337/diab.33.7.667. [DOI] [PubMed] [Google Scholar]
- 54.Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57:3069–3077. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pederson RA, Satkunarajah M, McIntosh CH, et al. Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor −/− mice. Diabetes. 1998;47:1046–1052. doi: 10.2337/diabetes.47.7.1046. [DOI] [PubMed] [Google Scholar]
- 56.De Leon DD, Deng S, Madani R, Ahima RS, Drucker DJ, Stoffers DA. Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes. 2003;52:365–371. doi: 10.2337/diabetes.52.2.365. [DOI] [PubMed] [Google Scholar]
- 57.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and s-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic s-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38:49–53. doi: 10.2337/diab.38.1.49. [DOI] [PubMed] [Google Scholar]
- 59.Kahn SE, Verchere CB, Andrikopoulos S, et al. Reduced amylin release is a characteristic of impaired glucose tolerance and type 2 diabetes in Japanese Americans. Diabetes. 1998;47:640–645. doi: 10.2337/diabetes.47.4.640. [DOI] [PubMed] [Google Scholar]
- 60.Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89:3629–3643. doi: 10.1210/jc.2004-0405. [DOI] [PubMed] [Google Scholar]
- 61.Porte D, Jr, Kahn SE. Hyperproinsulinemia and amyloid in NIDDM: clues to etiology of islet s-cell dysfunction? Diabetes. 1989;38:1333–1336. doi: 10.2337/diab.38.11.1333. [DOI] [PubMed] [Google Scholar]
- 62.Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA, Verchere CB. Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes. 2006;55:2192–2201. doi: 10.2337/db05-1566. [DOI] [PubMed] [Google Scholar]