

Abstract
Application of volatile anesthetics during the onset of reperfusion reduced ischemia-induced cardiac and brain injury (anesthetic postconditioning). This study was designed to evaluate whether volatile anesthetics induced a postconditioning effect in endothelial cells. Bovine pulmonary arterial endothelial cell (BPAEC) cultures were exposed to oxygen-glucose deprivation, a condition to simulate ischemia in vitro, for 3 h. The volatile anesthetics isoflurane and desflurane were applied during the early phase of simulated reperfusion. Cell injury was quantified by lactate dehydrogenase (LDH) release and flow cytometrical measurement after annexin V and propidium iodide staining. Oxygen-glucose deprivation and the subsequent simulated reperfusion increased LDH release and annexin V-positive staining cells, a characteristic of cell apoptosis. Posttreatment with isoflurane, but not desflurane, reduced this cell injury. This protection was apparent even when 2% isoflurane was applied at 60 min after the onset of reperfusion. The isoflurane postconditioning effect was abolished by glybenclamide, a general ATP sensitive K+ (KATP) channel blocker, 5-hydroxydecanoate, a mitochondrial KATP channel blocker, and chelerythrine, a protein kinase C inhibitor. Diazoxide, a mitochondrial KATP channel activator, applied at the onset of reperfusion also decreased oxygen-glucose deprivation-induced endothelial cell injury. This diazoxide-induced protection was abolished by chelerythrine and 5-hydroxydecanoate. We conclude that isoflurane induced a postconditioning effect in BPAEC. The effective time window of isoflurane postconditioning was from 0 to 60 min after the onset of reperfusion. This isoflurane postconditioning effect may be mediated by mitochondrial KATP channels and PKC. PKC may be downstream of mitochondrial KATP channels for this isoflurane effect.
Keywords: endothelial cells, postconditioning, volatile anesthetics
1. Introduction
Endothelial cells are an important component for all organs and systems. They represent a unique barrier to prevent free access of chemicals and cells in the blood to the vascular smooth muscle cells and parenchymal cells of organs. Endothelial cells also release various molecules, such as nitric oxide, to affect the functions of other cells (Nathan and Xie, 1994). Thus, it is critical to maintain structural and functional integrity of endothelial cells. However, various insults can threaten this integrity. For example, ischemia can injure endothelial cells.
One approach that has drawn significant attention recently to reduce ischemia-induced injury in various organs and cells is preconditioning. Preconditioning is a phenomenon in which a prior application of preconditioning stimulus induces a robust protection against the deleterious effects of subsequent insults (Murry et al., 1986). However, the use of preconditioning to protect organs and cells may be limited to a few clinical situations where ischemia or other detrimental insults can be predicted to occur. Recently, a phenomenon called postconditioning has been described (Na et al., 1996; Zhao et al., 2003). Postconditioning means to apply stimuli after ischemia or other detrimental insults to provide protection. Postconditioning with volatile anesthetics has been shown to reduce ischemia-induced injury in the brain and heart (Feng et al., 2005; Lee et al., 2008). This multiorgan protection may involve protection of endothelial cells. Thus, we hypothesize that volatile anesthetic postconditioning reduces ischemia-induced endothelial cell injury. We tested this hypothesis by using bovine pulmonary arterial endothelial cells (BPAEC) and subjected these cells to oxygen-glucose deprivation, a condition to simulate ischemia in vitro.
2. Materials and Methods
2.1. Cell culture
The bovine pulmonary arterial endothelial cell (BPAEC) was isolated and characterized as we described before (Zuo and Johns, 1997) and had been maintained by Dr. Lisa A. Palmer's laboratory in the Department of Pediatrics and Anesthesiology, University of Virginia. The cells were cultured in a T75 flask containing 12 ml of culture media composed of Dulbecco's Modified Eagle's Medium (DMEM) (containing 1,000 mg/l D-glucose, L-glutamine and pyridoxine HCl), 110 mg/l sodium pyruvate, 10% heat inactivated fetal bovine serum, 90 μg/ml thymidine, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were kept in a humidified atmosphere of 95% air-5% CO2 at 37°C. The medium was changed three times per week. When the cells were 70-80% confluent, they were exposed to 0.05% trypsin-EDTA solution and sub-cultured in a new flask. Experiments were performed on BPAECs between passage 8 and 20.
2.2. Exposure to oxygen-glucose deprivation and volatile anesthetics
The cells were placed into 6 well plates at a density of 5 × 103 cells/ml (2 ml/well) and cultured overnight (about 17 h). Glucose-free buffer contained 154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 2.3 mM CaCl2 and 5.0 mM HEPES. Glucose at 1.0 g/l was added to make glucose containing buffer. Oxygen-glucose deprivation buffer was prepared by bubbling the glucose-free buffer with 100% N2 for 30 min. The cells were divided into control, oxygen-glucose deprivation and oxygen-glucose deprivation plus isoflurane posttreatment groups. Cells in control group were washed with and incubated in glucose containing buffer. Oxygen-glucose deprivation was applied by washing cells with oxygen-glucose deprivation buffer three times and then placing 2 ml/well of this oxygen-glucose deprivation buffer. These plates were placed in an air tight chamber gassed with 100% N2 for 10 min. The oxygen content in the outlet of the chamber was monitored with a DatexTM infrared analyzer (Capnomac, Helsinki, Finland) and was below 2% at ∼3 min after the onset of gassing. This low oxygen content was maintained throughout the oxygen-glucose deprivation period. After closure of the inlet and outlet of the chamber, the chamber and the control cells were placed in a humidified atmosphere of 95% air-5% CO2 at 37°C for 3 h. After confirming that the oxygen content in the chamber was < 2%, the incubation solutions were replaced with new glucose containing solutions in all 3 groups. In a separate preliminary experiment, the O2 partial pressure in the incubation solutions during the oxygen-glucose deprivation exposure was measured to be < 10 mmHg. In volatile anesthetic posttreatment group, volatile anesthetics were delivered by air through an agent specific vaporizer. The glucose containing buffer was pregassed with these anesthetic containing gases for 10 min and 2 ml/well of this buffer was added to the cells. The cells were placed back into an air-tight chamber and this chamber was gassed with the anesthetic containing air for 10 min. The anesthetic concentrations in the outlet were also monitored by the DatexTM infrared analyzer and the target concentrations were reached in < 2 min. The chamber and cells without anesthetic treatment were placed in an incubator at 37°C for preset times. Cells in the chamber were then removed and placed in the incubator for the rest of the time to complete a total of 3-h duration after the oxygen-glucose deprivation (Fig. 1). The incubation buffer and cells were used for assay of lactate dehydrogenase (LDH) activity.
Fig. 1. Experimental protocol.

Bovine pulmonary arterial endothelial cells were exposed to or not exposed to a 3-h oxygen-glucose deprivation (OGD) and posttreated with or without 2% isoflurane (Iso-post) for 1 h immediately after the oxygen-glucose deprivation in the presence or absence of various reagents (R), such as glybenclamide, 5-hydroxydecanoate or chelerythrine. In some cells, 100 μM diazoxide (Di) was applied for 1 h immediately after oxygen-glucose deprivation in the presence or absence of reagents (R), such as 5-hydroxydecanoate or chelerythrine. Lactate dehydrogenase released into incubation buffer was determined 3 h after oxygen-glucose deprivation.
Application of various isoflurane concentrations (1, 2 and 3%) or desflurane concentrations (6 and 12%) for 60 min or different isoflurane exposure durations (2% isoflurane for 30, 60 and 90 min) immediately after the end of oxygen-glucose deprivation or application of 2% isoflurane for 60 min started at various times (10, 30, 60, and 90 min) after the oxygen-glucose deprivation was used in this study.
2.3. Application of chemicals
ATP sensitive K+ (KATP) channel blockers (0.3 μM glybenclamide, 500 μM 5-hydroxydecanoate) or protein kinase C (PKC) inhibitor (2 μM chelerythrine chloride) were added to the cells treated with or without 2% isoflurane for 1 h after oxygen-glucose deprivation to determine whether these inhibitors modulated the effects of isoflurane. After the incubation, the solutions were replaced with fresh glucose containing buffers without these reagents. The solutions in the control, oxygen-glucose deprivation only or oxygen-glucose deprivation plus isoflurane postconditioning groups in the same set experiments were also changed in the same way. In another set of experiments, a mitochondrial KATP channel activator (100 μM diazoxide) was applied for 1 h immediately after oxygen-glucose deprivation and chelerythrine chloride or 5-hydroxydecanoate was added in some of these cells for 1 h (Fig. 1).
2.4. Lactate dehydrogenase assay
LDH activity was determined using LDH cytotoxicity detection kit (Clontech Laboratory, La Jolla, CA). In which, NAD+ is reduced to NADH/H+ by the LDH-catalyzed conversion of lactate to pyruvate. In turn, yellow tetrazolium dye is reduced to a red formazan dye by the H/H+ from NADH/H+. Briefly, the solution from the post-oxygen-glucose deprivation 3-h incubation (the solution was from the last 2 h of simulated reperfusion in the set of experiments involving using PKC and KATP channel regulators) was centrifuged at 13,000 rpm for 10 min and the cell-free supernatant was transferred to 96 well plates. The 100 μl supernatant was incubated with the same amount of reaction mixture. LDH activity was determined by a colorimetric assay. The absorbance of samples was measured at 492 nm with the reference wavelength of 655 nm in a spectrophotometry (Bio-Rad Laboratories, Hercules, CA). Background absorbance from the cell-free buffer solution was subtracted from all absorbance measurements. After removal of the buffer from 6-well plates, 1% triton X-100 lysing solution was applied to the remaining cells. The percentage of LDH released to incubation buffer in total LDH was calculated: spontaneously released LDH in the buffer/(spontaneously released LDH in the buffer + intracellular LDH released by triton X-100). The LDH values from various treatment groups were then normalized by the values from control cells.
2.5. Annexin V and propidium iodide staining and flow cytometry
Almost 100% confluent cells in a T75 flask were divided into control, oxygen-glucose deprivation and oxygen-glucose deprivation plus isoflurane posttreatment groups. After treatments, the cells were stained with annexin V-fluorescein isothiocyanate conjugate and propidium iodide (PI) using an Alexa Fluor® 488 annexin V-PI staining kit (Molecular Probes, Eugene, OR) according to the manufacturer's protocol. Annexin V-fluorescein isothiocyanate conjugate labels apoptotic cells by binding phosphatidyl serine exposed on the outer leaflet of the plasma membrane. PI is impermeable to live cells and early phase of apoptotic cells, but stains dead cells with red fluorescence by binding tightly to the nucleic acids. After staining, the cells were analyzed with flow cytometry (FACSCalibur system; Becton, Dickinson and Company Biosciences, Franklin Lakes, NJ).
2.6. Materials
Isoflurane was purchased from Abbott Laboratories (North Chicago, IL). Chelerythrine chloride was obtained from Biomol (Plymouth Meeting, PA) and other chemicals were obtained from Sigma-Aldrich (ST Louis, MO), unless specified in the text.
2.7. Statistical Analyses
Data are expressed as mean ± S.D. with an N of > 8 for each experimental condition. One-way analysis of variance followed by the Tukey test for post hoc analysis was performed. A P value < 0.05 was considered statistically significant.
3. Results
3.1. Isoflurane postconditioning reduced oxygen-glucose deprivation-induced cell injury
LDH released to the incubation buffer in cells subjected to the 3-h oxygen-glucose deprivation was significantly higher than that in control cells (Fig. 2A), suggesting that oxygen-glucose deprivation and the subsequent re-oxygenation caused cell injury. Exposure of the cells to 2% isoflurane for 30, 60 and 90 min or to 1%, 2% or 3% isoflurane for 60 min immediately after the oxygen-glucose deprivation reduced the LDH release (Figs. 2 A and 2B). This reduced release occurred even when 2% isoflurane was applied at 10 min, 30 min and 60 min after the end of the oxygen-glucose deprivation. However, delaying the application of 2% isoflurane to 90 min after the oxygen-glucose deprivation did not attenuate the oxygen-glucose deprivation-induced LDH release (Fig. 2C). In contrast, the exposure of cells to 12% desflurane for 30, 60 or 90 min or to 6% or 12% desflurane for 60 min did not reduced oxygen-glucose deprivation-induced LDH release (Figs. 3A and 3B), suggesting that desflurane posttreatment may not protect these endothelial cells.
Fig. 2. Isoflurane postconditioning effect.
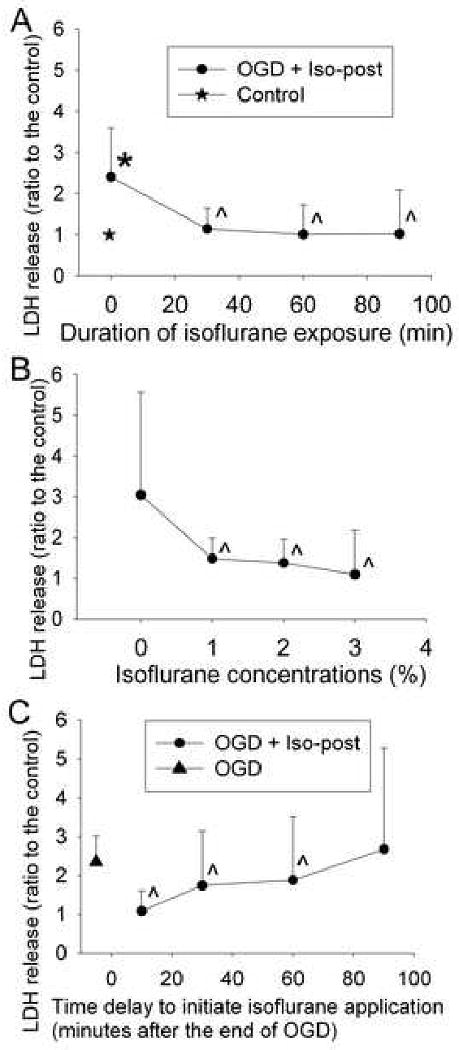
Bovine pulmonary arterial endothelial cells were exposed to or not exposed to a 3-h oxygen-glucose deprivation (OGD) and posttreated with or without 2% isoflurane (Iso-post) for 30, 60 or 90 min immediately after the oxygen-glucose deprivation (panel A), with or without various concentrations of isoflurane for 60 min immediately after the oxygen-glucose deprivation (panel B), or with or without 2% isoflurane for 60 min at 10, 30, 60 or 90 min after the oxygen-glucose deprivation (panel C). Results are means ± S.D. (n = 23 to 45 for panel A, 27 to 55 for panel B and 27 for panel C). * P < 0.05 compared with control. ˆ P < 0.05 compared with oxygen-glucose deprivation only.
Fig. 3. Desflurane post-treatment effect.
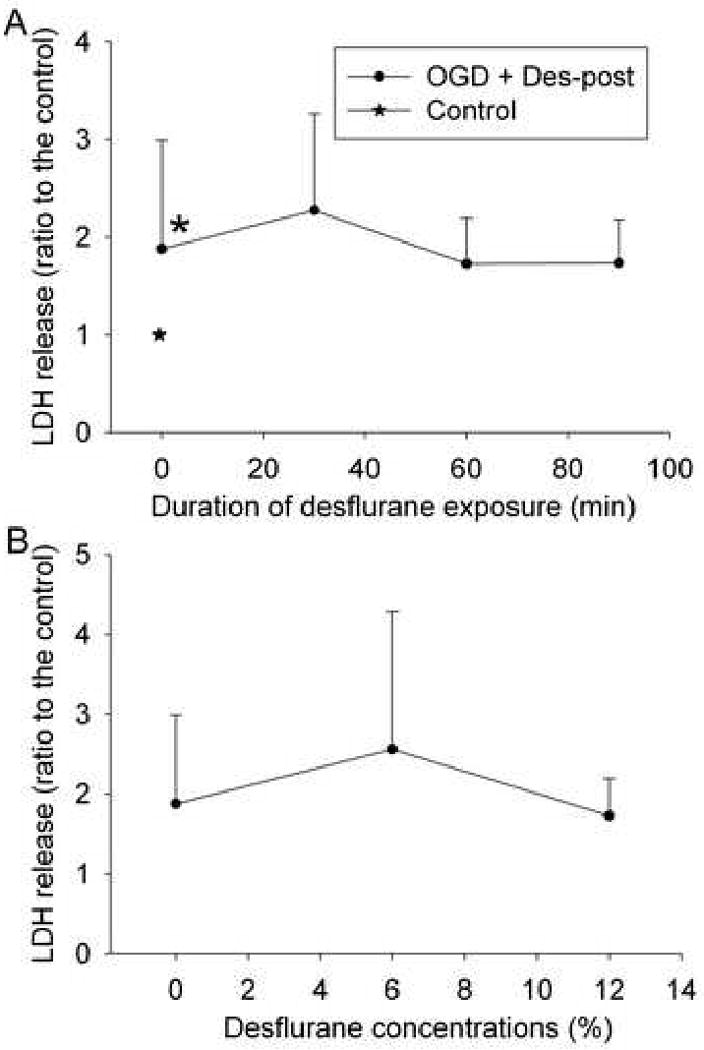
Bovine pulmonary arterial endothelial cells were exposed to or not exposed to a 3-h oxygen-glucose deprivation (OGD) and posttreated with or without 12% desflurane (Des-post) for 30, 60 or 90 min immediately after the oxygen-glucose deprivation (panel A) or with or without various concentrations of desflurane for 60 min immediately after the oxygen-glucose deprivation (panel B). Results are means ± S.D. (n = 28 to 36 for panel A and 35 to 36 for panel B). * P < 0.05 compared with control.
3.2. Isoflurane postconditioning reduced oxygen-glucose deprivation-induced cell apoptosis
There were 3.5 ± 0.8% and 11.6 ± 1.6% cells that were positively stained with annexin V only and PI, respectively, under control condition. Oxygen-glucose deprivation significantly increased the cells positively stained with annexin V only and did not change the PI-positive staining cells (Fig. 4), suggesting that our current oxygen-glucose deprivation protocol mainly induced endothelial cell apoptosis. Application of 2% isoflurane for 1 h immediately after the 3-h oxygen-glucose deprivation abolished the oxygen-glucose deprivation-induced increase of annexin V-positive staining cells. Similarly, oxygen-glucose deprivation increased the total dying cells that were positively stained with annexin V and/or PI. This increase also was attenuated by 2% isoflurane posttreatment (Fig. 4).
Fig. 4. Effects of isoflurane postconditioning on cell necrosis and apoptosis induced by oxygen-glucose deprivation.
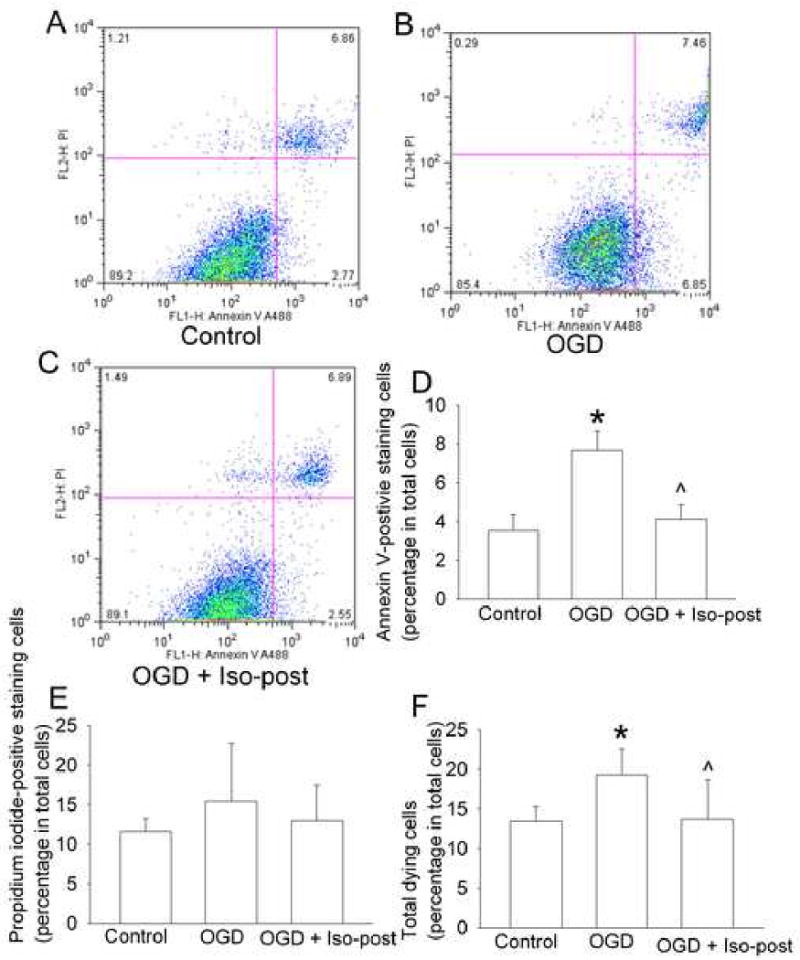
Bovine pulmonary arterial endothelial cells were exposed to or not exposed to a 3-h oxygen-glucose deprivation (OGD) and posttreated with or without 2% isoflurane (Iso-post) for 1 h immediately after the oxygen-glucose deprivation. These cells were stained with annexin V and propidium iodide (PI) 3 h after the oxygen-glucose deprivation and then were analyzed by flow cytometry. A representative of sorted cells by flow cytometry for each experimental condition is presented in panel A, B and C. These are two-parameter dot plots with the x-axis showing the intensity of annexin V staining and y-axis showing the intensity of PI staining. The location of a particular cell in the plot is determined by its intensities from those two stains. The pooled results from 8 flow cytometrical studies are presented in panel D, E and F. Total dying cells are cells stained positively with annexin V and/or PI. Results are means ± S.D. * P < 0.05 compared with control. ˆ P < 0.05 compared with oxygen-glucose deprivation only.
3.3. KATP channels and PKC may be involved in isoflurane postconditioning
While 0.3 μM glybenclamide, a general KATP channel blocker (Sato et al., 2000), or 500 μM 5-hydroxydecanoate, a mitochondrial KATP channel blocker (Lim et al., 2004; Sato et al., 2000), applied for 1 h immediately after oxygen-glucose deprivation did not affect the LDH release under oxygen-glucose deprivation only condition, the application of these KATP channel blockers abolished the protective effect of isoflurane posttreatment on the LDH release (Fig. 5A). Diazoxide, a mitochondrial KATP channel activator (Roseborough et al., 2006), applied for 1 h immediately after the oxygen-glucose deprivation reduced the oxygen-glucose deprivation-induced LDH release. This reduced release was blocked by 5-hydroxydecanoate and chelerythrine, a PKC inhibitor (Herbert et al., 1990). Chelerythrine also blocked the isoflurane posttreatment-induced protection. However, chelerythrine did not affect the oxygen-glucose deprivation-induced LDH release under oxygen-glucose deprivation only condition (Fig. 5B).
Fig. 5. Involvement of mitochondrial KATP channels and protein kinase C (PKC) in isoflurane postconditioning effect.
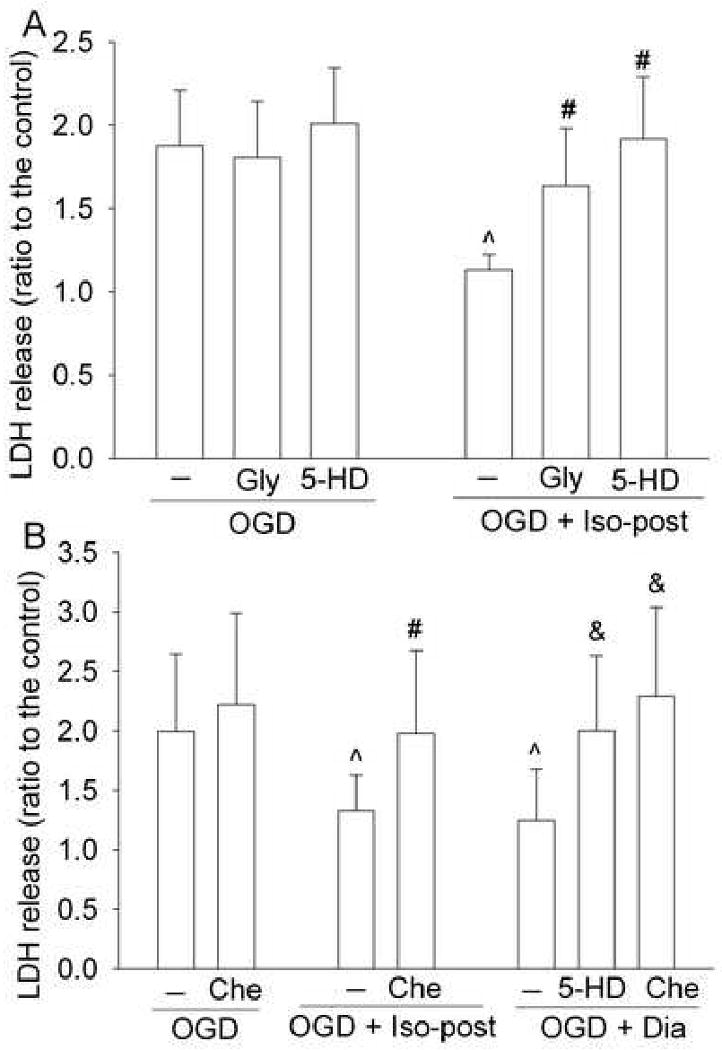
Bovine pulmonary arterial endothelial cells were injured by a 3-h oxygen-glucose deprivation (OGD) and posttreated with or without 2% isoflurane (Iso-post) for 1 h immediately after the oxygen-glucose deprivation in the presence or absence of 0.3 μM glybenclamide (Gly), 500 μM 5-hydroxydecanoate (5-HD) or 2 μM chelerythrine (Che) or posttreated with 100 μM diazoxide (Dia) for 1 h in the presence or absence of 500 μM 5-hydroxydecanoate or 2 μM chelerythrine. Results are mean ± S.D. (n = 12 for panel A and 10 for panel B). ˆ P < 0.05 compared with oxygen-glucose deprivation only. # P < 0.05 compared with oxygen-glucose deprivation + Iso-post. & P < 0.05 compared with oxygen-glucose deprivation + Dia.
4. Discussion
Postconditioning by short episodes of interruption of reperfusion to provide protection was named ischemic postconditioning (Na et al., 1996; Zhao et al., 2003). Ischemic postconditioning-induced protection has been shown in the heart and brain (Zhao et al., 2006; Zhao et al., 2003). Subsequently, it has been found that, in addition to short episodes of ischemia, drugs such as volatile anesthetics can also be used to postcondition organs and tissues (Feng et al., 2005; Lee et al., 2008). The drug-induced postconditioning may be very useful because well controlled short episodes of ischemia at the onset of reperfusion may be very difficult to achieve, especially in the case of brain. We now expand the drug-induced postconditioning to endothelial cells, a ubiquitous tissue existing in the vasculature of all organs. Our results showed that postconditioning with clinically relevant concentrations of isoflurane for 30 min or longer reduced oxygen-glucose deprivation-induced injury. This postconditioning-induced protection can be observed even when 2% isoflurane was applied at 60 min after the onset of simulated reperfusion. Consistent with our findings, a previous study showed that application of isoflurane, but not propofol, at 2 h after intravenous injection of lipopolysaccharide reduced pulmonary permeability in rats (Zhang et al., 2005).
Interestingly, posttreatment with desflurane in our study did not affect oxygen-glucose deprivation-induced injury. These results suggest that anesthetic postconditioning-induced endothelial cell protection is an agent-specific effect. Consistent with the idea that preconditioning- and postconditioning-induced protection is an agent-specific phenomenon, desflurane was less potent than isoflurane to provide protection against ischemia-reperfusion in rat kidney when the anesthetics were used during the ischemia and reperfusion (Lee et al., 2004). Also, we showed that desflurane induced an acute phase of preconditioning-induced neuroprotection (Wang et al., 2007). However, our results indicated that isoflurane (Zheng and Zuo, 2004), but not desflurane (unpublished preliminary results), induced a delayed phase of preconditioning-induced neuroprotection in rats. Of note, preconditioning and postconditioning with clinically relevant concentrations of desflurane have been shown to protect myocardium under in vitro conditions (Lemoine et al., 2008; Toller et al., 2000). The reason for these different findings with desflurane is not known. An obvious difference among these studies is that different cells/tissues are studied.
Our results showed that oxygen-glucose deprivation and simulated reperfusion conditions increased the annexin V-positive staining cells, suggesting that our experimental conditions induce endothelial cell apoptosis. Oxygen-glucose deprivation and simulated reperfusion have been shown to induce endothelial cell apoptosis (Zhang et al., 2007). Our results also showed that isoflurane postconditioning reduced annexin V-positive staining cells, suggesting that isoflurane postconditioning reduced endothelial cell apoptosis under the current experimental conditions.
The isoflurane postconditioning-induced endothelial cell protection was abolished by glybenclamide, a general KATP channel blocker (Sato et al., 2000), and 5-hydroxydecanoate, a mitochondrial KATP channel blocker (Lim et al., 2004; Sato et al., 2000). These results suggest that mitochondrial KATP channels are involved in this isoflurane postconditioning-induced protection. To support this idea, diazoxide, a mitochondrial KATP channel activator (Roseborough et al., 2006), used at the onset of the simulated reperfusion, also reduced the endothelial cell injury. Consistent with our findings, the involvement of mitochondrial KATP channels in volatile anesthetic postconditioning-induced protection in the heart and brain has been reported (Lee et al., 2008; Obal et al., 2005).
KATP channels, such as mitochondrial KATP channels, have been considered as a putative end-effector in organ protection induced by ischemic or anesthetic preconditioning (Zaugg et al., 2003). Under physiological conditions, KATP channels are usually not active. However, conditions, such as ischemia, that induce a low cellular ATP content activate KATP channels (Grover and Garlid, 2000). The activation of mitochondrial KATP channels reduces Ca++ influx into mitochondria and mitochondrial swelling under ischemia. The activation of these channels may also restore mitochondrial membrane potential and ATP production [reviewed in (Grover and Garlid, 2000; Zaugg et al., 2003)]. In addition, activation of mitochondrial KATP channels can produce signaling molecules, such as free radicals that can then activate PKC (Gopalakrishna and Anderson, 1989; Jabr and Cole, 1993; McPherson and Yao, 2001). Thus, mitochondrial KATP channels are also signaling molecules. In this study, we showed that the protection of diazoxide was abolished by 5-hydroxydecanoate, indicating that the effect of diazoxide is mediated by mitochondrial KATP channels. However, the diazoxide effect was also abolished by chelerythrine, a PKC inhibitor (Herbert et al., 1990). These results suggest that mitochondrial KATP channels may be a molecule upstream of PKC to induce endothelial cell protection. These results, together with the results that chelerythrine, glybenclamide and 5-hydroxydecanoate inhibited the isoflurane postconditioning-induced protection, indicate PKC may be a molecule downstream of mitochondrial KATP channels to mediate the isoflurane postconditioning-induced endothelial cell protection.
One of the mechanisms identified for ischemic postconditioning-induced cardioprotection is that ischemic postconditioning reduces the rapid restoration of pH during the early phase of reperfusion so that the reperfusion injury salvage kinases can have time to be activated. The activated reperfusion injury salvage kinases can then reduce/prevent the opening of mitochondrial permeability transition pores and, therefore, reduce reperfusion injury (Andreadou et al., 2008; Cohen et al., 2008). Future studies are needed to determine whether this mechanism identified for ischemic postconditioning plays a role in the anesthetic postconditioning and whether there is an interaction between this mechanism and the mechanism that we identified here (PKC and mitochondrial KATP channels) to induce the protection.
Our findings may have a broad implication. Postconditioning, compared with preconditioning, may have a broad application because of the elimination of the needed predication of the detrimental insults, such as ischemia, to occur for its application. Endothelial cells are ubiquitous in all organs and tissues and play a critical role in maintaining vasculature tone and normal coagulation status. In addition, endothelial cells are a physical barrier to prevent free access of chemicals and cells in the blood to the vascular smooth muscle cells and parenchymal cells of organs. Thus, maintaining the functional and structural integrity of endothelial cells is critical under physiological and pathophysiological conditions. Therefore, the postconditioning-induced endothelial cell protection as shown here may attenuate the pathophysiological process leading to parenchymal cell injury and organ dysfunction after ischemia or other detrimental insults and may contribute to the volatile anesthetic postconditioning-induced organ protection observed in the previous studies (Feng et al., 2005; Lee et al., 2008; Obal et al., 2005).
In conclusion, we have shown that isoflurane postconditioning reduced oxygen-glucose deprivation-induced endothelial cell apoptosis. This protection was apparent even when isoflurane was applied at 60 min after the onset of simulated reperfusion. Mitochondrial KATP channels and PKC may mediate this isoflurane postconditioning effect and PKC may be downstream of mitochondrial KATP channels for this isoflurane effect.
Acknowledgments
The authors thank Lisa A. Palmer, Ph.D., Department of Pediatrics and Anesthesiology, University of Virginia, for providing us with the BPAEC.
Grants: This study was supported by grants (GM065211 and NS045983 to Z Zuo) from the National Institute of Health, Bethesda, Maryland, by a grant from the International Anesthesia Research Society (Frontiers in Anesthesia Research Award to Z Zuo), Cleveland, Ohio, and by a Grant-in-Aid from the American Heart Association Mid-Atlantic Affiliate (0755450U to Z Zuo), Baltimore, Maryland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreadou I, Iliodromitis EK, Koufaki M, Kremastinos DT. Pharmacological pre- and post- conditioning agents: reperfusion-injury of the heart revisited. Mini Rev Med Chem. 2008;8:952–959. doi: 10.2174/138955708785132819. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol. 2008;103:464–471. doi: 10.1007/s00395-008-0737-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology. 2005;103:987–995. doi: 10.1097/00000542-200511000-00013. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys Res Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Jabr RI, Cole WC. Alterations in electrical activity and membrane currents induced by intracellular oxygen-derived free radical stress in guinea pig ventricular myocytes. Circ Res. 1993;72:1229–1244. doi: 10.1161/01.res.72.6.1229. [DOI] [PubMed] [Google Scholar]
- Lee HT, Ota-Setlik A, Fu Y, Nasr SH, Emala CW. Differential protective effects of volatile anesthetics against renal ischemia-reperfusion injury in vivo. Anesthesiology. 2004;101:1313–1324. doi: 10.1097/00000542-200412000-00011. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–1062. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine S, Beauchef G, Zhu L, Renard E, Lepage O, Massetti M, Khayat A, Galera P, Gerard JL, Hanouz JL. Signaling pathways involved in desflurane-induced postconditioning in human atrial myocardium in vitro. Anesthesiology. 2008;109:1036–1044. doi: 10.1097/ALN.0b013e31818d6b09. [DOI] [PubMed] [Google Scholar]
- Lim YJ, Zheng S, Zuo Z. Morphine preconditions purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100:562–568. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- McPherson BC, Yao Z. Morphine Mimics Preconditioning via Free Radical Signals and Mitochondrial K(ATP) Channels in Myocytes. Circulation. 2001;103:290–295. doi: 10.1161/01.cir.103.2.290. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Na HS, Kim YI, Yoon YW, Han HC, Nahm SH, Hong SK. Ventricular premature beat-driven intermittent restoration of coronary blood flow reduces the incidence of reperfusion-induced ventricular fibrillation in a cat model of regional ischemia. Am Heart J. 1996;132:78–83. doi: 10.1016/s0002-8703(96)90393-2. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Obal D, Dettwiler S, Favoccia C, Scharbatke H, Preckel B, Schlack W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth Analg. 2005;101:1252–1260. doi: 10.1213/01.ANE.0000181336.96511.32. [DOI] [PubMed] [Google Scholar]
- Roseborough G, Gao D, Chen L, Trush MA, Zhou S, Williams GM, Wei C. The mitochondrial K-ATP channel opener, diazoxide, prevents ischemia-reperfusion injury in the rabbit spinal cord. Am J Pathol. 2006;168:1443–1451. doi: 10.2353/ajpath.2006.050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O'Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- Toller WG, Gross ER, Kersten JR, Pagel PS, Gross GJ, Warltier DC. Sarcolemmal and mitochondrial adenosine triphosphate- dependent potassium channels: mechanism of desflurane-induced cardioprotection. Anesthesiology. 2000;92:1731–1739. doi: 10.1097/00000542-200006000-00033. [DOI] [PubMed] [Google Scholar]
- Wang C, Lee J, Jung H, Zuo Z. Pretreatment with volatile anesthetics, but not with the nonimmobilizer 1,2-dichlorohexafluorocyclobutane, reduced cell injury in rat cerebellar slices after an in vitro simulated ischemia. Brain Res. 2007;1152:201–208. doi: 10.1016/j.brainres.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551–565. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- Zhang H, Xue ZG, Jiang H. The effect of posttreatment with isoflurane versus propofol on pulmonary alveolar capillary barrier in endotoxemic rats. Zhonghua Yi Xue Za Zhi. 2005;85:1708–1713. [PubMed] [Google Scholar]
- Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol. 2007;292:H2573–2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of p38 mitogen-activated protein kinase. Mol Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Johns RA. Inhalational anesthetics up-regulate constitutive and lipopolysaccharide-induced inducible nitric oxide synthase expression and activity. Mol Pharmacol. 1997;52:606–612. doi: 10.1124/mol.52.4.606. [DOI] [PubMed] [Google Scholar]