

Abstract
Study Objectives:
The orexin-producing neurons are hypothesized to be essential for the circadian control of sleep/wake behavior, but it remains unknown whether these rhythms are mediated by the orexin peptides or by other signaling molecules released by these neurons such as glutamate or dynorphin. To determine the roles of these neurotransmitters, we examined the circadian rhythms of sleep/wake behavior in mice lacking the orexin neurons (ataxin-3 [Atx] mice) and mice lacking just the orexin neuropeptides (orexin knockout [KO] mice).
Design:
We instrumented mice for recordings of sleep-wake behavior, locomotor activity (LMA), and body temperature (Tb) and recorded behavior after 6 days in constant darkness.
Results:
The amplitude of the rapid eye movement (REM) sleep rhythm was substantially reduced in Atx mice but preserved in orexin KO mice. This blunted rhythm in Atx mice was caused by an increase in the amount of REM sleep during the subjective night (active period) due to more transitions into REM sleep and longer REM sleep episodes. In contrast, the circadian variations of Tb, LMA, Wake, non-REM sleep, and cataplexy were normal, suggesting that the circadian timekeeping system and other output pathways are intact in both Atx and KO mice.
Conclusions:
These results indicate that the orexin neurons are necessary for the circadian suppression of REM sleep. Blunting of the REM sleep rhythm in Atx mice but not in orexin KO mice suggests that other signaling molecules such as dynorphin or glutamate may act in concert with orexins to suppress REM sleep during the active period.
Citation:
Kantor S; Mochizuki T; Janisiewicz AM; Clark E; Nishino S; Scammell TE. Orexin neurons are necessary for the circadian control of REM sleep. SLEEP 2009;32(9):1127-1134.
Keywords: Ataxin-3, knock-out, narcolepsy, cataplexy, circadian rhythm, constant darkness, body temperature, REM sleep, REM sleep latency, REM sleep propensity
MANY RESEARCHERS HAVE HYPOTHESIZED THAT THE POORLY CONSOLIDATED WAKE AND INAPPROPRIATELY TIMED RAPID EYE MOVEMENT (REM) SLEEP of narcolepsy could be caused by disruption of the circadian rhythms of sleep and wakefulness.1–4 The suprachiasmatic nucleus (SCN) is essential for the timing and consolidation of sleep and wakefulness;5 and squirrel monkeys with SCN lesions lose the circadian rhythms of sleep and wakefulness and are unable to produce long bouts of wakefulness.6 Similarly, people with narcolepsy have difficulty maintaining wakefulness, and their naps often include bouts of REM sleep, regardless of the time of day.1 Most likely, this is not caused by a primary defect in the generation of circadian rhythms because the daily rhythms of body temperature and cortisol are essentially normal.7 Instead, people with narcolepsy may have a specific defect in the circadian control of sleep and wakefulness.
Considerable basic research supports this hypothesis. Narcolepsy with cataplexy is caused by a loss of the orexin/hypocretin-producing neurons.8–10 Since the orexin neurons receive both direct and indirect signals from the SCN and send projections to many state-regulatory brain regions, they are anatomically well positioned to mediate the circadian timing of sleep and wakefulness.11–17 Extracellular levels of orexin vary in a circadian pattern, with high levels during the waking period, and lesions of the SCN abolish this rhythm.18–20 Furthermore, nonspecific lesions of the orexin field or of the pathways between the SCN and the orexin neurons disrupt the timing of sleep/wake behavior.12,13,21 We therefore hypothesized that the orexin neurons are an essential relay for the circadian signals that time and consolidate sleep and wake.
To determine whether the orexin neurons are necessary for the circadian control of sleep and wakefulness, we examined free-running circadian rhythms in 2 different strains of orexin deficient mice: orexin/ataxin-3 (Atx) mice with an acquired, selective loss of the orexin/hypocretin neurons and orexin peptide knockout (KO) mice.22,23
MATERIALS AND METHODS
All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 8023, revised 1978). These studies were approved by the Institutional Animal Care and Use Committees of Beth Israel Deaconess Medical Center and Harvard Medical School.
Animals
We used 8 male, transgenic orexin/ataxin-3 mice (Atx), 6 orexin peptide knockout (KO) mice, and corresponding wild-type littermates (8 WTAtx and 8 WTKO). The orexin KO mice have a neomycin cassette inserted into the prepro-orexin gene and simply lack orexin-A and –B.22 In Atx mice, the human prepro-orexin promoter drives expression of ataxin-3, a toxic transgene that selectively kills the orexin neurons by around 12 weeks of age.23 Thus, these Atx mice are an excellent model of the loss of the orexin-producing neurons that occurs in people with narcolepsy with cataplexy9,10 and allow one to test the roles of other neurotransmitters (e.g., dynorphin and glutamate) produced in the orexin neurons.8,24 To maximize genetic homogeneity, we backcrossed the Atx and orexin KO lines with C57BL/6J mice for 6-8 generations.
Mice were identified by genotyping and immunostaining. PCR was performed on genomic DNA from tail biopsies. Primers used were 5’-CAT GAA GGA AGA AGG TCC TGG and 3’-CCT TGC ACC CAG GAA TCT GG against the orexin/ataxin-3 transgene and 5’-GAC GAC GGC CTC AGA CTT CTT GGG and 3’-TCA CCC CCT TGG GAT AGC CCT TCC against the endogenous mouse prepro-orexin gene. We identified orexin KO mice using a neo primer 5’-TAG TTG CCA GCC ATC TGT TG and a genomic primer 3’-ACT CTC CAC CCA CAG ACA GG. After the polysomnographic recordings, mice were deeply anesthetized and perfused. Immunostaining for orexin-A (1:2500; Santa Cruz Biotechnology, CA, USA, Lot# A2604, catalogue# sc-8070) confirmed a > 95% loss of the hypothalamic orexin neurons in all Atx mice and no orexin immunoreactivity in all KO mice (see17,22,25 for detailed description of immunostaining methods).
Surgery and EEG/EMG recordings
At the time of surgery, mice were 16-19 weeks old and weighed 35 to 40 g. Under anesthesia with ketamine-xylazine (100 and 10 mg/kg ip), we implanted mice with EEG and electromyogram (EMG) electrodes, as described previously.26 Briefly, stainless steel screw electrodes were implanted epidurally over the left frontal cortex (1.5 mm lateral and 1 mm anterior to bregma) and left parietal cortex (1.5 mm lateral and 1.0 mm anterior to lambda) for frontoparietal EEG recordings. EMG signals were acquired by a pair of multistranded stainless steel wires inserted into the neck extensor muscles. A telemetric body temperature (Tb) and locomotor activity (LMA) transmitter (TA-F20, Data Sciences International, St. Paul, MN) was placed in the peritoneal cavity. Transmitters were factory-calibrated to an accuracy of 0.1°C.
One week after surgery, mice were transferred to recording cages in a sound-attenuated chamber with a 12:12 h light-dark (LD) cycle (30 lux daylight-type fluorescent tubes with lights on at 07:00), constant temperature (23 ±1°C), and with food and water available ad libitum. The recording cable was attached to a low torque commutator, fixed above the cages that allowed free movement. Mice were habituated to the cables for 5 days before the experiments and remained connected throughout the study. After acclimating to the LD cycle for 10 days, baseline sleep/wake behavior was recorded (data not shown). Mice were then kept in constant darkness (DD) for 7 days. On days 6 and 7 of DD, we recorded the polysomnogram (EEG/EMG, infrared video), Tb, and LMA for 48-h and then analyzed 24-h recordings after determining the subjective day onset for each individual animal (see below).
The EEG/EMG signals were acquired using Grass Model 12 amplifiers (West Warwick, RI) and digitized at 128 Hz. The signals were digitally filtered (EEG: 0.3-30 Hz, EMG: 5-30 Hz) and scored in 10-s epochs as Wake, non-REM (NREM) sleep, or REM sleep using SleepSign (Kissei Comtec, Matsumoto, Japan). S.K. then visually inspected all scoring and made corrections when appropriate.
Behavior was scored as cataplexy using the new consensus definition of murine cataplexy.27 Specifically, an epoch was scored as cataplexy when: (1) the mouse had one or more epochs of EEG θ activity and muscle atonia immediately preceded and followed by active Wake; (2) at least 40 sec of Wake preceded cataplexy to exclude any REM sleep that might follow a brief awakening;28 and (3) the animal was immobile. Whenever behavior met criterion 1, we examined integrated infrared video recordings (SleepSign) to determine if all criteria were fulfilled.27,29
Period, Phase, and Amplitude Analysis
Tb and LMA were recorded at 5-min intervals by an antenna (RPC-1, Data Sciences International) below the recording cage and digitally acquired (Dataquest, Data Sciences International). Analysis of the Tb rhythm over 7 days showed that mice in all groups had the same phase angle of entrainment in LD. To measure the period of the free-running rhythm, we performed a chi-square periodogram analysis (Circadia, Behavioral Cybernetics, Cambridge, MA) on the Tb data of each mouse over the first week of constant darkness. A subset of mice (5 WTAtx, 4 Atx) continued in DD for a total of 27 days, and their free-running rhythms were very similar to that seen in the first week of DD (data not shown). The onset of subjective night was calculated for each animal based on the period of its free running Tb rhythm: subjective night onset = 19:00 + n*(period of Tb - 24 hr), where n is the number of days after onset of constant darkness.12,13
Sleep/wake rhythms in rodents are clearly not sinusoidal and have multiple peaks and abrupt changes near CT0 and CT12 that hinder analysis using techniques such as cosinor analysis. Thus to measure the amplitudes of circadian rhythms of sleep/wakefulness, we used the circadian index (CI) and the nocturnality ratio.30,31 The CIs of Wake, NREM sleep, REM sleep, LMA and Tb were calculated from the following formula: CI = (meannight - meanday)/mean24hr, where meanday is the average over the subjective day, meannight is the average over the subjective night, and mean24hr is the average over the entire day.12,13 CIs are plotted as positive values in which low values indicate little circadian variation. Nocturnality ratios were defined as the percentage of total amount of a behavior (Wake, NREM sleep, REM sleep, LMA, or Tb) occurring during the subjective night. For the nocturnality analysis of Tb, the values above the daily mean were counted. Thus, high nocturnality ratios represent high incidence of the behavior during the night, while ratios close to 50% indicate behavior evenly distributed between subjective day and night.
Analysis of REM Sleep Propensity
To measure the propensity for REM sleep, we analyzed REM sleep latency, REM sleep bout durations, and the probability of transitioning into REM sleep from NREM sleep. We defined the sleep cycle as beginning at the onset of NREM sleep and as ending at the offset of REM sleep, allowing for brief awakenings no longer than 10 sec. Cycles were excluded from analysis if 1) they lacked REM sleep; 2) the REM sleep episode was only one epoch long (10 sec), followed by NREM sleep; or 3) the NREM sleep episode prior to REM sleep was shorter than 30 sec.26 REM sleep latency was calculated for each sleep cycle and then averaged for the subjective day and night.
To analyze the probability of transitioning into REM sleep as a function of NREM sleep bout length, we first calculated the absolute probability of transitioning from NREM into REM sleep for each 10 sec epoch of NREM sleep and calculated the weighted average probability for bins of increasing duration (< 90, 90-180, 180-270, 270-360, and > 360 sec).
Statistical Analysis
We used ANOVA for repeated measures to analyze changes in each vigilance state as a function of strain (WTAtx, WTKO, Atx, and KO) and time. The same test was used with 2 main factors (strain and day-night cycle) to compare vigilance state parameters. CIs and nocturnality ratios of sleep/wake behavior were compared between strains using unpaired t-tests. To analyze the probability of NREM to REM sleep transitions and the distribution of REM sleep bout durations, we used ANOVA for repeated measures with strain as the between factor and bout length as the repeated measurement. The Bonferroni/Dunn test was used for post hoc comparisons. All results are expressed as means ± SE.
RESULTS
Impaired Circadian Control of REM Sleep in Atx Mice
In constant darkness, the amplitude of the circadian rhythm of REM sleep was much smaller in Atx mice than their WTAtx littermates (t = 3.19, P < 0.01; Figure 2A). Atx mice had nearly half (41%) of their total amount of REM sleep time during the subjective night; a considerably higher proportion than in WTAtx mice (26%) (t = 3.19, P < 0.01; Figure 2B). This reduction in circadian rhythmicity was due to a near doubling in the amount of REM sleep during the subjective night (F1,14 = 8.09, P < 0.05; Table 1). This increase was still apparent when REM sleep was viewed as a percentage of total sleep time (TST, F1,14 = 15.78, P < 0.01; Figure 3), demonstrating that the increase in REM sleep was not a consequence of more sleep in general. Atx mice had less REM sleep in the first 2 h of the subjective night, compared to the rest of the night, probably due to displacement of REM sleep by the increased amounts of Wake during this interval (Figure 1).
Figure 2.

Atx mice have little circadian variation in REM sleep rhythm, indicated by low circadian index (A) and a nocturnality ratio (B) close to 50%. In contrast, the circadian rhythms of Wake, NREM sleep, Tb and LMA are preserved in Atx mice. The circadian index is normalized to the mean circadian amplitude of WTAtx mice (100%). **P < 0.01 compared to WTAtx
Table 1.
Vigilance State Parameters Recorded from Orexin/Ataxin-3 Transgenic (Atx), Orexin Knockout (KO) and their Wild-Type Littermate Control (WT) Mice
| Variate | Subjective Day |
Subjective Night |
||||||
|---|---|---|---|---|---|---|---|---|
| WTAtx | Atx | WTKO | KO | WTAtx | Atx | WTKO | KO | |
| Wake | ||||||||
| Total time (min) | 242.4 ± 14.1 | 262.5 ± 8.7 | 302.1 ± 10.6 | 261.0 ± 9.0 | 438.7 ± 11.3 | 433.1 ± 9.8 | 421.4 ± 16.7 | 417.2 ± 14.3 |
| Mean duration (sec) | 109 ± 9 | 98 ± 3 | 152 ± 9 | 93 ± 6** | 248 ± 16 | 175 ± 10** | 252 ± 25 | 143 ± 12** |
| Number of bouts | 134 ± 4 | 160 ± 5** | 121 ± 5 | 170 ± 6** | 108 ± 5 | 150 ± 7** | 104 ± 6 | 178 ± 10** |
| NREM sleep | ||||||||
| Total time (min) | 429.0 ± 13.1 | 404.7 ± 9.5 | 372.7 ± 9.1 | 401.4 ± 8.8* | 263.5 ± 10.1 | 235.4 ± 7.6* | 275.9 ± 14.2 | 245.5 ± 13.9 |
| Mean duration (sec) | 183 ± 5 | 149 ± 7** | 180 ± 7 | 133 ± 5** | 144 ± 8 | 100 ± 3** | 158 ± 8 | 95 ± 3** |
| Number of bouts | 141 ± 4 | 164 ± 5** | 125 ± 5 | 181 ± 9** | 111 ± 6 | 141 ± 7** | 106 ± 7 | 157 ± 14** |
| REM sleep | ||||||||
| Total time (min) | 48.6 ± 2.8 | 48.2 ± 2.3 | 45.2 ± 2.4 | 49.7 ± 3.7 | 17.8 ± 2.9 | 33.8 ± 3.0** | 22.7 ± 2.7 | 23.8 ± 3.7 |
| Mean duration (sec) | 59 ± 3 | 53 ± 2 | 54 ± 4 | 36 ± 2** | 40 ± 4 | 49 ± 3 | 56 ± 5 | 43 ± 4 |
| Number of bouts | 50 ± 3 | 55 ± 4 | 51 ± 4 | 80 ± 4** | 26 ± 3 | 42 ± 4** | 24 ± 3 | 35 ± 7 |
| Cataplexy | ||||||||
| Total time (min) | 0 | 4.6 ± 1.0 | 0 | 8.0 ± 2.3 | 0 | 17.7 ± 2.5 | 0 | 33.3 ± 5.0 |
| Mean duration (sec) | - | 68 ± 4 | - | 80 ± 14 | - | 67 ± 6 | - | 78 ± 5 |
| Number of bouts | 0 | 4 ± 1 | 0 | 7 ± 2 | 0 | 17 ± 3 | 0 | 26 ± 4 |
| Tb (°C) | 36.3 ± 0.3 | 35.9 ± 0.1 | 36.0 ± 0.0 | 36.0 ± 0.0 | 37.5 ± 0.2 | 37.2 ± 0.0 | 37.0 ± 0.1 | 37.5 ± 0.0** |
Total time spent in each state, mean duration, number of bouts and Tb over the subjective day (rest) and night (active) periods. Results shown as mean ± SEM. Significant differences (repeated measures ANOVA) between Atx or KO and their WT littermates are indicated with asterisks (*P < 0.05; **P < 0.01).
Figure 3.
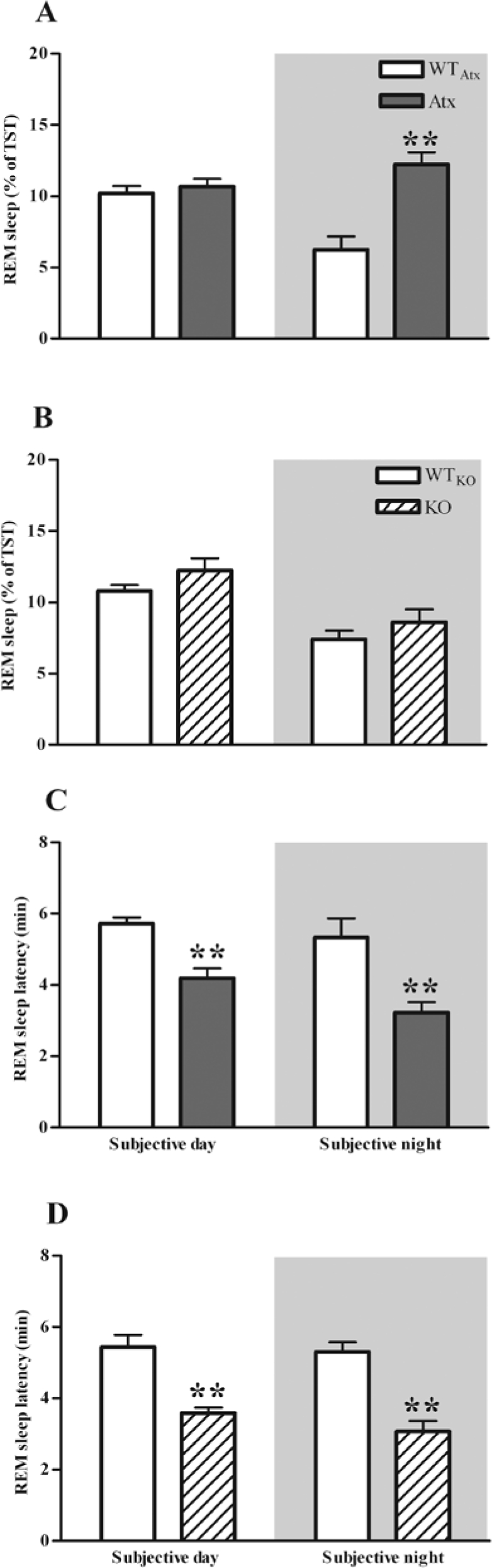
During the subjective night, Atx mice have an increased amount of REM sleep as a percentage of total sleep time (TST) (A) demonstrating that more REM sleep in these mice is not a consequence of more sleep in general. The average REM sleep latencies were shorter in both Atx and orexin KO mice than in WT littermates (C, D). **P < 0.01 compared to WTAtx or WTKO.
Figure 1.

In constant darkness, the hourly amounts of Wake (A) and NREM sleep (B) are similar in Atx (n = 8) and WTAtx (n = 8) mice. However, Atx mice have more REM sleep (C) than their WTAtx littermates during the subjective night. Nearly all cataplexy (D) occurs during the subjective night in Atx mice. *P < 0.05 compared to WTAtx
To determine whether the reduction in REM sleep rhythmicity was caused by orexin deficiency, we performed the same analyses in orexin KO mice and their WTKO littermates. In contrast to the Atx mice, the amounts and timing of REM sleep in orexin KO mice were very similar to WTKO littermates (Table 1). This was true for the absolute amounts of REM sleep as well as REM sleep as a percent of total sleep time (Figure 3).
In contrast, the circadian rhythms of Wake and NREM sleep were undisturbed in Atx and KO mice. The hourly amounts of Wake and NREM sleep were essentially normal, though the duration of Wake and NREM sleep bouts was much shorter in both Atx and KO mice than in WTAtxand WTKOmice, as reported previously.23,32 Cataplexy showed robust circadian variation in both Atx and KO mice, with nearly all cataplexy occurring during the subjective night. This was true for the absolute amounts of cataplexy as well as cataplexy as a percent of Wake time, indicating that circadian timing of cataplexy is not directly linked to the distribution of Wake (Supplemental Figure 1).
Increased REM Sleep Propensity in Atx Mice During the Subjective Night
To identify factors that contribute to the increase in REM sleep during the dark period, we examined REM sleep architecture in detail. Both Atx and KO mice had REM sleep latencies 30% to 40% shorter than their WTAtx and WTKO littermates (F1,14 = 29.07, P < 0.01 and F1,12 = 42.22, P < 0.01, respectively; Figure 3).
In addition, Atx mice were more likely to transition into REM sleep than WTAtx, WTKO, or orexin KO mice. In WTAtxand WTKO mice, the probability of entering REM sleep gradually rose across the duration of NREM sleep, but Atx mice had a much higher probability of entering REM sleep at any time during a NREM sleep episode (F1,14 = 8.30, P < 0.05; Figure 4). Interestingly, orexin KO mice had generally normal probabilities of entering REM sleep, suggesting that the frequent transitions into REM sleep in Atx mice are not simply due to orexin deficiency.
Figure 4.

In contrast to orexin KO mice (B, D), Atx mice have a generally higher probability of entering REM sleep (A) and have longer bouts of REM sleep (C) than their wild-type littermates during the subjective night (active period) suggesting that the orexin neurons may control the initiation and maintenance of REM sleep during the active period. *P < 0.05, **P < 0.01 compared to WTAtx or WTKO.
Atx mice also produced more bouts and longer bouts of REM sleep. During the subjective night, Atx mice had about twice as many long bouts (> 20-30 sec) of REM sleep as WTAtx littermates (F1,12 = 6.95, P < 0.05; Figure 4), and the mean duration of REM sleep bouts was increased by about 20% (Table 1). In contrast, the durations of REM sleep bouts appeared roughly normal in KO mice, with no increase in long REM sleep bouts and a slightly reduced mean duration of REM sleep bouts.
These findings demonstrate that two main factors contribute to the increased REM sleep of Atx mice: a higher probability of entering REM sleep, and better maintenance of REM sleep bouts than WTAtx mice.
Preserved Fundamental Circadian Rhythmicity in DD
To determine whether this reduction in the circadian rhythm of REM sleep was caused by a general decrease in circadian rhythmicity, we analyzed the periods of the Tb rhythms. Both WTAtx and Atx mice had robust free-running circadian Tb rhythms in DD with periods significantly shorter than 24.0 h (t = 3.7, P < 0.01; t = 2.6, P < 0.05, respectively). The mean periods of the Tb rhythms (23.92 ± 0.02 vs. 23.88 ± 0.05; t = 0.67, P = 0.5), as well as the amplitudes of the circadian rhythms of Tb (t = 0.69, P = 0.5) and LMA (t = 0.35, P = 0.7) were very similar in WTAtx and Atx mice, suggesting that even in the absence of the orexin neurons, fundamental circadian rhythmicity is preserved.
In mice with mutations in clock regulatory genes, it can take several weeks for rhythms to fully disappear,33 so we maintained a subset of mice (5 WTAtx, 4 Atx) in DD for 27 days. Their sleep/wake behavior and free-running rhythms were very similar to that seen after 6 days of DD (data not shown).
DISCUSSION
These results indicate that the orexin neurons are necessary for the circadian suppression of REM sleep during the active period. Specifically, we found that in constant darkness, Atx mice lacking the orexin neurons had much less circadian variation in REM sleep than their WTAtx littermates whereas orexin ligand KO mice had a normal rhythm of REM sleep. The reduction in the REM sleep rhythm in Atx mice was due to a near doubling in the amount of REM sleep during the subjective night related to a higher probability of transitioning into REM sleep and longer bouts of REM sleep. Since the circadian rhythms of Tb, LMA, and Wake were normal, it is likely that the circadian clock itself and the clock effector mechanisms responsible for timing wakefulness are intact in both Atx and KO mice.
Our results are in accord with and build upon prior studies of narcoleptic rodents and humans. The first studies of Atx and orexin KO mice as well as orexin deficient Atx rats described an increase in REM sleep during the dark (active) period,22,23,32,34,35 but these studies were limited in that they did not distinguish between REM sleep and cataplexy. A recent, detailed analysis that scored REM sleep and cataplexy separately revealed that Atx mice had twice the normal number of NREM to REM sleep transitions during the night and slightly longer REM sleep bouts.32 However, light/dark cycles can influence sleep patterns and mask any underlying defects in circadian regulation,36,37 and no prior studies examined orexin neuron deficient animals under free running conditions. In a study of people with narcolepsy, Dantz and colleagues examined the circadian control of sleep using a forced desynchrony protocol in which subjects had an opportunity to sleep for 30 minutes every 90 minutes over a 3-day period.1 They found that narcoleptic subjects had much less circadian variation in REM sleep than controls, but with such a short sleep period, many control subjects had difficulty obtaining enough REM sleep and had substantial rebound REM sleep on the subsequent recovery day. Despite these limitations, these studies, along with our new observations suggest that the orexin neurons are necessary for the circadian control of REM sleep.
REM sleep is clearly under strong circadian and homeostatic control, but the nature of this control is controversial.1,38–44 Specifically, it is unclear whether the circadian variation in REM sleep is due to active promotion of REM sleep during the rest period; inhibition of REM sleep during the active period; or passive gating of REM sleep by competing arousal states. Wurts and colleagues hypothesized that REM sleep is actively promoted by the SCN during the rest period because SCN-lesioned rats made fewer attempts to enter REM sleep after REM sleep deprivation than unlesioned controls.43 One might expect that less active promotion would result in less REM sleep overall, but the SCN-lesioned rats had normal amounts of REM sleep during baseline recordings.43 In our study, orexin neuron deficiency resulted in an increased amount of REM sleep during the active period with no change in REM sleep during the rest period. Therefore, we propose that circadian regulation of REM sleep is achieved, at least in part, by suppression of REM sleep during the active period by the orexin neurons. This hypothesis is supported by the anatomic observations that orexin neurons receive direct and indirect projections from the SCN and send projections to many state-regulatory nuclei of the brain including many regions that suppress REM sleep.11–16 Furthermore, the orexin neurons are most active during wakefulness and relatively silent during sleep45–48 and selective stimulation of the orexin neurons is sufficient to trigger awakenings from NREM and REM sleep.49 These data together with our results suggest that the orexin neurons mediate the circadian timing of REM sleep by suppressing REM sleep during the active period, though it remains possible that other mechanisms promote REM sleep during the rest period.
In contrast to Atx mice, we found that orexin KO mice had normal circadian rhythms of REM sleep, as reported previously.26 This difference suggests that the impaired circadian control of REM sleep in Atx mice is not simply due to orexin deficiency. The orexin neurons produce other signaling molecules including glutamate and dynorphin,8,24,50 and even in the absence of orexin, these neurotransmitters may still relay circadian timing signals that suppress REM sleep during the active period. The synergistic effects of orexin and dynorphin have been demonstrated in the wake-promoting neurons of the tuberomammillary nucleus (TMN): orexin directly excites TMN neurons and dynorphin disinhibits TMN neurons by presynaptically inhibiting GABAergic inputs.50 Similarly, brain regions that inhibit REM sleep (including the ventrolateral periaqueductal grey, lateral pontine tegmentum, locus coeruleus and dorsal raphe) receive excitatory inputs from the orexin neurons and inhibitory inputs from sleep-promoting neurons in the ventrolateral preoptic area.51–53 We speculate that orexins excite neurons that inhibit REM sleep, and dynorphin may enhance this effect by inhibiting GABAergic inputs from sleep-promoting brain sites. Together, the synergistic effects of these neuropeptides would strongly suppress REM sleep during the active period.
These mice provide novel perspectives on the circadian control of sleep and wakefulness, but some aspects of their behavior leave open other interpretations. First, KO mice lack orexin throughout development, and they may have more opportunity for developmental compensation than Atx mice in which the orexin neurons are lost postnatally. These compensatory changes may contribute to the milder REM sleep phenotype in orexin KO mice. In addition, it is possible that both Atx and KO mice have poor circadian suppression of REM sleep during the active period, but orexin KO mice express this as partial transitions into cataplexy whereas Atx mice are more prone towards complete transitions into REM sleep. Last, disinhibition of REM sleep in Atx mice could mask the REM sleep rhythm. Though this could be caused by abnormal REM sleep homeostasis, this seems unlikely as Atx mice had normal amounts of REM sleep during the subjective day. Additional studies, perhaps using selective REM sleep deprivation in narcoleptic patients, may help clarify whether the homeostatic control of REM sleep is abnormal in narcolepsy.
In addition to promoting wakefulness and regulating REM sleep, the orexin neurons increase autonomic tone, boost metabolism, and enhance feeding, reward-seeking and other motivated behaviors.23,54–58 In fact, many researchers view the orexin neurons as playing a central role in coordinating these functions so that an animal is alert and energetic when engaged in the active behaviors of wakefulness.59,60 Our finding that the orexin neurons are necessary for the circadian control of REM sleep fits well with this perspective. An animal would certainly be exposed to attack or injury during the atonia and unconsciousness of REM sleep, and by suppressing REM sleep during the active period, the orexin neurons may help ensure that this vulnerable state does not intrude into wakefulness.
DISCLOSURE STATEMENT
This study was partially supported by a research grant from Takeda Pharmaceuticals. The listed authors independently analyzed all data and wrote the entire manuscript. Dr. Scammell has received honoraria from Takeda Pharmaceuticals. Dr. Mochizuki received a research grant from Jazz Pharmaceuticals. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
This research was made possible by grants from Takeda Pharmaceuticals North America and the National Institute of Health (NS055367, HL60292). Elizabeth Clain provided invaluable help with data analysis. We are grateful for the insightful comments and suggestions provided by Elizabeth Klerman, Ralph Mistlberger, Janet Mullington, and David Weaver.
REFERENCES
- 1.Dantz B, Edgar DM, Dement WC. Circadian rhythms in narcolepsy: studies on a 90 minute day. Electroencephalogr Clin Neurophysiol. 1994;90:24–35. doi: 10.1016/0013-4694(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 2.Selbach O, Haas HL. Hypocretins: the timing of sleep and waking. Chronobiol Int. 2006;23:63–70. doi: 10.1080/07420520500545961. [DOI] [PubMed] [Google Scholar]
- 3.Broughton R, Krupa S, Boucher B, Rivers M, Mullington J. Impaired circadian waking arousal in narcolepsy-cataplexy. Sleep Res Online. 1998;1:159–65. [PubMed] [Google Scholar]
- 4.Lavie P. REM periodicity under ultrashort sleep/wake cycle in narcoleptic patients. Can J Psychol. 1991;45:185–93. doi: 10.1037/h0084284. [DOI] [PubMed] [Google Scholar]
- 5.Mistlberger RE. Circadian regulation of sleep in mammals: role of the suprachiasmatic nucleus. Brain Res Brain Res Rev. 2005;49:429–54. doi: 10.1016/j.brainresrev.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Edgar DM, Dement WC, Fuller CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation. J Neurosci. 1993;13:1065–79. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer G, Hellmann F, Leonhard E, Meier-Ewert K. Circadian temperature and activity rhythms in unmedicated narcoleptic patients. Pharmacol Biochem Behav. 1997;58:395–402. doi: 10.1016/s0091-3057(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 8.Crocker A, Espana RA, Papadopoulou M, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–8. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 11.Abrahamson EE, Leak RK, Moore RY. The suprachiasmatic nucleus projects to posterior hypothalamic arousal systems. Neuroreport. 2001;12:435–40. doi: 10.1097/00001756-200102120-00048. [DOI] [PubMed] [Google Scholar]
- 12.Lu J, Zhang YH, Chou TC, et al. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J Neurosci. 2001;21:4864–74. doi: 10.1523/JNEUROSCI.21-13-04864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;23:10691–702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deurveilher S, Semba K. Indirect projections from the suprachiasmatic nucleus to major arousal-promoting cell groups in rat: implications for the circadian control of behavioural state. Neuroscience. 2005;130:165–83. doi: 10.1016/j.neuroscience.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci. 2001;4:732–8. doi: 10.1038/89522. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida K, McCormack S, Espana RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2006;494:845–61. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deboer T, Overeem S, Visser NA, et al. Convergence of circadian and sleep regulatory mechanisms on hypocretin-1. Neuroscience. 2004;129:727–32. doi: 10.1016/j.neuroscience.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Zeitzer JM, Yoshida Y, et al. Lesions of the suprachiasmatic nucleus eliminate the daily rhythm of hypocretin-1 release. Sleep. 2004;27:619–27. doi: 10.1093/sleep/27.4.619. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida Y, Fujiki N, Nakajima T, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–81. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- 21.Gerashchenko D, Kohls MD, Greco M, et al. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci. 2001;21:7273–83. doi: 10.1523/JNEUROSCI.21-18-07273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–51. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 23.Hara J, Beuckmann CT, Nambu T, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–54. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 24.Chou TC, Lee CE, Lu J, et al. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumann CR, Clark EL, Pedersen NP, Hecht JL, Scammell TE. Do enteric neurons make hypocretin? Regul Pept. 2008;147:1–3. doi: 10.1016/j.regpep.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T, Scammell TE. Behavioral state instability in orexin knock-out mice. J Neurosci. 2004;24:6291–300. doi: 10.1523/JNEUROSCI.0586-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scammell TE, Willie JT, Guilleminault C, Siegel JM. A consensus definition of cataplexy in mouse models of narcolepsy. Sleep. 2009;32:111–6. [PMC free article] [PubMed] [Google Scholar]
- 28.Fujiki N, Cheng T, Yoshino F, Nishino S. Specificity of direct transitions from wake to REM sleep in orexin/ataxin-3 narcoleptic mice. Sleep. 2006;29:A225. doi: 10.1016/j.expneurol.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Espana RA, McCormack SL, Mochizuki T, Scammell TE. Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep. 2007;30:1417–25. doi: 10.1093/sleep/30.11.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seroka CD, Johnson CE, Heideman PD. Variation in nocturnality and circadian activity rhythms between photoresponsive F344 and nonphotoresponsive Sprague Dawley rats. J Circ Rhythms. 2008;6:8. doi: 10.1186/1740-3391-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landry GJ, Yamakawa GR, Mistlberger RE. Robust food anticipatory circadian rhythms in rats with complete ablation of the thalamic paraventricular nucleus. Brain Res. 2007;1141:108–18. doi: 10.1016/j.brainres.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 32.Zhang S, Zeitzer JM, Sakurai T, Nishino S, Mignot E. Sleep/wake fragmentation disrupts metabolism in a mouse model of narcolepsy. J Physiol. 2007;581:649–63. doi: 10.1113/jphysiol.2007.129510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vitaterna MH, King DP, Chang AM, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–25. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S, Lin L, Kaur S, et al. The development of hypocretin (orexin) deficiency in hypocretin/ataxin-3 transgenic rats. Neuroscience. 2007;148:34–43. doi: 10.1016/j.neuroscience.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beuckmann CT, Sinton CM, Williams SC, et al. Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J Neurosci. 2004;24:4469–77. doi: 10.1523/JNEUROSCI.5560-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deboer T, Ruijgrok G, Meijer JH. Short light-dark cycles affect sleep in mice. Eur J Neurosci. 2007;26:3518–23. doi: 10.1111/j.1460-9568.2007.05964.x. [DOI] [PubMed] [Google Scholar]
- 37.Miller AM, Obermeyer WH, Behan M, Benca RM. The superior colliculus-pretectum mediates the direct effects of light on sleep. Proc Natl Acad Sci U S A. 1998;95:8957–62. doi: 10.1073/pnas.95.15.8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beersma DG, Dijk DJ, Blok CG, Everhardus I. REM sleep deprivation during 5 hours leads to an immediate REM sleep rebound and to suppression of non-REM sleep intensity. Electroencephalogr Clin Neurophysiol. 1990;76:114–22. doi: 10.1016/0013-4694(90)90209-3. [DOI] [PubMed] [Google Scholar]
- 39.Benington JH, Heller HC. REM-sleep timing is controlled homeostatically by accumulation of REM-sleep propensity in non-REM sleep. Am J Physiol. 1994;266:R1992–2000. doi: 10.1152/ajpregu.1994.266.6.R1992. [DOI] [PubMed] [Google Scholar]
- 40.Endo T, Schwierin B, Borbely AA, Tobler I. Selective and total sleep deprivation: effect on the sleep EEG in the rat. Psychiatry Res. 1997;66:97–110. doi: 10.1016/s0165-1781(96)03029-6. [DOI] [PubMed] [Google Scholar]
- 41.Shea JL, Mochizuki T, Sagvaag V, Aspevik T, Bjorkum AA, Datta S. Rapid eye movement (REM) sleep homeostatic regulatory processes in the rat: changes in the sleep-wake stages and electroencephalographic power spectra. Brain Res. 2008;1213:48–56. doi: 10.1016/j.brainres.2008.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–38. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wurts SW, Edgar DM. Circadian and homeostatic control of rapid eye movement (REM) sleep: promotion of REM tendency by the suprachiasmatic nucleus. J Neurosci. 2000;20:4300–10. doi: 10.1523/JNEUROSCI.20-11-04300.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carskadon MA, Dement WC. Sleepiness and sleep state on a 90-min schedule. Psychophysiology. 1977;14:127–33. doi: 10.1111/j.1469-8986.1977.tb03362.x. [DOI] [PubMed] [Google Scholar]
- 45.Estabrooke IV, McCarthy MT, Ko E, et al. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–62. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–20. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–98. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi K, Lin JS, Sakai K. Neuronal activity of orexin and non-orexin waking-active neurons during wake-sleep states in the mouse. Neuroscience. 2008;153:860–70. doi: 10.1016/j.neuroscience.2008.02.058. [DOI] [PubMed] [Google Scholar]
- 49.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–4. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eriksson KS, Sergeeva OA, Selbach O, Haas HL. Orexin (hypocretin)/dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur J Neurosci. 2004;19:1278–84. doi: 10.1111/j.1460-9568.2004.03243.x. [DOI] [PubMed] [Google Scholar]
- 51.Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- 52.Fuller PM, Saper CB, Lu J. The pontine REM switch: past and present. J Physiol. 2007;584:735–41. doi: 10.1113/jphysiol.2007.140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8:302–30. doi: 10.1016/j.sleep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 54.Thorpe AJ, Kotz CM. Orexin A in the nucleus accumbens stimulates feeding and locomotor activity. Brain Res. 2005;1050:156–62. doi: 10.1016/j.brainres.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 55.Shirasaka T, Nakazato M, Matsukura S, Takasaki M, Kannan H. Sympathetic and cardiovascular actions of orexins in conscious rats. Am J Physiol. 1999;277:R1780–5. doi: 10.1152/ajpregu.1999.277.6.R1780. [DOI] [PubMed] [Google Scholar]
- 56.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–9. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 57.Boutrel B, Kenny PJ, Specio SE, et al. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102:19168–73. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lubkin M, Stricker-Krongrad A. Independent feeding and metabolic actions of orexins in mice. Biochem Biophys Res Commun. 1998;253:241–5. doi: 10.1006/bbrc.1998.9750. [DOI] [PubMed] [Google Scholar]
- 59.Scammell TE. Wakefulness: an eye-opening perspective on orexin neurons. Curr Biol. 2001;11:R769–71. doi: 10.1016/s0960-9822(01)00466-3. [DOI] [PubMed] [Google Scholar]
- 60.Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–81. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]