

Abstract
Alzheimer's disease (AD) is a common neurodegenerative disease characterized by both extra- as well as intracellular deposition of amyloid beta peptides (Aβ). The accumulation of Aβ in mitochondria is associated with mitochondrial dysfunction and oxidative stress in AD. Recent evidences suggest the involvement of Aβ interaction with mitochondrial proteins such as cyclophilin-D (CypD) in oxidative stress, mitochondrial permeability transition (MPT) and Alzheimer's associated neurodegeneration. The present study is an effort to elucidate the molecular interaction of Aβ with other proteins involved in MPT like adenine nucleotide translocase (ANT). Based on our prediction for sub-cellular localization using WolfPSORT and other experimental evidences, we suggest that Aβ molecules localize in mitochondrial inner membrane in close vicinity with ANT. Our simulation study for protein–protein interaction clearly suggests that the ANT-Aβ interaction is stronger than CypD-Aβ interaction. Further the lipophilic nature and evidences regarding the localization of Aβ in the mitochondrial inner-membrane also support the possibility of strong interaction between ANT and Aβ. Interaction between ANT and Aβ may affect normal physiological function of ANT i.e. transport of ATP and ADP. Since both the CypD-Aβ as well as ANT-Aβ interaction are energetically favorable and both CypD and ANT are associated with the regulation of MPT, the functional impact of both these interactions warrants more in-depth investigations for elucidating the mechanisms involved in Aβ-induced oxidative stress.
Keywords: Alzheimer, amyloid beta peptide, adenine nucleotide translocase, voltage dependent anion channel, mitochondrial permeability
Abbreviations
AD - Alzheimer's disease, Aβ - amyloid beta peptide, APP - Amyloid precursor protein, MPT - Mitochondrial permeability transition, MPTP - Mitochondrial permeability transition pore, ANT - adenine nucleotide translocase, CypD - cyclophilin-D, VDAC - voltage dependent anion channel.
Background
Alzheimer's disease (AD) is a common neurodegenerative disease that affects cognitive functions of the brain. The cardinal features of AD include the formation of extracellular protein deposits in the brain consisting predominantly of aggregates of amyloid beta (Aβ), intracellular neurofibrillary tangles, and disturbances in calcium homeostasis and degeneration of synapses and neurons [1]. Despite these observable changes in the morphology and physiology of the brain the mechanism of neurodegeneration in AD is not clearly understood. An inflammatory process in the central nervous system is believed to play an important role in the pathways leading to neuronal cell death. In addition to inflammatory pathway (extracellular pathway of neurodegeneration), the Aβ mediated mitochondrial dysfunction and induction of intrinsic cell death has been recently realized as an important contributor in AD related neurodegeneration. Mitochondria play central role in the bioenergetics of cell and mitochondrial permeability transition pore (MPTP) has a central role in the regulation of both necrotic and apoptotic modes of neuronal cell death. Mitochondrial permeability transition (MPT) is described as an abrupt increase of inner membrane permeability to solutes with molecular mass of <1500 Da, which leads to dissipation of the mitochondrial membrane potential and influx of cytosolic solutes, causing expansion of matrix and finally the release of cytochrome-c leading to caspase activation and cell death [2]. Structurally the MPTP is thought to involve VDAC (voltage dependent anion channel) in the outer membrane, the ANT (adenine nucleotide translocase) in the inner membrane and CypD (Cyclophilin D) in the mitochondrial matrix. MPTP spans through both the inner and outer mitochondrial membranes. VDAC is a porin known by its role in metabolite transport across mitochondria and acts as entry for molecules from cytosol to mitochondria. Administration of specific anti-VDAC antibodies prior to Aβ-peptide uptake has been observed to prevent neurotoxicity, which suggests the role of MPTP in regulation of Aβ toxicity [3]. The other two major proteins of MPTP, CypD and ANT, have regulatory functions. CypD, which is a peptidyl prolyl cis-trans isomerase has been reported to regulate the MPTP. Oxidative and other stress promote CypD translocation to the inner membrane and this translocation acts as a key factor to trigger the opening of calcium-sensitive non-specific channels and subsequent change in mitochondrial calcium level. CypD deficiency has been recently reported to attenuate Aβ- induced mitochondrial and neuronal perturbation, suggesting that its interaction with Aβ contributes to development of AD [5]. The other regulatory molecule ANT is located at the inner membrane and is known to switch its function from ADP/ATP transporter to a poreforming channel during apoptosis. ANT is also proposed as a target for oxidative radicals that may finally lead to induction of pore opening [4]. Advancements in the molecular, cellular and animal model studies have revealed that Aβ enters mitochondria and induces the generation of free radicals. Aβ is a small peptide derived from regulated proteolysis of amyloid precursor protein (APP) in ER/Golgi compartments as well as in lysosomes and on the cell surfaces. Two complex proteases β- and γ- secretase are found involved in the generation of these toxic fragment [6]. Aβ molecules at higher concentration can directly disrupt mitochondrial function and the interaction of Aβ with mitochondrial proteins (e.g. CypD) significantly enhances the accumulation and production of mitochondrial reactive oxygen species. Our present study offers an important advancement in the present understanding of MPTP opening and mitochondrial dysfunction in AD. In this study we have clearly demonstrated the predicted processing of APP as well as the interacting partners of Aβ in the cytoplasm, mitochondria and extracellular matrix. Our study strongly suggests that Aβ can interact with ANT (besides CypD) in the mitochondrial inner membrane, and this interaction is crucial for AD related pathophysiology.
Methodology
Data set and Sequence retrieval
Amyloid beta keyword was used as input in the STITCH (protein-protein interaction resource database, http://stitch.embl.de). STITCH is a sister project of proteinprotein interaction server STRING and is a resource to explore known and predicted interactions over 1.5 million proteins from 373 species. ’Homo sapiens‘ was selected as the species of interest. The default setting for 20 outputs was changed to 50 interacting partners. We selected amyloid-beta A4 protein precursor. Out of the 50 interacting proteins, 12 proteins (with >0.910 STITCH Score) related to the processing of APP with known role in AD development were selected for further study. We selected the interacting proteins based on their involvement in APP processing and proteolysis (Evin and Weidemann 2002 [6] and http://www.uniprot.org). We also confirmed the interactions of these proteins with the database of predicted human protein-protein interactions PIPs (http://www.compbio.dundee.ac.uk/www-pips). In addition to these proteins, two proteins of MPTP, ANT and CypD were also selected and used for in silico interaction study. The related protein sequences were retrieved from the Entrez protein database (available at http://www.ncbi.nlm.nih.gov) using the protein name as keyword. Accession number for each sequence used for analysis is given (Table 2 in supplementary).
Prediction of sub-cellular localization
All selected proteins were subjected to sub-cellular localization prediction using WolfPSORT server (http://wolfpsort.org) to know the relative abundance of these proteins in different sub-cellular compartments. The protein sequences retrieved from Entrez protein database were taken as input sequence. WolfPSORT predicts the sub-cellular localization sites of proteins based on both known sorting signal motifs and some corrective sequence features such as amino acid content with prediction accuracy of >80%. Further, the relative localization of different AD related proteins was correlated with proteinprotein interaction information available from STITCH.
Protein-Protein interaction studies
The structural details of interactions between Aβ (1IYT_A), ANT (NP_001142) and CypD (NP_005029) were elucidated by molecular docking experiments. 3D models were constructed by automated protein structure homology-modeling Swissmodel server model 8.05 (http://swissmodel.expasy.org/workspace). Protein-protein interaction simulation (docking) experiments were performed using RosettaDock server (http://rosettadock.graylab.jhu.edu). RosettaDock is a structure-prediction-based program based on SVM (support vector machine), which searches the rigid-body and side-chain conformational space of two interacting proteins to find a minimum free-energy complex structure. Further we also used Hex-5.1 software using solid surface docking simulations. In these docking calculations, each molecule is modeled using 3D expansions of real orthogonal spherical polar basis functions to encode both surface shape and electrostatic charge and potential distributions. All docking experiments were replicated 1000 times and best Etotal for the interaction was taken as key parameter to evaluate the significance of interaction. Etotal was represented in terms of KJ/mol.
Discussion
Prediction for presence and synthesis of Aβ in the mitochondria
First we performed sequence-based subcellular localization of some important proteins involved in APP processing, interaction and Aβ production. List of these proteins was obtained from STITCH database. WolfPSORT-based prediction for the sub-cellular localization indicated that neither amyloid precursors (APP) nor Aβ fragments are localized in the mitochondria (Table 1 in supplementary material). Interestingly, presence of secretase-β (beta-A and beta-B) involved in Aβ processing is predicted in mitochondria, indicating a possibility of Aβ processing inside the mitochondria. It was previously suggested that APP interacts with TOM and TIM proteins and gets a partial entry into the mitochondria via dual leader sequence [7]. Partially internalized APP may produce reduction of permeability of mitochondrial membranes due to engagement of TIM and TOM systems and it is suggested as a possible mechanism of mitochondrial dysfunction. It was also shown that amyloid portion of the APP resides outside of mitochondria, thus there is no internal generation of Aβ. Recently Hansson-Petersen et al. have shown that Aβ molecules are imported to mitochondria directly via TOM mechanism [8]. Presence of Aβ in the insoluble fraction suggests the integration of Aβ with lipid bilayer of inner membrane. It was suggested that its import is driven by hydrophobic interaction with receptors of TOM complex. As it has no classical signaling sequence it may interact with its hydrophobic domain but could not pass through inner membrane to mitochondrial matrix. Aβ1-42 has α-helical hydrophobic c-terminus sequence (residues 29-42) and is partially integrated in lipid layer. Integration of Aβ in the lipid bilayer forms ion channel like structures which are considered important in disrupting membrane potential. In vitro studies suggest that its integration into lipid layer is dependent on length and helix forming potential of hydrophobic sequence and is also regulated by metal ions, pH and constituents of lipid layer [9]. Due to this the possibility of incorporation of Aβ1-42 is greater than Aβ1-40. In many experiments the Aβ1-42 has been reported more toxic than other amyloid fragments [10]. Further, we also wanted to study the localization of Aβ interacting proteins into the mitochondria, and we found the significantly higher score for mitochondrial localization of insulin protease, TGF-β, CypD and ANT (Table 1 in supplementary material). Since the CypD and ANT are known to regulate the mitochondrial membrane potential, we further explored the interaction of CypD, ANT with Aβ.
Interaction between Aβ, CypD and ANT
We hypothesized that amyloid beta may interact with ANT, an inner membrane transporter protein and may disrupt its function. We used interaction simulation with docking program RosettaDock server. The RosettaDock server performs a local docking search for a set of conformations for the optimal fit between the two proteins. Plot of 1000 simulated complexes shows a hallmark of a successful run in which low-energy structures clustered around a single position (Figure 1). We also performed solid surface based docking simulation with software Hex 5.1 and found that Aβ forms a very stable complex with ANT (Etotal= -577.65 kJ/mol) (Figure 1). Hydrophobic helix of Aβ chain is interacting with the hydrophobic transmembrane region of ANT. Also Aβ is abundant in the inner membrane, the localization site of ANT, indicating that possibility of its interaction with ANT is high. Interestingly, in our Hex interaction simulation, the binding of ANT (Etotal= -577.65 kJ/mol) with Aβ is stronger than that of CypD (Etotal= -454.21 kJ/mol) suggesting that binding of Aβ with ANT may even have pathogenic consequences. Our computer simulation based study shows that Aβ interacts with both ANT and CypD. Recently it was reported that stress induced by Aβ induces ROS generation and translocation of CypD to inner membrane, which results in mitochondrial swelling and cell death [5]. Genetic removal of CypD significantly protected the cells in this study. Authors suggested that Aβ interacts with CypD although its role in ROS generation was not clear. We sought to analyze the interaction with Aβ to understand role of Aβ in translocation of CypD and the opening of MPTP. Interestingly, our predictions of subcellular localization as well as the results of Hansson- Peterson et al. clearly rule out the presence of Aβ in the soluble or matrix fraction [8]. Therefore, this interaction is most likely possible in the inner membrane only.
Figure 1.
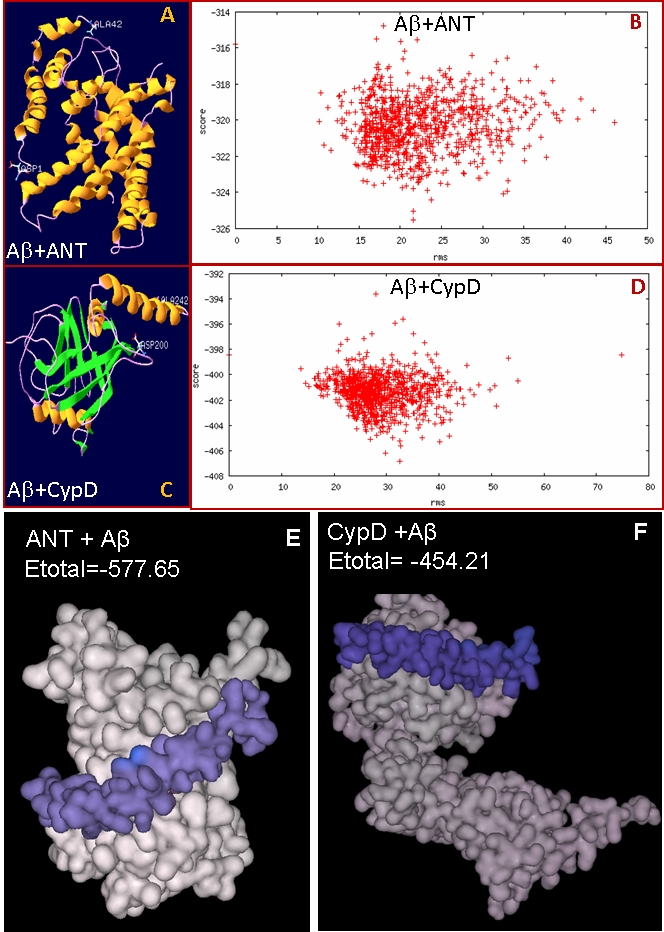
Models of ANT-Aβ (A) and CypD-Aβ (C) complexes predicted by RosettaDock server along with plots of scores (energy) versus distance from the starting structure of full set of 1000 structures created in docking run of Aβ with ANT (B) and CypD (D). In both the cases low energy structures are clustered around a single position as shown in dot plot. Most stable complex models of ANT- Aβ (E) and CypD-Aβ (F) predicted by Hex 5.1. Aβ is bound in hydrophobic region of ANT and low Etotal of ANT-Aβ complex showing its stability.
It is reported that under oxidative stress CypD is translocated to inner membrane and induces opening of MPTP [11], although the regulation of its translocation not clear. CypD and Aβ interaction is reportedly involved in the induction of oxidative stress and it is suggested that oxidative stress induced through CypD-Aβ may inhibit functioning of mitochondria and also induce cell death by other mechanisms such as Ca++ independent signaling pathways, oxidative stress induced kinase system and activation of transcription factors. High abundance of Aβ in the inner membrane and its interaction with CypD may facilitate its anchoring in inner membrane, and in consequence its interaction with ANT may result in disturbance of mitochondrial membrane potential, mitochondrial swelling and cell death. Current evidences show that regulation of MPTP is very complex. Studies reveal that CypD binds tightly to VDAC-ANT and it seems more likely that the VDAC-ANT-CypD complex is a permanent feature of healthy cells and several mechanisms have been postulated to explain the MPTP opening [2, 11]. These mechanisms include changes in protein-protein interactions such as Bcl-2 or Bax binding, modulation of expression levels of pro- or anti-apoptotic components of MPTP, and modifications of the lipid microenvironment in mitochondrial membranes. Recent reports show that ANT has regulatory function rather than only the formation of structural transmembrane pore [12]. Our results show that Aβ tightly binds with ANT and may alter the interactions with pro- or anti-apoptotic factors. Also its localization and possible interaction with other proteins in the inner membrane may affect the lipid microenvironment. Aβ interaction with mitochondrial proteins namely ABAD (amyloid binding alcohol dehydrogenase) [13] and CypD [5] were found to produce reactive oxygen species (ROS) which can also alter the fluidity and permeability of membrane and may enhance mitochondrial dysfunction and synaptic dysfunction [14].
ANT transports ADP/ATP through an exchange-diffusion mechanism with a one-to-one stoichiometry and maintains the adenine nucleotide pool at a constant level in the matrix. During oxidative phosphorylation, external ADP enters into matrix and in exchange ATP is released into the inter-membrane space. Nucleotides are transported in free form, thus the charge is not compensated and transport is driven by membrane potential at the cost of high energy consumption [15]. Interaction of Aβ with ANT may affect the physiological function of ANT and leads to dysfunction in energy metabolism prior to induction of cell death. Also ATP is directly involved in long term potentiation, a phenomenon involved in synaptic plasticity [16]. Our hypothesis is in line with the observation that mitochondrial dysfunction and in consequence synaptic dysfunction occurs much earlier than the neuronal loss [1, 17].
Conclusion
In this study we predicted the interaction between Aβ, CypD and ANT. We found that Aβ may bind tightly with ANT which suggests that the interaction may alter regulation of MPTP and is involved in mitochondrial dysfunction. We also hypothesize that Aβ would interact with CypD and ANT in the inner mitochondrial membrane. Our prediction shows that the Etotal for ANT-Aβ interaction is significantly less than CypD-Aβ interaction, which shows that binding of Aβ with ANT is likely stronger than that with CypD, possibly affecting the normal physiological function of ANT. Hence we conclude that interaction of Aβ with ANT alone may induce mitochondrial dysfunction independent of MPTP. Dysregulation in ADP/ATP transport directly affects the physiological processes of cells and may further result in synaptic dysfunction.
Supplementary material
Acknowledgments
PS acknowledges Senior Research Fellowship provided by Council of Scientific and Industrial Research, New Delhi, India.
Footnotes
Citation:Singh et al, Bioinformation 3(10): 440-445 (2009)
References
- 1.Selkoe DJ. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 2.Kumarswamy R, Chandna S. Mitochondrion. 2009;9:1–8. doi: 10.1016/j.mito.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Marin R, et al. Mol Membr Biol. 2007;24:148–160. doi: 10.1080/09687860601055559. [DOI] [PubMed] [Google Scholar]
- 4.Brookes PS, et al. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 5.Du H, et al. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evin G, Weidemann A. Peptides. 2002;23:1285–1297. doi: 10.1016/s0196-9781(02)00063-3. [DOI] [PubMed] [Google Scholar]
- 7.Lin MT, Beal MF. Nat Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 8.Hansson Petersen CA, et al. Proc Natl Acad Sci USA. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curtain CC, et al. J Biol Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 10.Findeis MA. Pharmacol Ther. 2007;116:266–286. doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Kroemer G, et al. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 12.Leung AW, Halestrap AP. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Lustbader JW, et al. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 14.Keating DJ. J Neurochem. 2008;104:298–305. doi: 10.1111/j.1471-4159.2007.04997.x. [DOI] [PubMed] [Google Scholar]
- 15.Duszynski J, et al. Biochim Biophys Acta. 1981;673:2170–2223. [Google Scholar]
- 16.Lim W, Issac JT. Neuropharmacology. 2005;48:949–955. doi: 10.1016/j.neuropharm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Rowan MJ, et al. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.