

Abstract
Store-operated Ca2+ entry through calcium release–activated calcium channels is the chief mechanism for increasing intracellular Ca2+ in immune cells. Here we show that mouse T cells and fibroblasts lacking the calcium sensor STIM1 had severely impaired store-operated Ca2+ influx, whereas deficiency in the calcium sensor STIM2 had a smaller effect. However, T cells lacking either STIM1 or STIM2 had much less cytokine production and nuclear translocation of the transcription factor NFAT. T cell–specific ablation of both STIM1 and STIM2 resulted in a notable lymphoproliferative phenotype and a selective decrease in regulatory T cell numbers. We conclude that both STIM1 and STIM2 promote store-operated Ca2+ entry into T cells and fibroblasts and that STIM proteins are required for the development and function of regulatory T cells.
Calcium is a universal second messenger1. In T cells, mast cells and other cells of the immune system, Ca2+ entry occurs mainly through the specialized store-operated Ca2+ entry channels known as calcium release–activated Ca2+ (CRAC) channels2–5. CRAC channels are low-conductance Ca2+ channels with an unusually high selectivity for Ca2+ and a characteristic inwardly rectifying current-voltage relationship6,7. CRAC channels open in response to signaling cascades initiated by immunoreceptors such as T cellantigen receptors (TCRs) and mast cell Fc receptors. Stimulation of these immunoreceptors induces the recruitment and activation of protein tyrosine kinases and the formation of large adaptor protein complexes and ultimately results in tyrosine phosphorylation and activation of phospholipase C-γ (PLC-γ). In T cells and mast cells, activated PLC-γ1 generates the second messenger inositol-1,4,5-trisphosphate, which by binding to receptors in the endoplasmic reticulum membrane causes the release of endoplasmic reticulum Ca2+ stores. In turn, the depletion of endoplasmic reticulum Ca2+ stores induces the opening of CRAC channels, which permit sustained influx of Ca2+ into the cell. Increases in the intracellular Ca2+ concentration promote rapid responses such as mast cell degranulation, as well as long-term responses that involve new gene transcription5,8.
Two chief participants in the pathway connecting the depletion of endoplasmic reticulum stores to the opening of CRAC channels have been identified. The endoplasmic reticulum Ca2+ sensor STIM1 (stromal cell—interaction molecule) has been identified by two limited RNA-mediated interference screens in drosophila and HeLa cells9,10. The drosophila protein Orai (encoded by olf186-F), identified as an essential regulator of store-operated Ca2+ influx by a series of genome-wide drosophila RNA-mediated interference screens11–13, and its three human homologs, ORAI1—ORAI3 (also called CRACM1—CRACM3), are small proteins with four transmembrane domains whose amino and carboxyl termini are both located in the cytoplasm11,12,14. Mutational and electrophysiological analyses have identified drosophila Orai and human ORAI1 as pore subunits of the CRAC channel15–17.
The importance of Ca2+ influx through CRAC channels is emphasized by the existence of at least three families with severe combined immunodeficiency associated with defects in CRAC channel function18–20. The underlying genetic defect responsible for the impaired CRAC channel function in one such family is a cytosine-to-thymine transition leading to a missense mutation in the gene encoding ORAI1 (ref. 11). T cells from patients of all three families show almost no store-operated Ca2+ entry or CRAC channel function and, as a result, fail to proliferate in response to TCR stimulation in vitro18–22 and fail to activate the Ca2+-responsive transcription factor NFAT or produce appreciable amounts of NFAT-dependent cytokines18,21,23.
STIM1 and STIM2 are single-pass transmembrane proteins with paired amino-terminal ‘EF hands’ located in the endoplasmic reticulum lumen and several protein-protein—interaction domains located in the endoplasmic reticulum lumen and the cytoplasm4,9,10,24. The first EF hand of each pair binds Ca2+ with low affinity matched to the high concentration of Ca2+ in the endoplasmic reticulum (200–600 μM)25,26. After store depletion, STIM proteins form oligomers in the endoplasmic reticulum membrane and then move to regions of endoplasmic reticulum—plasma membrane apposition that coincide with sites of Ca2+ entry and contain small clusters (puncta) of STIM1 and ORAI1 localized together10,24,27–30. STIM2 differs from STIM1 in that it is already partially active at basal endoplasmic reticulum Ca2+ concentrations and becomes activated earlier during endoplasmic reticulum store depletion, before substantial decreases in endoplasmic reticulum Ca2+ concentrations24. STIM proteins with EF-hand alterations that impair Ca2+ binding are constitutively present in puncta, even in resting cells with full Ca2+ stores10,24,31. The propensity of STIM proteins to undergo a conformational change and form oligomers when their EF hands are not bound by Ca2+ has also been demonstrated with short recombinant amino-terminal fragments containing only the EF hands and the adjacent sterile α-motif domains25.
Here we investigated the physiological functions of STIM1 and STIM2 in mice with conditionally targeted alleles of Stim1 and Stim2. Each protein promoted store-operated Ca2+ entry. In T cells, deficiency of either Stim1 or Stim2 impaired sustained Ca2+ influx, nuclear translocation of NFAT and cytokine production. However, mice lacking both Stim1 and Stim2 in T cells developed a lymphoproliferative syndrome characterized by splenomegaly, lymphadenopathy, infiltration of leukocytes into many organs and a selective decrease in the number and function of regulatory T cells (Treg cells) expressing the lineage-specific transcription factor Foxp3. This syndrome was ameliorated by adoptive transfer of wild-type Treg cells, and the Treg cell defect was cell intrinsic rather than being the result of the lack of interleukin 2 (IL-2) production by STIM-deficient T cells32–34. Our findings are consistent with published studies demonstrating ‘cooperation’ of NFAT and the signal transducer Smad3 at the Foxp3 promoter35 and a requirement for NFAT in Foxp3-mediated transcription and suppressor function36–38.
RESULTS
Conditional ablation of Stim1 or Stim2
As both STIM1 and STIM2 are ubiquitously expressed in vivo39, we generated mice bearing loxP-flanked alleles of Stim1 and Stim2 (Supplementary Fig. 1 online). We bred these mice with a ‘CMV-Cre deleter strain’ (expressing a transgene encoding Cre recombinase under control of the cytomegalovirus promoter)40 to examine the effects of deleting Stim1 and Stim2 in all tissues. STIM1-deficient mice on the C57BL/6 background were alive at the expected mendelian ratios at embryonic day 18.5 but died perinatally, with 75% of the pups born dead and most of the remaining pups dying within 2 d; in contrast, mice lacking STIM2 survived until 4 weeks after birth but showed slight growth retardation and died at 4–5 weeks of age (Supplementary Table 1a,b online). To ‘rescue’ the STIM1-deficient mice from perinatal death, we crossed these mice with the outbred ICR mouse strain. Although perinatal death was still high (38%), about half of the outbred STIM1-deficient offspring survived past day 2 with severe growth retardation and died of unknown causes within the next 2 weeks (Supplementary Table 1c).
To examine the effect of STIM deficiency in T cells, we crossed mice with loxP-flanked Stim1 (Stim1fl/fl) or Stim2 (Stim2fl/fl) with mice expressing a Cre transgene under control of a Cd4 enhancer-promoter-silencer cassette (CD4-Cre) that causes deletion at the double-positive (CD4+CD8+) stage of thymocyte development41. Thymic cellularity and T cell development seemed normal in Stim1fl/fl CD4-Cre+ and Stim2fl/fl CD4-Cre+ mice (data not shown), which permitted analysis of peripheral CD4+ and CD8+ T cells (Fig. 1). Unless otherwise indicated, we used T cells from mice in which Stim1 and/or Stim2 were (was) conditionally deleted with CD4-Cre for T cell experiments, whereas we used mouse embryonic fibroblasts (MEFs) from Stim1fl/fl CMV-Cre+ and Stim2fl/fl CMV-Cre+ mice for fibroblast experiments.
Figure 1.

STIM1 is a predominant effector of store-operated Ca2+ entry into T cells. (a) Store-operated Ca2+ influx in littermate control (Stim1+/+ CD4-Cre or Stim1fl/fl) (black lines) and Stim1fl/fl CD4-Cre (gray lines) naive CD4+ T cells in response to 1 μM thapsigargin (TG; left) or crosslinking with anti-CD3 (α-CD3) followed by 1 μM ionomycin (Iono; right) in the presence of 0.2 or 2 mM extracellular Ca2+. (b) IL-2 production by naive CD4+ T cells stimulated for 6 h with PMA and ionomycin, assessed by intracellular cytokine staining. CTRL, control (Stim1+/+ CD4-Cre or Stim1fl/fl). (c) Store-operated Ca2+ entry in response to 1 μM thapsigargin (top) or crosslinking with anti-CD3 followed by 1 μM ionomycin (center and bottom) in naive CD4+ T cells from wild-type mice (black lines) and Stim2–/– mice (gray lines), both obtained by intercrossing of Stim2+/- CMV-Cre– mice. (d) IL-2 production by naive wild-type (WT) and Stim2–/– CD4+ T cells stimulated for 6 h with PMA and ionomycin, assessed by intracellular cytokine staining. (e) [Ca2+]i responses of control (Stim2+/+ CD4-Cre or Stim2fl/fl; black lines) and Stim2–/– (gray lines) helper T cells differentiated for 7 d in vitro in nonpolarizing conditions, in response to high (1 μM) or low (10 nM) concentrations of thapsigargin or anti-CD3 followed by ionomycin. (f) Production of IL-2 and IFN-γ by wild-type and Stim2–/– helper T cells differentiated for 7 d in vitro in nonpolarizing conditions, then restimulated for 6 h with PMA and ionomycin. T cells from Stim1+/+ or Stim2+/+ CD4-Cre mice were compared with T cells from Stim1fl/fl or Stim2fl/fl mice in initial experiments to confirm that Cre expression had no toxic or other deleterious effects on proliferation or cytokine expression; in subsequent experiments, Stim1+/+ CD4-Cre and Stim1fl/fl mice or Stim2+/+ CD4-Cre and Stim2fl/fl mice, respectively, were used interchangeably as controls. Data are representative of at least three independent experiments.
STIM proteins regulate store-operated Ca2+ influx
STIM1-deficient CD4+ T cells showed almost no Ca2+ influx after passive depletion of endoplasmic reticulum Ca2+ stores with thapsigargin, an inhibitor of the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase, or after crosslinking of TCRs with antibody to CD3 (anti-CD3; Fig. 1a). Resting control and STIM1-deficient T cells had similar expression of surface CD3 and TCRβ and similar depletion of endoplasmic reticulum Ca2+ stores and expression of the activation markers CD25 and CD69 after stimulation with anti-CD3 or with anti-CD3 and anti-CD28 (Supplementary Fig. 2a—c online). STIM1-deficient CD4+ T cells failed to produce IL-2 after stimulation with the phorbol ester PMA and ionomycin (Fig. 1b) or with anti-CD3 and anti-CD28 (Supplementary Fig. 2d). These results collectively provide genetic evidence that STIM1 controls store-operated Ca2+ influx and the production of Ca2+-dependent cytokines in primary mouse T cells.
In contrast, STIM2-deficient primary CD4+ T cells from Stim2fl/fl CMV-Cre+ mice showed little or no impairment in Ca2+ influx or IL-2 production relative to control CD4+ T cells in response to treatment with thapsigargin, ionomycin or anti-CD3 (Fig. 1c,d). A possible explanation for this discrepancy is that naive CD4+ T cells had much lower expression of STIM2 than STIM1 (Supplementary Fig. 3a online). Although T cell activation led to a substantial increase in STIM2 expression, which was maintained after 3–7 d of differentiation into T helper type 1 or T helper type 2 cells, this greater quantity of STIM2 nevertheless amounted to only a small proportion (3–10%) of total STIM protein (Supplementary Fig. 3a,b). Consistent with the fact that even in differentiated T cells, STIM2 constitutes a minor fraction of total STIM protein, STIM2-deficient helper T cells differentiated for 1 week in nonpolarizing conditions showed slightly less Ca2+ influx in response to acute low-dose thapsigargin or anti-CD3 stimulation (Fig. 1e). However, these cells were much less able to produce IL-2 and interferon-γ (IFN-γ) after sustained stimulation with PMA and ionomycin (Fig. 1f) or anti-CD3 and anti-CD28 (Supplementary Fig. 2e). Likewise, STIM2-deficient T helper type 1 and T helper type 2 cells had less production of IL-2, IL-4 and IFN-γ (data not shown).
We also examined store-operated Ca2+ influx in MEFs from Stim1fl/fl CMV-Cre+ and Stim2fl/fl CMV-Cre+ mice. We confirmed that Ca2+ influx induced by treatment of wild-type MEFs with thapsigargin was due to depletion of endoplasmic reticulum Ca2+ stores, as we noted no Ca2+ influx in the absence of thapsigargin treatment (data not shown). As in T cells, STIM1 deficiency abrogated Ca2+ influx; moreover, in MEFs, STIM2 deficiency also resulted in less Ca2+ influx (Fig. 2a). These experiments collectively show that STIM1 deficiency results in a complete loss of store-operated Ca2+ entry in T cells and fibroblasts, whereas STIM2 deficiency has a smaller effect.
Figure 2.

Both STIM1 and STIM2 reconstitute store-operated Ca2+ entry and cytokine production in STIM1-deficient T cells and MEFs. (a) Ca2+ influx into wild-type, Stim1–/– and Stim2–/– MEFs stimulated with 1 μM thapsigargin in Ringer’s solution containing 20 mM Ca2+ (top), and reconstitution of store-operated Ca2+ entry by retroviral transduction of Stim1–/– MEFs with Myc-tagged STIM1 or STIM2 or empty vector (bottom). Expression vectors contain an internal ribosome entry site–GFP cassette and only GFP+ cells are analyzed here. (b) Ca2+ influx in response to treatment with 1 μM thapsigargin in control (Stim1+/+ CD4-Cre or Stim1fl/fl) and Stim1fl/fl CD4-Cre helper T cells differentiated for 7 d in vitro in nonpolarizing conditions and transduced with retroviral vector encoding Myc-tagged STIM1 or STIM2 or empty vector (only GFP+ cells are analyzed). (c) Flow cytometry (left) and averaged data (right) of cytokine production by control and Stim1fl/fl CD4-Cre cells transduced with retroviral vectors (right margin and below bars); only GFP+ cells are analyzed. Numbers in quadrants (left) indicate percent cells in each. Stim1+/+ or Stim2+/+ CD4-Cre mice were used initially as controls, after which both Stim1+/+ CD4-Cre and Stim1fl/fl mice or Stim2+/+ CD4-Cre and Stim2fl/fl mice, respectively, were used. Data are representative of three independent experiments (error bars, s.d.).
To confirm that STIM2 is a functional endoplasmic reticulum Ca2+ sensor, we reconstituted STIM1-deficient T cells and MEFs with Myc-tagged STIM1 or STIM2 then tested Ca2+ influx and cytokine production (Fig. 2a,b). We used retroviral vectors permitting stable expression of small amounts of protein to avoid artifacts resulting from overexpression. The introduced STIM1 and STIM2 were expressed in similar amounts in both cell types, as shown by immunoblot analysis with anti-Myc (Supplementary Fig. 3b and data not shown). STIM1 robustly reconstituted store-operated Ca2+ influx in STIM1-deficient MEFs (Fig. 2a) and in STIM1-deficient helper T cells differentiated for 1 week in nonpolarizing conditions and treated with thapsigargin (Fig. 2b); in contrast, we noted weaker Ca2+ influx in cells reconstituted with STIM2 (Fig. 2a,b). Nevertheless, STIM2 was unexpectedly effective in restoring cytokine expression to STIM1-deficient T cells stimulated with PMA and ionomycin (Fig. 2c). These results collectively indicate that endogenous STIM2 is a positive regulator of Ca2+ signaling in T cells and MEFs rather than being an inhibitor of STIM1 function, as proposed before42.
Aborted Ca2+ entry and nuclear transport of NFAT
To reconcile the slightly lower store-operated Ca2+ influx with the (relatively) much lower cytokine expression noted in STIM2-deficient T cells, we examined Ca2+ influx on a longer time scale by loading the cells with Fura-PE3, a calcium indicator that is well retained in the cytoplasm43. STIM2-deficient T cells had less store-operated Ca2+ entry than did wild-type T cells and attained a lower plateau of sustained intracellular free Ca2+ concentration ([Ca2+]i) after 20 min (Fig. 3a). To confirm that Ca2+ signaling is lower in STIM2-deficient cells, we monitored the nuclear translocation of the calcium-dependent transcription factor NFAT1 (ref. 8; A000024). We quantified nuclear translocation with the MetaXpress program (Supplementary Fig. 4 online) and differentiated helper T cells from wild-type, STIM1-deficient and STIM2-deficient mice for 1 week in nonpolarizing conditions and then stimulated them with PMA and ionomycin in the same conditions of stimulation used for the cytokine assay, so that we could directly compare nuclear translocation of NFAT1 (Fig. 3b,c) and cytokine expression (Fig. 3d,e).
Figure 3.

Loss of STIM2 affects sustained Ca2+ influx and the late phase of NFAT1 nuclear localization. (a) Averaged peak and steady-state (60 min after stimulation) [Ca2+]i and initial rates of change in [Ca2+]i in control CD4+ T cells (CTRL; n = 8; Stim2+/+ CD4-Cre or Stim2fl/fl) or STIM2-deficient CD4+ T cells (2KO; n = 7; Stim2fl/fl CD4-Cre) differentiated for 5 d in nonpolarizing conditions, labeled with Fura-PE3 and stimulated with PMA and ionomycin. P values, unpaired Student’s t-test. (b,c) STIM1-deficient (b), STIM2-deficient (c) and control cells with nuclear NFAT1 after stimulation as described in a, calculated with the data in Supplementary Figure 4. (d,e) Expression of IL-2 and IFN-γ by the cells in b (d) and c (e). In b,d, control is Stim1+/+ CD4-Cre or Stim1fl/fl; in c,e, control is Stim2+/+ CD4-Cre or Stim2fl/fl. Stim1+/+ or Stim2+/+ CD4-Cre mice were used initially as controls, after which both Stim1+/+ CD4-Cre and Stim1fl/fl mice or Stim2+/+ CD4-Cre and Stim2fl/fl mice, respectively, were used. Data are representative of three independent experiments (error bars, s.e.m. (a) or s.d. (b,c)), with at least 300 cells per well analyzed for each time point (b–e).
The results unambiguously showed that in physiological conditions both STIM1 and STIM2 contributed to the sustained Ca2+ influx and nuclear translocation of NFAT required for high expression of cytokine genes8. In STIM1-deficient helper T cells differentiated for 1 week in nonpolarizing conditions, NFAT was transiently imported into the nucleus, presumably because of the transient increase in [Ca2+]i that accompanies the depletion of endoplasmic reticulum Ca2+ stores, but it was imported into the nucleus in only a fraction of cells and was rapidly re-exported (Fig. 3b). In contrast, almost as many STIM2-deficient as control cells (70–75% versus 85–95%, respectively) had nuclear NFAT1 at 10 min, but this response was sustained in control but not STIM2-deficient cells (Fig. 3c). These results point to an important function for STIM2 in T cell signaling and explain the much lower cytokine expression in STIM2-deficient T cells (Figs. 1f and 3e).
CRAC current impairment in STIM1-deficient cells
We used whole-cell patch-clamp recordings to determine whether deletion of STIM1 and STIM2 affected the CRAC current (ICRAC). In response to depletion of endoplasmic reticulum Ca2+ stores by thapsigargin, control mouse CD4+ T cells had Ca2+ currents with properties similar to those of ICRAC of human T cells (Fig. 4). These properties included an inwardly rectifying current-voltage relationship with a very positive reversal potential in the presence of 20 mM Ca2+ (Fig. 4a), fast inactivation in 20 mM Ca2+ (Supplementary Fig. 5c online), depotentiation of the Na+ current in divalent cation—free solutions (Fig. 4b), low Cs+ permeability (Cs+ permeability/Na+ permeability = 0.2 ± 0.04; n = 14 samples), blockade of the Na+ current by micromolar concentrations of extracellular Ca2+ (Supplementary Fig. 5b) and potentiation and inhibition by low and high concentrations of 2-aminoethoxydiphenyl borate44 (although potentiation by 2-aminoethoxydiphenyl borate in mouse cells did not seem as robust as that in Jurkat or human T cells; Supplementary Fig. 5d and data not shown). Furthermore, inclusion of calcium-specific chelator BAPTA in the patch pipette at a concentration of 10 mM caused the slow development of an inward current in an extracellular calcium concentration of 20 mM after whole-cell break-in, reminiscent of the development of ICRAC in response to store depletion (data not shown). These results collectively indicated that mouse T cells have a Ca2+ current with properties indistinguishable from those of ICRAC.
Figure 4.

STIM1-deficient but not STIM2-deficient T cells lack ICRAC. (a) Current-voltage relations recorded in individual differentiated CD4+ T cells in 20 mM extracellular Ca2+ or divalent cation—free solution (DVF), assessing the Ca2+ current (left) and monovalent cation current (right) elicited by 1 μM thapsigargin. (b) Single-cell recordings of depotentiating Na+ current elicited by replacement of 20 mM Ca2+ in Ringer’s solution (filled bars) with divalent cation—free solution (open bars), measured during hyperpolarizing pulses to –100 mV applied every 1 s. (c) Summary of peak current densities averaged over all cells analyzed in a,b (n, number of cells). CTRL, Stim1+/+ CD4-Cre, Stim1fl/fl or Stim2fl/fl, or Stim1fl/flStim2fl/fl; STIM1-KO, Stim1fl/fl CD4-Cre; STIM2-KO, Stim2fl/fl CD4-Cre; DKO, Stim1fl/flStim2fl/fl CD4-Cre. Stim1+/+ or Stim2+/+ CD4-Cre mice were used initially as controls, after which both Stim1+/+ CD4-Cre and Stim1fl/fl mice or both Stim2+/+ CD4-Cre and Stim2fl/fl mice, respectively, were used. Data are representative of individual single-cell measurements (a,b) or are the mean ± s.e.m. of all single-cell measurements (c).
Consistent with the Ca2+ imaging results, STIM1-deficient T cells showed no detectable ICRAC in either 20 mM Ca2+ or divalent cationfree medium; in contrast, STIM2-deficient cells had an ICRAC of slightly smaller magnitude than that of wild-type cells that did not, however, reach statistical significance with the number of cells examined (Fig. 4c). The properties of ICRAC were unaltered in STIM2-deficient T cells in terms of Ca2+ and Cs+ selectivity, fast inactivation, depotentiation and responsiveness to high and low concentrations of 2-aminoethoxydiphenyl borate (Fig. 4, Supplementary Fig. 5 and data not shown). These results indicate that endogenous STIM1 is required for ICRAC in primary mouse CD4+ T cells but that endogenous STIM2 makes little or no contribution to the recorded ICRAC in the same conditions, possibly because it constitutes a very low fraction of total STIM protein even in differentiated T cells (Supplementary Fig. 3b).
Complex phenotype of mice with double-knockout T cells
To analyze the consequences of combined deletion of Stim1 and Stim2 in T cells in vivo, we generated Stim1fl/flStim2fl/fl CD4-Cre+ double-knockout mice. These mice showed no defect in conventional thymic development, as assessed by thymic cellularity and the numbers and proportions of CD4–CD8– double-negative cells, CD4+CD8+ double-positive cells and CD4+ or CD8+ single-positive cells (data not shown). Two possible explanations are that STIM proteins are long-lived and thus residual STIM1 and STIM2 protein may be present and functional well after gene deletion has occurred at the double-positive stage, or that thymocytes use STIM-independent Ca2+ influx mechanisms that differ from those used by peripheral T cells. The functions of STIM proteins in T cell development and thymic selection remain to be identified with mice in which Cre expression is initiated early during T cell or hematopoietic cell development.
As expected from the severely deleterious phenotype of STIM1-deficient T cells, peripheral CD4+ T cells lacking both STIM proteins showed essentially no Ca2+ influx in response to stimulation with thapsigargin or anti-CD3 (Fig. 5a and Supplementary Fig. 6 online). The small amount of residual Ca2+ influx in the averaged curves of [Ca2+]i (Fig. 5a) and the small residual ICRAC (Fig. 4c) stemmed from a small number of individual cells with normal Ca2+ influx and ICRAC (single-cell images, Fig. 5a, right). Of 15 double-knockout cells examined, two had normal ICRAC in the recordings in Figure 4c and the remaining cells had no ICRAC. These may have represented contaminating non—CD4+ cells that ‘came through’ the purification procedure or a small number of CD4+ T cells that escaped Cre-mediated deletion of STIM1 or STIM2 (discussed below). The double-knockout T cells produced almost no IL-2, although they did produce small amounts of tumor necrosis factor in response to primary stimulation (Fig. 5b), possibly because of PMA-induced activation of the transcription factor NF-κB, which proceeded normally in the double-knockout cells (M.O., unpublished data). The double-knockout T cells did upregulate expression of the activation markers CD69 and CD25, albeit to a lesser extent than did control T cells (Fig. 5c), and they underwent proliferation, albeit to a much smaller extent, after TCR stimulation (Fig. 5d,e).
Figure 5.

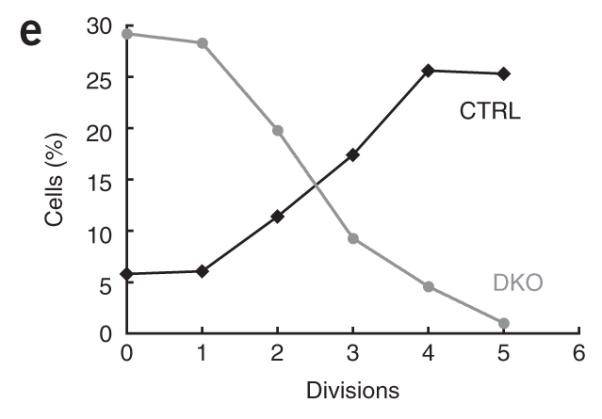
Impaired Ca2+ influx, cytokine production and proliferation in double-knockout T cells. (a) Store-operated Ca2+ influx in naive CD4+ T cells from littermate control mice (CTRL; Stim1fl/flStim2fl/fl) and double-knockout mice (DKO; Stim1fl/flStim2fl/fl CD4-Cre), stimulated with thapsigargin (top) or with anti-CD3 followed by crosslinking with streptavidin (SA; bottom) in nominally Ca2+-free Ringer’s solution, followed by perfusion with 2 mM Ca2+ in Ringer’s solution to induce Ca2+ influx (left). Middle, quantification of peak [Ca2+]i in 2 mM Ca2+ in Ringer’s solution. Right, single-cell ‘false-color’ images showing [Ca2+]i at the peak of the Ca2+ influx response. Original magnification, ×20. (b) Production of IL-2 and tumor necrosis factor (TNF) by naive CD4+ T cells from control (Stim1+/+ CD4-Cre or Stim1fl/flStim2fl/fl) or double-knockout mice, stimulated for 6 h with PMA and ionomycin. (c) Expression of CD25 and CD69 on naive CD4+ T cells from control (Stim1+/+CD4-Cre or Stim1fl/flStim2fl/fl) or double-knockout mice, left unstimulated (Unstim; control cells) or stimulated for 16 h with anti-CD3 and anti-CD28 (control (CTRL) or double-knockout (DKO) cells). (d) Proliferation of naive CD4+CD25– T cells from control (Stim1fl/flStim2fl/fl) or double-knockout mice, stimulated for 72 h with anti-CD3 and anti-CD28 and assessed by CFSE labeling. Open histograms, stimulated cells; shaded histograms, unstimulated cells. Numbers above bracketed lines indicate number of cell divisions. (e) Cells undergoing zero to five cell divisions (from data in d). Stim1+/+ or Stim2+/+ CD4-Cre mice were used initially as controls, after which Stim1+/+ CD4-Cre, Stim2+/+ CD4-Cre and Stim1fl/flStim2fl/fl mice were used. Data are representative of three (a) or two (b—d) independent experiments (error bars (a), s.e.m.).
Unexpectedly, double-knockout mice older than 8 weeks of age developed a notable phenotype of splenomegaly, lymphadenopathy, dermatitis and blepharitis (Supplementary Fig. 7a online and data not shown). Histological analysis showed infiltration of leukocytes into many organs, including lung and liver (Fig. 6a and data not shown). Mice lacking STIM1 alone in CD4+ T cells had a milder version of the lymphoproliferative phenotype (Supplementary Fig. 7a and data not shown). The double-knockout mice also had more CD4+ T cells with a surface phenotype characteristic of memory or effector status (CD62LloCD44hiCD69hiCD45RBloCD5hi), more germinal centers in the spleen, more B cells with a germinal center phenotype (CD95hiCD38lo) and more differentiated immunoglobulin E—positive (IgE+) B cells, large numbers of CD11b+IL-5R+ eosinophils or basophils and much higher serum concentrations of IgG1 (Supplementary Fig. 7b—d and data not shown). CD4+ T cells from double-knockout mice produced IL-5 but not IL-4 in response to stimulation with PMA and ionomycin (data not shown). Notably, the phenotype of double-knockout mice was similar but not identical to that of mice with a substitution (Y136F) in the T cell transmembrane adaptor Lat that eliminates the docking site for PLC-γ1 and results in lower Ca2+ influx in response to TCR crosslinking45,46.
Figure 6.

Double deficiency in STIM1 and STIM2 disrupts peripheral T cell homeostasis. (a) Hematoxylin and eosin staining of lung sections from a 4-month-old control mouse (Stim1fl/flStim2fl/fl) and a double-knockout mouse (Stim1fl/flStim2fl/fl CD4-Cre). Scale bar, 100 μm. (b) Staining of CD4, CD25 and Foxp3 in thymocytes and splenocytes from 5- to 6-week-old (left) or 3-month-old (right) control mice (Stim1+/+ CD4-Cre or Stim1fl/flStim2fl/fl) and double-knockout mice. Numbers above outlined areas indicate percent cells in gate. (c) Total cells (left) and CD4+CD25+ Treg cells (right) in thymi and spleens of 5- to 6-week-old control mice (Stim1+/+ CD4-Cre or Stim1fl/flStim2fl/fl) or double-knockout mice (n = 3 mice). (d) Peak Ca2+ response induced by CD3 crosslinking (left) or treatment with 1 μM thapsigargin (right) in CD4+CD25+ cells isolated from control (Stim1fl/flStim2fl/fl) or double-knockout mice. Measurements were made in Ringer’s solution with 2 mM Ca2+. Stim1+/+ or Stim2+/+ CD4-Cre mice were used initially as controls to confirm that Cre expression did not result in toxicity or other adverse effects, after which Stim1fl/flStim2fl/fl mice were used to control for other factors, such as sex, age and breeding environment. Data are representative of results from two independent experiments with three control or double-knockout mice (a); three independent experiments (b,c; mean ± s.d., c); or three (anti-CD3) and two (thapsigargin) independent experiments with 110–170 cells analyzed in each (d).
STIM in Treg cell differentiation and function
The autoreactive phenotype of the Lat (Y136F) mutant mice described above has been attributed to impaired negative selection allowing escape of autoreactive T cells into the periphery47 and to a lower number of Treg cells48. Direct examination of the function of STIM proteins in positive and negative selection of thymocytes will require mice in which Stim1 and Stim2 are ablated at an earlier stage of T cell development with Lck-Cre and breeding of these mice with HY TCR-transgenic mice. Meanwhile, here we documented many fewer Treg cells in the thymi, spleens and lymph nodes of 5- to 6-week-old Stim1fl/flStim2fl/fl CD4-Cre mice (Fig. 6b,c and data not shown). The proportion of Treg cells in the spleens and lymph nodes of double-knockout mice increased with age but nevertheless remained between 10% and 20% of that in control mice (Fig. 6c); these increases were probably the result of age-dependent increases in the size of peripheral lymphoid organs. Mice lacking either STIM1 or STIM2 had normal numbers of CD4+CD25+Foxp3+ Treg cells (data not shown). The number of cells expressing GITR, another marker of Treg cells, was also lower in double-knockout mice (data not shown). We noted complete deletion of Stim1 and Stim2 in CD25– and CD25+ T cells from older (8-week-old) double-knockout mice (Supplementary Fig. 8a online). Like CD4+CD25– T cells, CD4+CD25+ Treg cells from double-knockout mice showed impaired Ca2+ influx in response to treatment with thapsigargin or anti-CD3 (Fig. 6d and Supplementary Fig. 8b).
The much lower percentages and absolute numbers of Treg cells in double-knockout mice could have reflected defective Treg cell development, survival in the periphery or both. Given the finding that double-knockout T cells made very little IL-2, one possibility was that Treg cell development was defective in part because of a lack of IL-2 production by ‘bystander’ T cells32–34. To address those possibilities, we generated mixed—bone marrow chimeras. We reconstituted sublethally irradiated recipient mice deficient in recombination-activating gene 1 (Rag1–/– mice) with T cell—depleted bone marrow from Thy-1.2+ control mice alone or Thy-1.2+ double-knockout mice alone or bone marrow from Thy-1.2+ double-knockout mice and Thy-1.1+ wild-type mice mixed at a ratio of 2:1. As expected, mice given only double-knockout bone marrow had many fewer Treg cells in both thymus and lymph nodes than did mice reconstituted with control bone marrow (Fig. 7) and developed the same severe lymphoproliferative phenotype as that of unmanipulated double-knockout mice (Supplementary Fig. 9a online). In contrast, mixed chimeras reconstituted with both double-knockout and wild-type bone marrow did not show any signs of lymphoproliferative disease and remained as healthy as control chimeric mice (Supplementary Fig. 9a). In these chimeric mice, the wild-type bone marrow gave rise to normal numbers of peripheral Treg cells, whereas the double-knockout precursors yielded far fewer Treg cells both in thymus and in the periphery (Fig. 7). These results collectively indicate that the double-knockout mice have a cell-intrinsic defect in Treg cell development that is not restored by IL-2 produced by ‘bystander’ T cells derived from wild-type bone marrow32–34.
Figure 7.

Absence of STIM1 and STIM2 impairs the development of Treg cells. Flow cytometry of thymus and lymph node cells from sublethally irradiated Rag1–/– mice reconstituted with T cell—depleted bone marrow from Thy-1.2+ control littermates (Stim1fl/flStim2fl/fl) alone (3 × 106 cells), from Thy-1.2+ double-knockout mice alone (3 × 106 cells), or from both Thy-1.2+ double-knockout mice (3 × 106 cells) and congenic B6 Thy-1.1+ wild-type mice (WT; 1.5 × 106 cells), stained with anti-CD4, anti-Thy-1.1 and anti-Thy-1.2, together with anti-CD25 and anti-Foxp3, at 10–12 weeks after reconstitution. Numbers in quadrants and above outlined areas indicate percent cells in each. SP, single-positive. As no Cre toxicity was noted in previous experiments, Stim1fl/flStim2fl/fl mice were used as controls to control for other factors, such as sex, age and breeding environment. Data are representative of results from two independent experiments with three mixed chimeric mice.
Next we determined whether we could prevent onset of the lymphoproliferative phenotype of double-knockout mice by injecting young double-knockout mice with Treg cells from wild-type mice. Injection of 2-week-old double-knockout mice with wild-type Treg cells prevented the development of lymphoadenopathy and splenomegaly 8 weeks later, whereas injection with phosphate-buffered saline or with non–Treg cells did not (Fig. 8a and Supplementary Fig. 9b). Because double-knockout mice had very few endogenous Treg cells and we transferred only 3 × 105 wild-type CD4+CD25+ T cells into each double-knockout mouse, the injected mice continued to have very few Treg cells at 8 weeks after injection (Fig. 8b). We distinguished Thy-1.1+ Treg cells derived from the transferred wild-type Treg population from endogenous Thy-1.2+ Treg cells by flow cytometry (Fig. 8b,c). As expected, endogenous Thy-1.2+ Treg cells accounted for most of the CD4+CD25+Foxp3+ cells in double-knockout mice injected with wild-type CD4+CD25– T cells; these mice showed lymphadenopathy and splenomegaly at 10 weeks of age (Fig. 8c). In contrast, mice injected with wild-type CD4+CD25+ Treg cells and ‘cured’ of the lymphopro-liferative phenotype had a Treg cell population derived approximately equally from Thy-1.1+ donor and Thy-1.2+ endogenous cells (Fig. 8c). These data indicate that even though some Treg cells develop in double-knockout mice, they function poorly (if at all) relative to Treg cells from wild-type mice. Moreover, the numbers of endogenous Treg cells were lower in the presence of transferred wild-type Treg cells (Fig. 8c, left versus right), which suggested that Treg cells from the double-knockout mice were at a competitive disadvantage in vivo. We confirmed the defective function of the residual Treg cells in double-knockout mice with an in vitro suppression assay (Fig. 8d and Supplementary Fig. 10 online). These data collectively suggest that loss of both STIM1 and STIM2 impaired the development and function of Foxp3+ Treg cells.
Figure 8.


Adoptive transfer of wild-type Treg cells suppresses the lymphoproliferative phenotype of double-knockout mice. (a–c) CD4+CD25+ (CD25+) or CD4+CD25– (CD25–) Thy-1.1+ T cells from wild-type mice (3 × 105 cells) were transferred into 2-week-old Thy-1.2+ double-knockout mice, and cells in recipient mice were analyzed 8 weeks after transfer. CTRL, control littermates (Stim1fl/flStim2fl/fl) injected with PBS; PBS, double-knockout mice injected with PBS; CD25–, double-knockout mice injected with CD25– cells; CD25+, double-knockout mice injected with CD25+ cells. (a) Spleen and lymph node (LN) cells from recipient mice. P values, paired Student’s t-test. (b) Antibody staining of spleen and lymph node cells from recipient mice. Numbers above outlined areas indicate percent cells in gate. (c) Staining of cells with anti-Thy-1.1, anti-Thy-1.2, anti-CD4, anti-CD25 and anti-Foxp3 for analysis of donor cell engraftment, presented as endogenous Treg cells (endog; Thy-1.2+) and transferred Treg cells (transf; Thy-1.1+) in mice that received CD25– (Non) or CD25+ T cells. (d) In vitro suppression assay of CD4+CD25+ T cells purified from control (Stim1fl/flStim2fl/fl) or double-knockout mice and cultured for 72 h at a ratio of 1:1 with CFSE-labeled responder CD4+CD25– T cells in the presence of mitomycin C–treated T cell—depleted splenocyte samples and anti-CD3 (0.3 μg/ml). As no Cre toxicity was noted in previous experiments, Stim1fl/flStim2fl/fl mice were used as controls to control for other factors, such as sex, age and breeding environment. Data are representative of results from two independent experiments with three mice (a–c; error bars (a,c), s.d.), or at least three independent experiments (d).
DISCUSSION
Here we have examined the physiological functions of STIM1 and STIM2 in mice with conditionally targeted alleles of Stim1 and Stim2. We found that STIM1 is a critical activator of store-operated Ca2+ entry and the function of CRAC channels, in agreement with published studies in which STIM1 was depleted by RNA-mediated interference9,10. In the absence of STIM1, T cells and fibroblasts showed almost no Ca2+ influx in response to depletion of endoplasmic reticulum Ca2+ stores. T cells from STIM1-deficient mice also lacked detectable ICRAC and consequently were severely compromised in their ability to produce the cytokines IL-2, IFN-γ and IL-4 in response to TCR stimulation. These results are consistent with published analyses of immunodeficient patients with a mutation in the gene encoding the ORAI1 subunit of the CRAC channel11,21 and confirm the prevailing idea that STIM1 and ORAI1 are critical components of the pathway of store-operated Ca2+ entry2,3. STIM1 is also crucial in mediating store-operated Ca2+ entry, degranulation and cytokine production by mast cells stimulated through the Fc receptor FcεRI (ref. 49).
Our work has also clarified the biological function of endogenous STIM2. STIM2-deficient fibroblasts had much less store-operated Ca2+ entry, which indicated a ‘positive’ function for STIM2 in this cell type. STIM2-deficient CD4+ T cells differentiated for 3–7 d in vitro in nonpolarizing conditions consistently showed slightly less store-operated Ca2+ entry in response to treatment with low-dose thapsigargin, ionomycin or anti-CD3. Furthermore, STIM2 compensated partially for the absence of STIM1, as STIM1-deficient T cells and fibroblasts regained a moderate degree of store-operated Ca2+ influx after ectopic expression of STIM2. These data collectively show that endogenous STIM2 promotes Ca2+ influx in both T cells and fibroblasts, consistent with published studies demonstrating coupling of both STIM1 and STIM2 to store-operated Ca2+ entry in HeLa cells10,24.
The moderately diminished store-operated Ca2+ entry and CRAC channel function noted in differentiated STIM2-deficient cells contrasts with their more notably diminished cytokine expression. We traced the cytokine deficiency to the inability of STIM2-deficient cells to sustain nuclear translocation of NFAT. Many studies have established that optimal cytokine production by activated T cells requires sustained Ca2+ influx and prolonged nuclear residence of NFAT5,8,50. Consistent with those findings, the amount of cytokines produced by wild-type, STIM1-deficient and STIM2-deficient T cells correlated precisely with their ability to sustain increased [Ca2+]i and to maintain NFAT for long periods in the nucleus. Wild-type T cells retained substantial NFAT in the nucleus for more than 6 h during continuous stimulation and had robust production of cytokines. STIM1-deficient T cells showed only a small early transient increase in nuclear NFAT, which returned to baseline within 30 min even during continuous stimulation, and they produced almost no cytokines. STIM2-deficient T cells had an almost normal early phase of NFAT nuclear residence, which presumably reflected the residual function of STIM1, but were unable to maintain nuclear translocation of NFAT to the extent that wild-type T cells did and produced much smaller amounts of cytokines than did wild-type cells. These findings are consistent with the demonstration that STIM2 is active at higher endoplasmic reticulum Ca2+ concentrations than is STIM1 and therefore would maintain store-operated Ca2+ entry and NFAT nuclear residence during the late stages of a response when STIM1 has been inactivated by partial refilling of the endoplasmic reticulum stores24.
Mice with targeted deletion of both STIM1 and STIM2 in T cells had a notable lymphoproliferative phenotype characterized by splenomegaly, lymphadenopathy, infiltration of leukocytes into many organs and signs of autoimmunity (or infection secondary to immune deficiency) in the form of blepharitis and dermatitis. This syndrome was associated with many fewer Treg cells (approximately 10% of wild-type control numbers), which distinguished it from other lymphopro-liferative disorders such as autoimmune lymphoproliferative syndrome (mutation in the gene encoding the cytokine receptor Fas) and X-linked lymphoproliferative disease (mutation in the gene encoding the adaptor protein SAP)51,52. Mixed—bone marrow chimera experiments showed that this syndrome was also distinct from Treg cell deficiencies arising from loss of IL-2 or transforming growth factor-β signaling33,34,53,54. In addition, both in the mixed—bone marrow chimeras and in double-knockout mice that received wild-type CD4+CD25+ Treg cells at a young age, the development of splenomegaly and lymphade-nopathy was mostly prevented, which indicated that the small population of residual Treg cells in double-knockout mice was functionally impaired. In support of that conclusion, double-knockout Treg cells were far less able than wild-type Treg cells to suppress the proliferation of cocultured CD4+CD25– responder T cells in vitro. These findings indicate that store-operated Ca2+ entry through the STIM-Orai1 pathway is essential for the development and function of Treg cells.
We also found that Foxp3-expressing natural Treg cells were fully capable of activating store-operated Ca2+ influx in response to CD3 crosslinking, which indicated that the steps of PLC-γ1 activation, generation of inositol-1,4,5-trisphosphate, store-operated Ca2+ influx and (presumably) nuclear translocation of NFAT were entirely functional in this cell type. On the basis of those data and published studies indicating NFAT-Foxp3 ‘cooperation’ is essential for the suppressive function of Foxp3-transduced T cells37,38, we propose that NFAT activation and possibly NFAT-Foxp3 ‘cooperation’ are also essential for the development and function of thymus-derived Treg cells. Indeed, it has been shown that when thymi from 1-week-old heterozygous nude mice (nu/+), treated from birth with the NFAT inhibitor cyclosporin A, are transplanted into recipient homozygous nude mice (nu/nu), the recipient mice develop diverse organ-specific autoimmunity55. In this model, autoimmunity can be prevented by inoculation of the recipient mice with suspensions of normal thymocytes, leading the authors of that study to suggest that cyclosporin A interferes selectively with thymic production of ‘suppressor T cells’ whose responsibility it is to control self-reactive T cells. The defect in Treg cell development noted in that study and here could also reflect the reported ‘cooperation’ between Smad3 and NFAT at an enhancer element in the Foxp3 locus35. It is notable that Treg cell development is also impaired in mice with other mutations that affect TCR signaling, including mice lacking Bcl-10, which show defective activation of NF-κB56.
Aspects of the lymphoproliferative phenotype of the double-knockout mice resembled the phenotype of mice with other hypomorphic mutations in the Ca2+-NFAT pathway. For example, mice with a Y136F substitution of the transmembrane adaptor Lat (which when phosphorylated forms a docking site for PLC-γ1; refs. 57–59) also have a block in Treg cell development but can be distinguished from STIM double-knockout mice in that they also have a substantial block in the double-negative—to—double-positive stage of conventional T cell development45,46. Similarly, mice lacking NFAT1 and NFAT4 show hyperproliferation and hyperactivation of T cells and B cells60 but normal development and function of Treg cells; indeed, T cells in these mice are not effectively inhibited by wild-type Treg cells61. Finally, mice with early Lck-Cre—mediated deletion of the gene encoding calcineurin B1 in the thymus show a block at the double-positive—to—single-positive stage of thymocyte development, but autoimmune and/or hyperproliferative syndromes and Treg cell dysfunction have not been reported62. Further studies are needed to elucidate the complex effects of mutations in the Ca2+-calcineurin-NFAT signaling pathway on the development of conventional and regulatory T cells.
METHODS
Animals and conditional gene targeting
Stim1 and Stim2 were targeted by homologous recombination in Bruce-4 embryonic stem cells derived from C57BL/6 mice as described63. Chimeric mice with targeted Stim1 or Stim2 alleles were generated by blastocyst injection of heterozygous Stim1neo/+ or Stim2neo/+ embryonic stem cell clones (Supplementary Fig. 1; ‘neo’ indicates the neomycin-resistance gene). Stim1–/– or Stim2–/– mice were generated by intercrossing of the progeny of founder Stim1neo/+ or Stim2neo/+ mice after they were bred with CMV-Cre (‘Cre deleter’) transgenic mice40. For establishment of Stim1+/- or Stim2+/- mice without the transgene encoding Cre, Stim1+/- CMV-Cre+ mice or Stim2+/- CMV-Cre+ mice were bred with C57BL/6 mice. For generation of the conditional Stim1fl/+ or Stim2fl/+ alleles, founder Stim1neo/+ or Stim2neo/+ chimeric mice were bred with ‘Flp deleter’ transgenic mice64 for removal of the neomycin-resistance cassette from the targeted Stim1 or Stim2 alleles. Rag1–/– and B6.Cg (Igha, Thy-1.1, Gpi1a) mice were from the Jackson Laboratory. For the generation of mice with T cell—specific disruption of Stim1 and/or Stim2, CD4-Cre transgenic mice41 were bred with each founder Stim1fl/+ or Stim2fl/+ mouse and the progeny were intercrossed. All mice were maintained in specific pathogen—free barrier facilities at Harvard Medical School and were used in accordance with protocols approved by the Center for Animal Resources and Comparative Medicine of Harvard Medical School.
T cell differentiation, retroviral transductions and stimulation
Purification of CD4+ T cells from spleen and lymph nodes, induction of T helper cell differentiation, stimulation with 10 nM PMA and 1 μM ionomycin or with plate-bound anti-CD3 (purified from supernatants of the 2C11 hybridoma) and anti-CD28 (37.51; BD Pharmingen), and assessment of cytokine production by intracellular staining and flow cytometry were done as described65. Foxp3 expression was assessed by intracellular staining with anti-Foxp3 (FJK-16s; eBioscience) according to the manufacturer’s protocol and was analyzed by flow cytometry. Retroviral transduction was done as described37 with KMV retroviral expression plasmids (a modified moloney murine leukemia virus vector), either empty or containing Stim1 or Stim2 cDNA, followed by cDNA encoding green fluorescent prptein (GFP) under control of an internal ribosome entry site. Despite their defect in Ca2+ influx, STIM1-deficient T cells upregulated CD25 (IL-2 receptor α-chain) normally. Thus, because differentiating cultures were maintained in IL-2, almost equivalent cell numbers were recovered (total number of differentiated STIM1-deficient cells at 7 d was 60–70% that of wild-type) and STIM-deficient cells could be retrovirally transduced with the same efficiency as wild-type cells.
Establishment of MEF cell lines
Stim1-/- and Stim2-/- MEFs were established with standard protocols from embryos at embryonic day 14.5 obtained by intercrossing of Stim1+/- or Stim2+/- mice. MEFs were immortalized by retroviral transduction with SV40 large T antigen in a plasmid carrying the hygromycin-resistance gene, followed by hygromycin selection.
Antibodies and immunoblot
Cell extracts were prepared by resuspension of cells in PBS, followed by lysis in a buffer of 50 mM NaCl, 50 mM Tris-HCl, pH 6.8, 2% (wt/vol) SDS and 10% (vol/vol) glycerol (final concentrations). Protein concentrations were determined with the BCA Protein Reagent kit (Pierce), then 2-mercaptoethanol was added to a final concentration of 100 μM and samples were boiled. Standard protocols were used for immunoblot analysis. Polyclonal anti-STIM1 (Open Biosciences) was generated against a carboxy-terminal peptide of human STIM1 (CDNGSIGEETDSSPGRKKFPLKIFKKPLKK-COOH, in which the cysteine at the amino terminus was introduced for the purpose of coupling the peptide with a carrier protein) and was used at a dilution of 1:2,000. Affinity-purified polyclonal antibodies were generated against a carboxy-terminal peptide of human STIM2 (CKPSKIKSLFKKKSK, in which the cysteine at the amino terminus was introduced for the purpose of coupling the peptide with a carrier protein) and were used at a concentration of 2 μg/ml. Polyclonal anti-actin (I-19; SC-1616; Santa Cruz) was used at a dilution of 1:500. Monoclonal antibody to the Myc epitope tag was purified from supernatants of 9E10 hybridoma cell lines. All of the following fluorescence-conjugated antibodies used for flow cytometry were from eBioscience or BD Pharmingen: Pacific blue—conjugated anti-CD4 (RM4-5); fluorescein isothiocyanate—conjugated anti-CD8 (53-6.7), anti-Thy-1.1 (HIS51), anti-IgE (R35-72) and anti-CD11c (M1/70); phycoerythrin-conjugated anti-IL-2 (JES6-5HA), anti-IL-4 (11B11), anti-Foxp3 (FJK- 16s), anti-CD19 (1D3), anti-CD125 (T21.2), anti-CD44 (IM7) and anti-CD95 (Jo2); peridinine chlorophyll protein complex—conjugated anti-B220 (RA3-6B2); peridinine chlorophyll protein complex—cyanine 5.5—conjugated anti-CD4 (RM4-5); phycoerythrin-indotricarbocyanine—conjugated anti-Thy-1.2 (53-2.1); allophycocyanin-conjugated anti-IFNγ (XMG1.2), anti-IL-10 (JES5-16E3), anti-body to tumor necrosis factor (anti-TNF; MP6-XT22), anti-CD25 (PC61.5), anti-TCRβ (H57-597), anti-CD38 (90) and anti-CD62L (MEL-14); biotinylated anti-CD5 (53-7.3) and anti-CD69 (H1.2F3); and phycoerythrin-streptavidin.
Single-cell [Ca2+]i imaging
CD4+ T cells were isolated as described65, were incubated overnight in loading medium (10% (vol/vol) FBS in RPMI 1640 medium) and were loaded for 30 min at 22–25 °C with calcium indicator Fura-2-AM (1 μM; Invitrogen) at a density of 1 × 106 cells per ml. Before measurements, T cells were attached for 15 min to poly-L-lysine-coated cover-slips. For anti-CD3 stimulation, T cells were incubated for 15 min at 22–25 °C with biotin-conjugated anti-CD3 (5 μg/ml; 2C11; BD Pharmingen), and anti-CD3 crosslinking was achieved by perfusion of cells with streptavidin (10 μg/ml; Pierce). For long-term Ca2+ imaging, differentiated CD4+ T cells were loaded with 1 μM Fura-PE3, then were stimulated with 10 nM PMA and 0.5 μM ionomycin in Ringer’s solution containing 2 mM Ca2+ supplemented with 2% (vol/vol) FCS. During image acquisition, cells were constantly perfused with buffer warmed to 37 °C. [Ca2+]i was measured and analyzed as described11. Ca2+ influx rates were inferred from the maximum rate of the initial increase in [Ca2+]i in 0.2–2 mM extracellular Ca2+, expressed as the ratio ‘Δ[Ca2+]i/Δt’, where ‘Δ[Ca2+]i’ is the maximum difference in [Ca2+]i over a 20-second time interval (Δt) between the readdition of extracellular Ca2+ and the peak of the Ca2+ influx response. For each experiment, 100–150 T cells or at least 30 individual MEFs were analyzed with Igor Pro analysis software (Wavemetrics).
NFAT1 nuclear-translocation assay
CD4+ T cells were cultured in nonpolarizing conditions and were collected at day 5, then were stimulated for various times with 10 nM PMA plus 1 μM ionomycin at a density of 1 × 105 cells per well in a volume of 200 μl in 96-well plates and were attached to poly-L-lysine-coated wells in 384-well plates (5 × 103 to 8 × 103 cells per well; three wells per sample) by centrifugation for 3 min at 149g. Cells were fixed with 3% (vol/vol) paraformaldehyde then were stained with anti-NFAT1 (purified rabbit polyclonal antibody to the ‘67.1’ peptide of NFAT1)66 and indocarbocyanine-conjugated anti-rabbit (secondary antibody) and counterstained with the DNA-intercalating dye DAPI (4,6-diamidino-2-phenylindole). Images were acquired with the ImageXpress Micro automated imaging system (Molecular Devices) with a 20 × objective and were analyzed with the translocation application module of MetaXpress software version 6.1 (Molecular Devices). Cytoplasmic-to-nuclear translocation was assessed by calculation of a correlation of the intensity of indocarbocyanine—anti-NFAT1 staining and DAPI staining; T cells were considered to have nuclear NFAT1 when over 90% of the indocarbocyanine—anti-NFAT1 staining coincided with the fluorescence signal from DAPI. Each data point represents an average of at least 300 individual cells per well.
Patch-clamp measurements
CD4+ T cells were cultured in nonpolarizing conditions and were collected at day 5. An Axopatch 200 amplifier (Axon Instruments) interfaced to an ITC-18 input/output board (Instrutech) and an iMac G5 computer were used for patch-clamp recordings. Currents were filtered at 1 kHz with a four-pole Bessel filter and were sampled at 5 kHz. Recording electrodes were ‘pulled’ from 100-μl pipettes, were coated with Sylgard and were fire-polished to a final resistance of 2–5 MΩ. ‘In-house’ routines developed on the Igor Pro platform (Wavemetrics) were used for stimulation and data acquisition and analysis. All data were corrected for the liquid-junction potential of the pipette solution relative to that of Ringer’s solution in the bath (–10 mV) and for leak currents collected in 20 mM extracellular Ca2+ plus 25 μM La3+. The standard extracellular Ringer’s solution was 130 mM NaCl, 4.5 mM KCl, 20 CaCl2, 1 mM MgCl2, 10 mM D-glucose and 5 mM Na-HEPES, pH 7.4. In some experiments, 2 mM CaCl2 was used in the standard extracellular solution and the NaCl concentration was increased to 150 mM. Standard divalent cation—free Ringer’s solutions were 150 mM NaCl, 10 mM tetraacetic acid, 1 mM EDTA and 10 mM HEPES, pH 7.4. Charybdotoxin (25 nM; Sigma) was added to all extracellular solutions to eliminate contamination from Kv1.3 channels. The standard internal solution was 145 mM cesium aspartate, 8 mM MgCl2, 10 mM BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid) and 10 mM Cs-HEPES, pH 7.2. Averaged results are presented as the mean value ± s.e.m. Curve fitting was done by the least-squares methods with built-in functions in Igor Pro 5.0. The permeability of Cs+ relative to that of Na+ was calculated from the bionic reversal potential with the equation
where PCs and PNa are the permeability of Cs+ (the test ion) and Na+, respectively; [Cs]i and [Na]o are the ionic concentrations; Erev is the reversal potential; F is the Faraday constant (9648 C mol–1); R is the gas constant (8.314 J K–1 mol–1); and T is the absolute temperature.
Hematoxylin and eosin staining
Tissues from 3- to 4-month-old mice were fixed in 10% (vol/vol) formalin. A standard procedure was used for hematoxylin and eosin staining.
Purification and adoptive transfer of Treg cells
CD4+CD25+ and CD4+CD25– T cells derived from Thy-1.1+ congenic mice were sorted with a FACSVantage after purification of CD4+ T cells with Dynabeads Mouse CD4 and DETA-CHaBEAD Mouse CD4 (Invitrogen). Wild-type Thy-1.1+ CD4+CD25+ or CD4+CD25– T cells (3 × 105) were injected intraperitoneally into 2-week-old mice. Injected mice were analyzed at 8 weeks after adoptive transfer.
Mixed—bone marrow transfer
T cell—depleted bone marrow from Thy-1.2+ double-knockout mice (3 × 106 cells) was mixed with that of Thy-1.1+ congenic wild-type mice (1.5 × 106 cells) and the mixture was injected through the retro-orbital sinus into sublethally irradiated Rag1–/– mice (450 rads). Reconstituted mice were analyzed 10–12 weeks after bone marrow transfer.
Proliferation and in vitro suppression assays
CD4+CD25– or CD4+CD25+ T cells were positively selected with MACS CD25 microbeads (Miltenyi Biotec) after purification of CD4+ T cells. Purified CD4+CD25– T cells (2 × 107 cells per ml) were incubated for 10 min at 37 °C with 1.25 μM CFSE (carboxyfluorescein diacetate succinimidyl diester). Cells were stimulated for 72 h with anti-CD3 and anti-CD28 and the number of cell divisions was assessed by flow cytometry. In vitro suppression assays were done by coculture of 5 × 104 CFSE-labeled CD4+CD25– T cells at various ratios with CD4+CD25+ T cells purified from control littermates or double-knockout mice; cells were cocultured for 72 h at 37 °C in round-bottomed plates in the presence of mitomycin C—treated T cell—depleted splenocyte samples (5 × 104 cells) and anti-CD3 (0.3 μg/ml; 2C11).
Accession code
UCSD-Nature Signaling Gateway (http://www.signalinggateway.org): A000024.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Rajewsky and members of the Rajewsky lab for help with blastocyst injection of embryonic stem cells; M.E. Pipkin and A.Y. Rudensky for comments and discussions; Y. Gwack for purification of anti-STIM2; and B. Baust for help in establishing the NFAT-translocation assay. Supported by the National Institutes of Health (A.R., S.F. and M.P.), Juvenile Diabetes Research Foundation (A.R.), March of Dimes Foundation (S.F.), Uehara Memorial Foundation (M.O.) and Canadian Institutes of Health Research (S.S.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING INTERESTS STATEMENT The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/natureimmunology/.
Reprints and permissions information is available online at http://npg.nature.com/ reprintsandpermissions
References
- 1.Carafoli E. The calcium-signalling saga: tap water and protein crystals. Nat. Rev. Mol. Cell Biol. 2003;4:326–332. doi: 10.1038/nrm1073. [DOI] [PubMed] [Google Scholar]
- 2.Putney JW., Jr. New molecular players in capacitative Ca2+ entry. J. Cell Sci. 2007;120:1959–1965. doi: 10.1242/jcs.03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 4.Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem. Sci. 2007;32:235–245. doi: 10.1016/j.tibs.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Feske S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 6.Parekh AB, Putney JW., Jr. Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 7.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium. 2003;33:311–321. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 8.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 9.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 12.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang SL, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 15.Prakriya M, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 16.Yeromin AV, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vig M, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feske S, et al. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur. J. Immunol. 1996;26:2119–2126. doi: 10.1002/eji.1830260924. [DOI] [PubMed] [Google Scholar]
- 19.Le Deist F, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–1062. [PubMed] [Google Scholar]
- 20.Partiseti M, et al. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- 21.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 22.Feske S, Okamura H, Hogan PG, Rao A. Ca2+/calcineurin signalling in cells of the immune system. Biochem. Biophys. Res. Commun. 2003;311:1117–1132. doi: 10.1016/j.bbrc.2003.09.174. [DOI] [PubMed] [Google Scholar]
- 23.Feske S, Draeger R, Peter HH, Eichmann K, Rao A. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID Tcells. J. Immunol. 2000;165:297–305. doi: 10.4049/jimmunol.165.1.297. [DOI] [PubMed] [Google Scholar]
- 24.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 26.Zheng L, Stathopulos PB, Li GY, Ikura M. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem. Biophys. Res. Commun. 2007 December 31; doi: 10.1016/j.bbrc.2007.12.129. published online. (doi:10.1016/j.bbrc.2007.12.129) [DOI] [PubMed] [Google Scholar]
- 27.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu P, et al. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem. Biophys. Res. Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 30.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of storeoperated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+CD25+CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 34.D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- 35.Tone Y, et al. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2007;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 36.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated Tcells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. USA. 2005;102:5138–5143. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 38.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams RT, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem. J. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 42.Soboloff J, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr. Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 43.Vorndran C, Minta A, Poenie M. New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes. Biophys. J. 1995;69:2112–2124. doi: 10.1016/S0006-3495(95)80082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. (Lond.) 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sommers CL, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 46.Aguado E, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- 47.Sommers CL, et al. Mutation of the phospholipase C-γ1-binding site of LAT affects both positive and negative thymocyte selection. J. Exp. Med. 2005;201:1125–1134. doi: 10.1084/jem.20041869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koonpaew S, Shen S, Flowers L, Zhang W. LAT-mediated signaling in CD4+CD25+ regulatory T cell development. J. Exp. Med. 2006;203:119–129. doi: 10.1084/jem.20050903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba Y, et al. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat. Immunol. 2008;9:81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 50.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 51.Fisher GH, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 52.Sayos J, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- 53.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 55.Sakaguchi S, Sakaguchi N. Thymus and autoimmunity. Transplantation of the thymus from cyclosporin A-treated mice causes organ-specific autoimmune disease in athymic nude mice. J. Exp. Med. 1988;167:1479–1485. doi: 10.1084/jem.167.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt-Supprian M, et al. Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-kappaB activation. Proc. Natl. Acad. Sci. USA. 2004;101:4566–4571. doi: 10.1073/pnas.0400885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang W, et al. Association of Grb2, Gads, and phospholipase C-γ1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 2000;275:23355–23361. doi: 10.1074/jbc.M000404200. [DOI] [PubMed] [Google Scholar]
- 58.Lin J, Weiss A. Identification of the minimal tyrosine residues required for linker for activation of T cell function. J. Biol. Chem. 2001;276:29588–29595. doi: 10.1074/jbc.M102221200. [DOI] [PubMed] [Google Scholar]
- 59.Paz PE, et al. Mapping the Zap-70 phosphorylation sites on LAT (linker for activation of T cells) required for recruitment and activation of signalling proteins in T cells. Biochem. J. 2001;356:461–471. doi: 10.1042/0264-6021:3560461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ranger AM, Oukka M, Rengarajan J, Glimcher LH. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity. 1998;9:627–635. doi: 10.1016/s1074-7613(00)80660-3. [DOI] [PubMed] [Google Scholar]
- 61.Bopp T, et al. NFATc2 and NFATc3 transcription factors play a crucial role in suppression of CD4+ T lymphocytes by CD4+CD25+ regulatory T cells. J. Exp. Med. 2005;201:181–187. doi: 10.1084/jem.20041538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity. 2004;20:255–266. doi: 10.1016/s1074-7613(04)00052-4. [DOI] [PubMed] [Google Scholar]
- 63.Muljo SA, et al. Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodriguez CI, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 65.Ansel KM, et al. Deletion of a conserved Il4 silencer impairs T helper type 1-mediated immunity. Nat. Immunol. 2004;5:1251–1259. doi: 10.1038/ni1135. [DOI] [PubMed] [Google Scholar]
- 66.Ho AM, Jain J, Rao A, Hogan PG. Expression of the transcription factor NFATp in a neuronal cell line and in the murine nervous system. J. Biol. Chem. 1994;269:28181–28186. [PubMed] [Google Scholar]
- 67.Prakriya M, Lewis RS. Regulation of CRAC channel activity by recruitment of silent channels to a high open-probability gating mode. J. Gen. Physiol. 2006;128:373–386. doi: 10.1085/jgp.200609588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.