

Summary
Gene valD, encodes a large Vicinal Oxygen Chelate (VOC) superfamily protein, has been identified in the validamycin biosynthetic gene cluster. Inactivation of valD significantly reduced validamycin A production, which was fully restored with the full-length valD and partially restored with either N-terminal or C-terminal half by complementation. Heterologously expressed ValD catalyzed the epimerization of 2-epi-5-epi-valiolone to 5-epi-valiolone. This metalloenzyme is a homodimer with a metal ion-binding ratio of 0.73 mol/mole protein towards Fe2+, Mn2+, Ni2+, and Zn2+. Individual and combined site-directed mutations of eight putative active site residues revealed that the N-terminal H44/E107 and the C-terminal H315/E366 are more critical for the activity than the internal H130, E183, H229, and E291. Our data has established ValD as one of the largest proteins of the VOC superfamily, catalyzing an alternative epimerization for C7N-aminocyclitol biosynthesis.
Introduction
The C7N-aminocyclitol family of natural products, exemplified by the α-glucosidase inhibitor acarbose, the trehalase inhibitors validamycin and salbostatin, the anti-tumor cetoniacytone A, and the antibacterial agent pyralomicin A, are increasingly gaining recognition due to their significant clinical and agricultural uses (Figure 1)(Mahmud, 2003). Validamycin A is produced industrially using developed strains of Streptomyces hygroscopicus var. limoneus or S. hygroscopicus subsp. jinggangensis 5008, and is widely used in Asia as a crop protectant (Iwasa et al., 1970; Xia and Jiao, 1986). Structurally, it consists of the unsaturated aminocyclitol unit valienamine, the saturated aminocyclitol unit validamine, and glucose. Through feeding experiments with synthesized precursors and/or in vitro enzymatic catalysis, it has been established that the valienamine biosynthesis is initiated by the conversion of sedoheptulose 7-phosphate to 2-epi-5-epi-valiolone, which undergoes further different modifications to give final diverse products (Choi et al., 2008; Dong et al., 2001; Mahmud et al., 1999; Naganawa et al., 2002; Wu et al., 2007). By comparing the stereochemistry of the final cyclitol moiety and the first intermediate 2-epi-5-epi-valiolone, an epimerization reaction is found to be required at the C-2 position in at least four out of the five abovementioned C7N-aminocyclitols.
Figure 1.
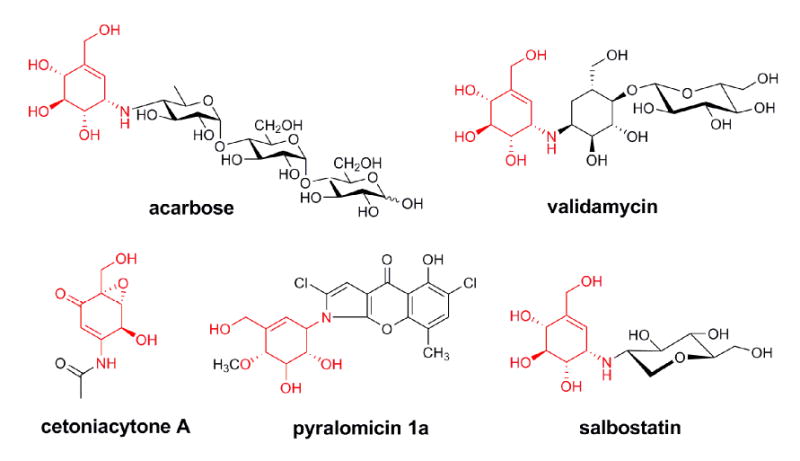
Chemical structures of bioactive C7N-aminocyclitols. The shared valienamine moiety is in red.
Recently, the biosynthetic gene clusters of acarbose, validamycin, salbostatin, and cetoniacytone A have been cloned and sequenced (Choi et al., 2008; Singh et al., 2006; Stratmann et al., 1999; Wu et al., 2007; Yu et al., 2005). In the first isolated acarbose biosynthetic gene cluster from Actinoplanes sp. SE50/110, the AcbO protein was found to have no similarity with any known proteins in the database and was initially predicted to be an epimerase (Zhang et al., 2002). After phosphorylation of 2-epi-5-epi-valiolone by AcbM, subsequent conversion of 2-epi-5-epi-valiolone 7-phosphate to 5-epi-valiolone 7-phosphate catalyzed by AcbO unambiguously established its role as the first epimerase involved in C7N-aminocyclitol biosynthesis (Zhang et al., 2003). Moreover, SalO from the salbostatin biosynthetic gene cluster in S. albus ATCC 21838 showed a 61% identity with AcbO, and was proposed to be responsible for the epimerization of 2-epi-5-epi-valiolone 7-phosphate to 5-epi-valiolone 7-phosphate in salbostatin biosynthesis (Choi et al., 2008).
However, so far no AcbO homolog has been found in the biosynthetic gene clusters of validamycin and cetoniacytone A (Accession No. EF120454) (Bai et al., 2006). Moreover, 2-epi-5-epi-valiolone was significantly incorporated into validamycin but not able to be phosphorylated by the required kinase ValC for validamycin biosynthesis (Dong et al., 2001; Minagawa et al., 2007), suggesting a direct conversion of unmodified 2-epi-5-epi-valiolone to 5-epi-valiolone, catalyzed possibly by a different epimerization reaction. In silico analysis of genes within the validamycin cluster revealed a gene (valD), located downstream of the kinase gene valC, that encodes a protein similar to members of the Vicinal Oxygen Chelate (VOC) superfamily (Bai et al., 2006). The VOC superfamily includes a set of structurally related proteins that are able to catalyze a large range of divalent metal-ion-dependent reactions. Members of this family include the glyoxalases I (GLO)(Cameron et al., 1997), the extradiol dioxygenases (DHBD)(Eltis and Bolin, 1996), the bleomycin resistance proteins (BRP)(Dumas et al., 1994), the fosfomycin resistance proteins (FOS)(Pakhomova et al., 2004), and the methylmalonyl-CoA epimerases (MMCE) involved in the epimerization of (2S)-methylmalonyl-CoA to its (2R)-stereoisomer (Leadlay, 1981).
In this paper, we report the requirement of ValD for validamycin biosynthesis through in vivo inactivation and complementation, and provide evidence that ValD is a large protein of the VOC superfamily with a novel catalytic activity responsible for the conversion of 2-epi-5-epi-valiolone to 5-epi-valiolone.
Results
Inactivation of valD in S. hygroscopicus 5008 Drastically Reduced the Production of Validoxylamine A and Validamycin A
To assess the role of valD in validamycin biosynthesis, a 1278-bp internal DNA fragment, coding for 426 aa, was replaced by the 1.4-kb aac(3)IV cassette. This was achieved by using a pHZ1358-derived plasmid pJTU713, in which aac(3)IV was inserted between a 4.8-kb left flanking and a 1.7-kb right flanking sequence of the 1278-bp DNA fragment to be replaced (Figure 2A). pJTU713 was introduced into wild-type S. hygroscopicus 5008 through conjugation (Yu et al., 2005) and two thiostrepton-sensitive apramycin-resistant derivatives (ZYR-4-1 and ZYR-4-2) were obtained. Total DNA from these two strains and from the wild-type 5008 was used as templates for PCR analysis using two primers, ValD-Det-F and ValD-Det-R. The wild-type 5008 DNA gave a 1.6-kb PCR product, while the mutants gave a 1.7-kb PCR product, which confirmed that a 1278-bp DNA fragment internal to valD has been replaced by the 1.4-kb aac(3)IV cassette (Figure 2B). HPLC analysis of the culture broths of ZYR-4-1 and ZYR-4-2 revealed that the production of validamycin A in the two valD mutants was drastically reduced to less than 5% of the wild-type production level, indicating the important role of ValD in validamycin biosynthesis (Figure 2C).
Figure 2.
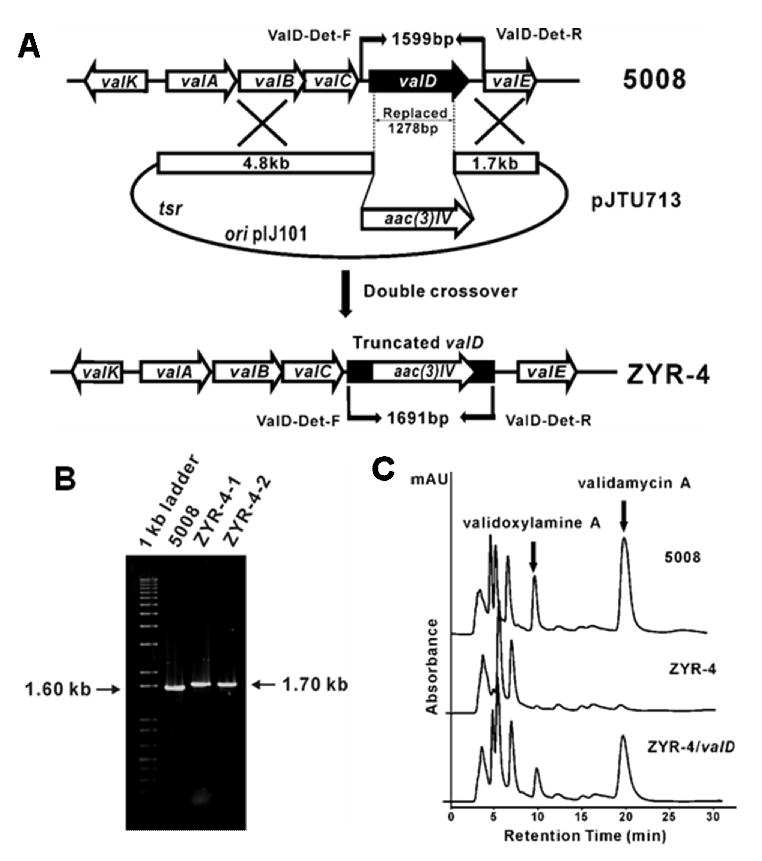
Construction and analysis of valD inactivated mutant strains of S. hygroscopicus 5008. A, Schematic representation for the replacement of a 1278-bp fragment of valD with apramycin resistance gene aac(3)IV. In shuttle plasmid pJTU713, aac(3)IV was inserted between 4.80-kb and 1.70-kb genomic fragments originally flanking the deleted 1278-bp region. While wild-type 5008 should give a 1.60-kb expected PCR-amplified product, mutant ZYR-4 should yield a 1.70-kb product using a pair of primers (ValD-Det-F and ValD-Det-R). B, Gel electrophoresis of PCR products of the wild-type and the mutant strains. C, HPLC analysis of the wild-type (5008), the valD mutant strain (ZYR-4), and the valD mutant strain complemented with a plasmid harboring an intact valD gene (ZYR-4/valD).Validamycin A and validoxylamine A are indicated with filled arrows.
To further confirm that ValD is instrumental in validamycin biosynthesis, the PCR-amplified valD gene was cloned downstream of PermE* promoter and subsequently introduced into a valD mutant ZYR-4-1 by conjugation. HPLC analysis of the fermentation extracts from the resulting S. hygroscopicus ZYR-4 complemented with valD clearly demonstrated that validoxylamine A and validamycin A production was restored to about 80% of that of the wild-type (Figure 2C).
Enzymatic Epimerization of 2-epi-5-epi-valiolone to 5-epi-valiolone by Heterologously Expressed ValD
To explore the exact biochemical function of ValD, the gene was amplified by PCR and cloned into expression vector pRSET B and introduced into E.coli BL21(DE3)pLysS. Expression of ValD was induced by IPTG to give a 51.8 kDa soluble His6-tagged protein, which was then purified on a BD TALON™ affinity column.
Characterization of ValD was carried out using the coupled reactions of ValA and ValD with sedoheptulose 7-phosphate as substrate. ValA is a 2-epi-5-epi-valiolone synthase that catalyzes the conversion of sedoheptulose 7-phosphate to 2-epi-5-epi-valiolone (Yu et al., 2005). The reaction was carried out for 3-6 h in the presence of NAD+ and Co2+, and the product was checked by TLC (Figure 3A). The results clearly demonstrated that sedoheptulose 7-phosphate was converted into 2-epi-5-epi-valiolone with ValA alone, and in the presence of ValD, this product was further converted into 5-epi-valiolone.
Figure 3.
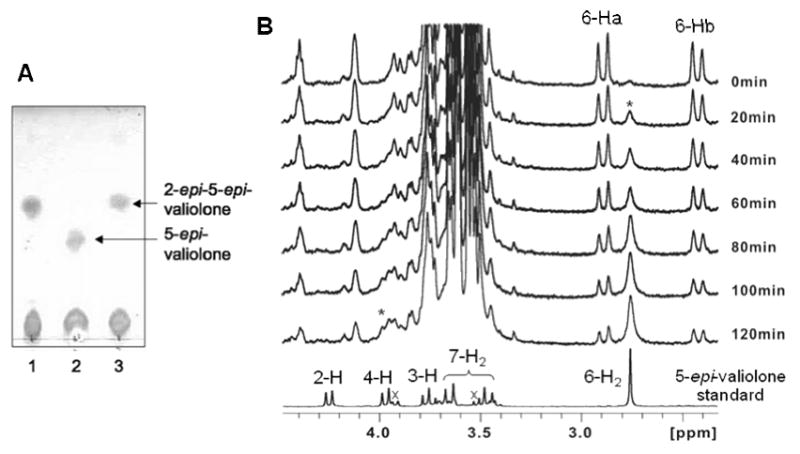
TLC and 1H NMR analyses of ValD-catalyzed reactions. A, TLC analysis of ValD-catalyzed reactions: lane 1, sedoheptulose 7-phosphate with 2-epi-5-epi-valiolone synthase ValA only; lane 2, sedoheptulose 7-phosphate with ValA and ValD; lane 3, sedoheptulose 7-phosphate with ValA and boiled ValD. B, 1H NMR analyses of ValD reaction. A solution of 2-epi-5-epi-valiolone in D2O was placed in a 5 mm NMR tube and the reaction was started by adding potassium phosphate buffer (pH 7.4, 25 mM) and purified ValD to a final concentration of 0.018 mg/ml. After one hour, additional ValD was added into the reaction mixture to a final concentration of 0.054 mg/ml protein. The geminal H-6a and H-6b of 2-epi-5-epi-valiolone appear as doublets, whereas those of 5-epi-valiolone appear as a singlet. * indicates the growing signal corresponding to the increased production of 5-epi-valiolone over the period of NMR measurement; × indicates signals of impurities.
To further identify this conversion, a time-course 1H NMR experiment was conducted. However, as the activity of ValA requires NAD+ and Co2+ cofactor, a direct 1H NMR experiment resulted in an unsatisfactory outcome due to perturbation of the spectrum by cobalt. Therefore, 2-epi-5-epi-valiolone was first enzymatically synthesized by a coupled enzyme reaction using transketolase (EC 2.2.1.1) (Zhang et al., 2002) and ValA. A decreased intensity of the proton signals (Ha-6, Hb-6) of 2-epi-5-epi-valiolone and an increased intensity of those of 5-epi-valiolone (H-4, H-6) were observed in the NMR spectra, confirming the function of ValD as a 2-epi-5-epi-valiolone epimerase. In addition, the lack of H-2 signal of the product indicates that the proton at this position has been replaced by deuterium, which confirmed the epimerization occurs at the C-2 position (Figure 3B).
Dimerization and Metal Ion Binding of ValD
In silico comparison and secondary structure analyses of ValD revealed that the protein contains two metal-binding sites built up from four βαβββ modules common to the VOC superfamily (Figure 4). Most members of the VOC superfamily exist as homodimers; each monomer contains two βαβββ modules that form a single metal-binding/active site (Bergdoll et al., 1998). While each has a different metal preference, members of the VOC superfamily share almost identical metal-binding sites consisting of four conserved residues, including the two Glu residues for catalysis. Sequence comparisons of ValD with CetB, a homologous protein from the cetoniacytone pathway (Wu et al., 2007), and MMCE from Propionibacterium shermanii showed that all of these proteins contain the conserved residues (Figure 4). Because the ValD primary sequence contains two repeating and highly homologous glyoxalase-like units, both the N-terminal half (G35-E194) and the C-terminal half (G220-A382) of the protein were individually analyzed as a separate entry in the alignment.
Figure 4.
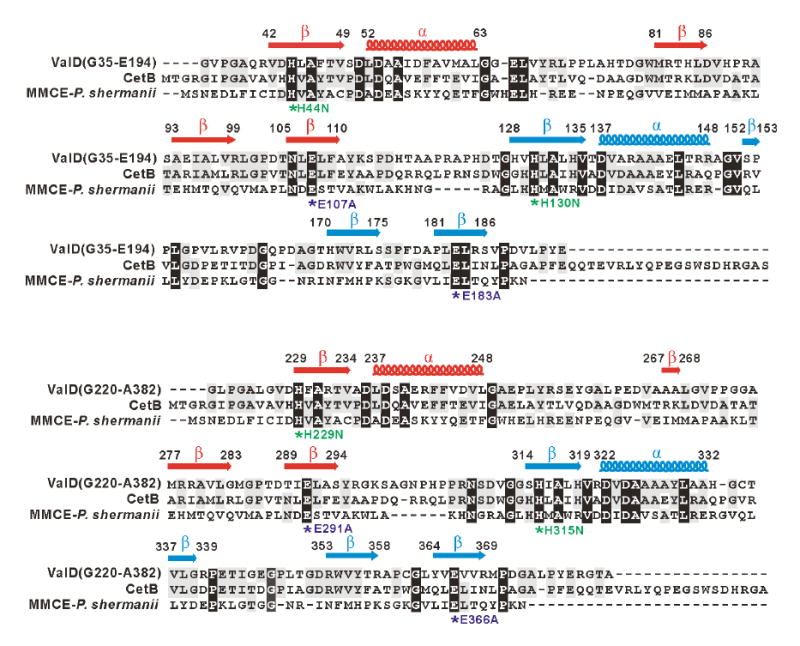
Amino acid alignments of the N-terminal half (G35-E194) and C-terminal half (G220-A382) of ValD with CetB and methylmalonyl-CoA epimerase and secondary structure predictions. Deduced amino acid sequences are from the following organisms: CetB (183 aa), Actinomyces sp. Lu 9419 (Accession No. EF120454); MMCE (148 aa), Propionibacterium shermanii (Accession No. AAL57846). Based on the analysis of the three-dimensional structure of the MMCE from Propionibacterium shermanii, a functional attribution of the aa residues probably involved in metal-ion binding and/or catalysis is indicated as asterisks below the alignment. In ValD mutants, conserved His residues are individually replaced with Asn, whereas conserved Glu residues are individually changed to Ala. The secondary structure of ValD was predicted using Jpred, and β sheets are labeled as arrows and α-helix are labeled as coils above the alignment.
Similar to ValD, several members of the VOC superfamily, e.g., large glyoxalases I from Saccharomyces cerevisiae (accession number CAC16163) and large extradiol dioxygenase from Pseudomonas cepacia (accession number 1HAN_A), also have four βαβββ modules. However, most of them, if not all, adopt a monomer conformation. To find out whether ValD forms a monomer or a dimer, the protein was purified and analyzed with a DynaPro™ dynamic light scattering instrument. The protein gave a radius of 4.0 nm and the calculated molecular weight is 83.5 kD based on the standard curve, and 103 kD based on the volume shape hydration model, both of which suggested that ValD forms a dimer. The result is consistent with that obtained from high performance gel filtration chromatography (see Figure S1).
To investigate the metal ion cofactor of ValD, inductively-coupled plasma atomic emission spectrometry (ICP-AES) was used to analyze samples of freshly purified Histag-free ValD for the presence of bound zinc, manganese, cobalt, nickel, iron, magnesium, calcium, or copper. The results indicated the presence of iron (0.24 mol), manganese (0.22 mol), nickel (0.15 mol), and zinc (0.12 mol) per mole of protein, which suggests that ValD, similar to MMCE and GLO, can bind a number of different metal ions (Clugston et al., 2004; Leadlay, 1981). However, the total amount of metals was consistently about 0.73 mol/mole protein, suggesting that the two putative metal binding sites in ValD are not fully occupied by metal ions and/or some of the protein present in its apo-form. In addition, the Histag-free protein was treated with 0.5 mM EDTA for 20 min to remove the metal ion, and dialyzed against metal ion-free buffer. Strong binding between ValD and metal ions was observed with the presence of iron (0.15 mol), manganese (0.22 mol), nickel (0.14 mol), and zinc (0.10 mol) per mole of protein by ICP-AES analysis of the EDTA-treated Histag-free ValD. The EDTA treatment slightly reduced the total amount of metals to 0.61 mol/mole protein mostly through the chelation of iron. In another metal ion depletion attempt with 0.1 mM 1,10-phenanthroline, the treated His6-tagged ValD still retained an epimerase activity when tested using 2-epi-5-epi-valiolone as substrate.
The N-terminal or C-terminal Half of ValD Retained Reduced Epimerization Activity
To investigate whether each individual half of ValD would independently have the same function as the full-length ValD, a complementation experiment was carried out through the introduction of plasmid pJTU946, a shuttle plasmid harboring the recombinant N-terminal half of valD under the control of the PermE* promoter, into ZYR-4 through conjugation. Validamycin production was partially restored ca. 20% of that of the wild-type. A similar complementation experiment with the C-terminal half of ValD was also performed, resulting in ca. 20% of validamycin production as well (Figure 5A). These results are consistent with those of the in vitro enzymatic assays using recombinant N-terminal half and C-terminal half of ValD, showing that the truncated proteins have a significantly reduced capacity to convert 2-epi-5-epi-valiolone to 5-epi-valiolone (data not shown). It is noteworthy that the recombinant N-terminal and C-terminal halves of ValD expressed poorly in E. coli and appeared to be less stable than the full-length protein.
Figure 5.
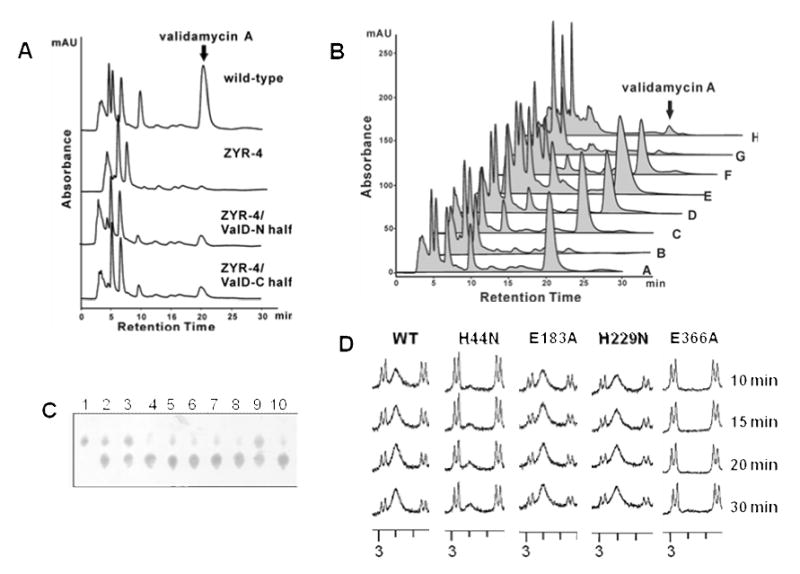
A, HPLC analysis of validamycin production in the wild-type strain, the valD disruption mutant ZYR-4, and ZYR-4 complemented with the N-terminal half and the C-terminal half of ValD. B, HPLC analysis of validamycin production in: (A) wild-type 5008; (B) valD disruption mutant ZYR-4;(C) ZYR-4 complemented with valD (H44N); (D) ZYR-4 complemented with valD (E184A); (E) ZYR-4 complemented with valD (H229N); (F) ZYR-4 complemented with valD (E366A); (G) ZYR-4 complemented with valD (H44N/H229N); (H) ZYR-4 complemented with valD (E184A/E366A). Peaks corresponding to validamycin A are indicated. C, partial TLC of enzyme assays with single-residue mutated ValD after 30 min of incubations. Lane 1, 2-epi-5-epi-valiolone standard; lanes 2 – 9, reactions with mutated proteins: H44N (2); E107A (3); H130N (4); E183A (5); H229N (6); E291A (7); H315N (8); E366A (9); 10, reaction with the wild-type ValD. D, Partial 1H NMR spectra of enzyme assays with a selected number of mutated ValD.
Investigation on the Involvement of Conserved Residues in Validamycin Production and Epimerization Through Site-directed Mutation
To investigate the effect of the conserved residues in ValD on its catalytic activity, a number of site-directed mutants of ValD were generated, and their in vivo activities were initially analyzed by complementation experiments. As shown in Figure 4, multiple sequence alignment of ValD and its homologs suggested that H44, E107, H130, and E183 are the putative active site residues in the N-terminal half, and H229, E291, H315, and E366 are the putative active site residues in the C-terminal half of ValD. Initially, only two His residues specific for metal ion binding and two Glu residues for catalysis were individually altered – H44 and H229 were replaced by Asn and E183 and E366 were replaced by Ala – to give single-residue mutants H44N, E183A, H229N, and E366A. Also, combined mutations of H44N and H299N were performed to remove the two putative metal binding residues, and combined mutations of E183A and E366A were carried out to eliminate the two putative catalytic residues, resulting in double-site mutants H44N/H299N and E183A/E366A. All mutants were individually cloned into the vector pPM927 and were introduced into ZYR-4 by conjugation. The production of validamycin A as a result of the complementation experiments was analyzed by HPLC. Surprisingly, all of the four mutated ValD proteins tested (H44N, E183A, H299N, and E366A) could restore the validamycin production of ZYR-4 to a comparable level of the wild-type, whereas the H44N/H299N and the E183A/E366A ValD proteins were only able to restore low levels of validamycin production (Figure 5B).
As results of the in vivo complementation experiments were inconclusive, we set out to characterize the in vitro catalytic activity of the mutated ValD. In this experiment, all eight putative active site residues were individually altered – His was replaced by Asn and Glu was replaced by Ala – to give single-residue mutants H44N, E107A, H130N, E183A, H229N, E291A, H315N, and E366A. The mutants were produced as His6-tagged proteins and their epimerase activity was tested using 2-epi-5-epi-valiolone as substrate. As kinetic studies of these proteins were hampered by the lack of feasible analytical methods, we chose to monitor the relative catalytic activity of the proteins using TLC and 1H NMR. The results, as monitored by TLC, indicated that the H130N, E183A, H229N, and E291A proteins had a comparable epimerase activity to that of the wild-type, whereas the H44N, E107A, and E366A proteins had significantly reduced activities (Figure 5C). Mutation at H315 appeared to have moderate effect on the epimerase activity. Comparisons of the epimerase activity of H44N, E183A, H229N, and E366A by 1H NMR experiments (Figure 5D) confirmed that the H229N and the E183A single mutation proteins retain an epimerase activity comparable to the wild-type enzyme, whereas the H44N and the E366A proteins had significantly reduced activities, consistent with the TLC results. The results suggested that four (the N-terminal residues H44 and E107 and the C-terminal residues H315 and E366) of the eight putative active site residues in ValD may play more significant roles in its catalytic activity, whereas the other four (the inner residues H130, E183, H229, and E291) may contribute less to its activity.
Discussion
Previously, valD had been considered to be a gene of unknown function with no direct involvement in the biosynthesis of validamycin. The encoded protein is similar to the glyoxalase/bleomycin resistance proteins; however, no clear function of this protein could be envisioned in the proposed validamycin pathway. In addition, heterologous expression of a cassette harboring a selected number of the val genes, but without valD, in S. lividans 1326 resulted in the production of validamycin A, albeit in minute amounts and just detectable by mass spectrometry (Bai et al., 2006). It was not until more recently that the wide distribution of valD homologs in other C7-aminocyclitol/C7-cyclitol biosynthetic pathways, e.g., in the cetoniacytone and BE-40644 pathways, became obvious (Wu et al., 2007). Results of in silico comparisons of these clusters suggested that they may share the first two biochemical steps: cyclization of sedoheptulose 7-phosphate and epimerization of the product to yield 5-epi-valiolone, and the latter step might be catalyzed by the glyoxalase/bleomycin resistance protein-like enzyme, such as ValD. In this study, the involvement of ValD in validamycin biosynthesis has been unambiguously demonstrated by gene inactivation in S. hygroscopicus 5008. ValD was further confirmed to be a 2-epi-5-epi-valiolone epimerase by in vitro characterization of the recombinant protein, site-directed mutations, and complementation experiments.
Despite the fact that ValD is directly involved in the biosynthesis of validamycin, mutant strains of S. hygroscopicus 5008 generated from valD inactivation still produce trace amounts of the antibiotic (Figure 2). It is possible that the conversion of 2-epi-5-epi-valiolone into 5-epi-valiolone may slowly occur non-enzymatically, as trace amounts of 5-epi-valiolone were occasionally detected in purified 2-epi-5-epi-valiolone samples. This low level non-enzymatic conversion may explain the production of a tiny amount of validamycin A detected by mass spectrometry in the heterologous experiments (without valD) in S. lividans 1326 (Bai et al., 2006). Alternatively, a VOC homolog may be present in S. lividans that accounts for the epimerase activity and subsequent production of validamycin A.
ValD is related to members of the divalent metal-ion-dependent Vicinal Oxygen Chelate (VOC) superfamily and shows its similarity to large GLOs and DHBDs, which are commonly present as monomers. However, ValD forms a dimer, making it one of the largest members of the VOC superfamily known to date. Each monomer contains four modules that correspond to two metal-binding sites. Site-directed mutagenesis of the individual putative active amino acid residues of ValD resulted in mutant proteins with various degrees of epimerase activity; the site-directed mutations at H44, E170, H315 and E366 reduced the activity of the epimerase, whereas mutations at H130, E183, H229, and E291 did not seem to affect the activity of the enzymes. Based on the four-module dimeric nature of ValD, the two monomers are probably arranged in anti-parallel. Moreover, the first two active residues H44 and E107 of one monomer may form a metal-binding site with the last two active H315 and E366 from the other monomer, creating two active peripheral metal-binding sites within the dimer.
Therefore, the epimerization reaction catalyzed by ValD is predicted to take place via a metal ion complex, involving deprotonation of 2-epi-5-epi-valiolone from one side of the complex by E107 or E366 to form an enolate intermediate, and re-protonation from the other side of the complex by E366 or E107 to give 5-epi-valiolone (see Figure S2).
Interestingly, in vivo complementation experiments with all single point mutation proteins resulted in full restoration of validamycin A production. Even though other unknown factor(s) probably involved in this build-up, it can be explained by the notion that even if some active site has been disrupted, the unaffected active sites may facilitate the production of validamycin A, albeit slowly, during the course of several days of fermentation to a comparable level of the wild-type production yield. However, when both putative active sites are disturbed, such as in the H44N/H299N and E183A/E366A mutants, the productivity of validamycin is severely compromised.
In summary, most C7N-cyclitol-containing secondary metabolites are derived from a shunt pathway of the pentose phosphate pathway involving a novel sugar phosphate cyclase (AcbC, CetA, SalQ, ValA or their homologs), which mediates the cyclization of sedoheptulose 7-phosphate to 2-epi-5-epi-valiolone (Figure 6). In acarbose and salbostatin biosynthetic pathways, 2-epi-5-epi-valiolone is proven or suggested to be converted to 5-epi-valiolone 7-phosphate through phosphorylation by AcbM or SalL first and then through epimerization by AcbO or SalO (Choi et al., 2008; Zhang et al., 2003). In validamycin biosynthesis, ValD establishes a branching point for valienamine biosynthesis through the alternative epimerization of 2-epi-5-epi-valiolone to 5-epi-valiolone, which is probably shared by CetB in cetoniacytone A biosynthesis. With the identification of additional C7N-aminocyclitols and the cloning of their biosynthetic gene clusters, further biosynthetic diversity for this class of natural products is expected.
Figure 6.
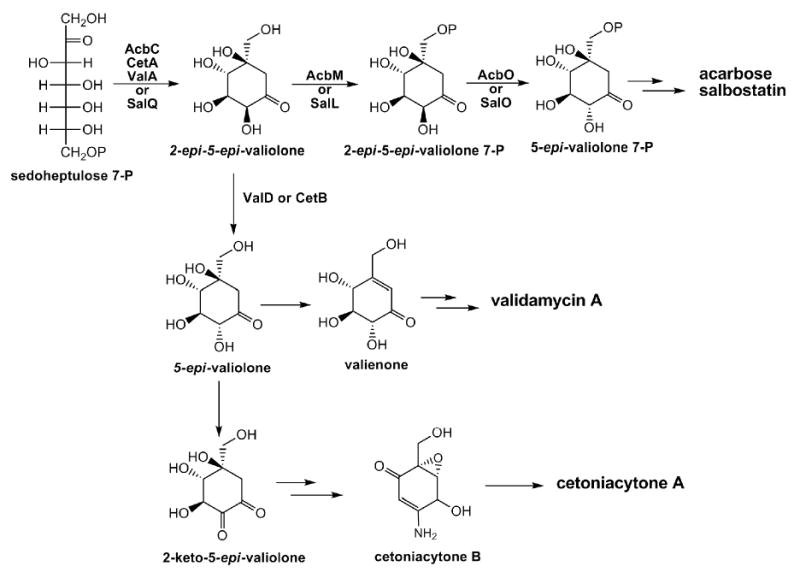
Proposed biosynthetic pathways to the valienamine moiety of C7N-aminocyclitol acarbose (Acb), salbostatin (Sal), validamycin A (Val), and cetoniacytone A (Cet).
Significance
Well-studied biosynthetic mechanisms are crucial for increasing the validamycin productivity and generating new validamycin derivatives with better biological activities using biosynthetic approaches. The data presented here provide new insights into the biosynthesis of validamycin, particularly the catalytic function of ValD, which catalyzes the conversion of free 2-epi-5-epi-valiolone to 5-epi-valiolone and represents an alternative epimerization involved in C7N-aminocyclitol biosynthesis. The dimeric ValD is one of the largest proteins found in the Vicinal Oxygen Chelate (VOC) superfamily, and displays a unique distribution of active site residues as revealed by site-directed mutation. It also shows a strong binding capacity towards several metal ions. Therefore in addition to the requirement of cobalt by the cyclase ValA, the presence of metalloenzyme ValD suggests possible productivity enhancement through metal ion optimization for validamycin. The characterization of ValD also enriches the still limited enzymatic toolbox for C7N-aminocyclitol biosynthetic pathway engineering. Furthermore, degenerate primers could be designed based on ValD/CetB and used for the screening of more validamycin-type biosynthetic gene clusters, whereas AcbO/SalO could be used for the screening of acarbose-type biosynthetic gene clusters.
Experimental Procedures
Bacterial Strains and Plasmids
Bacterial strains and plasmids used in this study are listed in Tables S1 and S2. S. hygroscopicus 5008, the wild-type producer of validamycin, was used for validamycin isolation, bioassay, and generation of mutant strains. E. coli DH10B was used as cloning host, and E. coli BW25113 was used as the host for λ-Red-mediated recombination (Gust et al., 2003). pBlueScript KS(-) or pMD-T vector (Takara) was used for cloning of PCR fragments. pHZ1358 (an E. coli and Streptomyces shuttle vector) was used for gene replacement in 5008 (Sun et al., 2002). pJTU968 (a pRSET B derivative with PermE* promoter and a polylinker) was used as a transitional vector. pPM927 was used for mutant complementation (Smokvina et al., 1990).
Reagents and General Procedures
Restriction enzymes, T4 DNA ligase, Taq polymerase and KOD-plus polymerase were purchased from various companies (New England Biolabs, Takara, MBI Fermentas, Toyobo, Shanghai Sangon). Synthesis of oligonucleotide primers and DNA sequencing of PCR products were performed by Shanghai Sangon and Invitrogen Co., Ltd. Primers used are listed in Table S3. Gel Recovery Kit (Tiangen) was used for DNA purification from agarose gels. Stratagene QuickChange II XL Site-Directed Mutagenesis Kit was used for site-directed mutagenesis. TSBY liquid medium (per liter: TSB 30 g, yeast extract 10 g, sucrose 103 g, pH 7.2) was used for growth of mycelia and isolation of total DNA. SFM medium (2% agar, 2% mannitol, 2% soybean powder, pH 7.2) was used for sporulation and conjugation. ISP2 liquid medium (0.4% yeast extract, 1% malt extract, 0.4 % glucose, pH 7.3) was used for the fermentation of S. hygroscopicus 5008 and its derivatives. For Streptomyces, apramycin and thiostrepton were used at 30 mg/ml and 25 mg/ml, respectively, both in SFM agar and in liquid media. E. coli strains were cultured as described (Sambrook et al., 1989).
Construction of pJTU713 for Targeted Replacement of a 1278-bp DNA Fragment Internal to valD
A 7.8-kb BclI fragment from pHZ2239 was cloned into the BamHI site of pHZ1358 to generate pJTU712. Using the gel-purified 1384-bp EcoRI/HindIII fragment from pIJ773 as template (Gust et al., 2003), a 1.4-kb disruption cassette was obtained by PCR amplification with the primer pairs ValD-PCR-F and ValD-PCR-R (Table S3). Amplification was performed with Taq DNA polymerase in a 50 μl reaction with 100 ng template DNA, 10 mM dNTPs, 50 pmol each primer, and 5% DMSO in a thermocycler (BioRad). After initial denaturation at 94°C for 2 min, 10 cycles were performed with denaturation at 94°C for 45 s, annealing at 50°C for 45 s, and extension at 72°C for 90 s, followed by 15 cycles with the annealing temperature increased to 55°C. A last elongation step was done at 72°C for 5 min. This fragment was used to replace the 1278-bp DNA internal to valD in pJTU712 using the λ-Red-mediated recombination. The resulting plasmid pJTU713 was used for targeted replacement of a 1278-bp DNA fragment internal to valD with the 1.4-kb aac(3)IV cassette in the wild-type strain 5008. The two oligonucleotide primers used for valD mutant (ZYR-4) confirmation were ValD-Det-F and ValD-Det-R. PCR amplification was performed with Taq DNA polymerase under similar conditions. The PCR product was analyzed by gel electrophoresis and enzymatic digestion.
Construction of pJTU926 for the Complementation of ZYR-4 with Full-length Recombinant valD
A 1.4-kb NdeI/EcoRI fragment from the expression plasmid harboring a full-length valD was ligated to the NdeI/EcoRI-digested pJTU968 to give pJTU925. Then the 1.7-kb MfeI/EcoRI fragment, containing PermE* promoter and valD, was cleaved from pJTU925 and ligated into EcoRI-digested pPM927 to give pJTU926. The plasmid was introduced into ZYR-4 through conjugation as previously described (Yu et al., 2005). The exconjugants were selected with thiostrepton.
Construction of Recombinant N-terminal Half of valD and Complementation of ZYR-4
The N-terminal half of valD gene encoding aa 1-194 of ValD was amplified by PCR with KOD-plus DNA polymerase using pJTU712 as template and primers ValD-N-F and ValD-N-R. The PCR amplification was carried out under the following conditions: initial denaturation at 94°C for 5 min, then 10 cycles with denaturation at 94°C for 1 min, annealing at 63°C for 45 s, and extension at 68°C for 2 min, followed by 20 cycles with denaturation at 94°C for 45 s, annealing at 60°C for 45 s, and extension at 68°C for 2min. A last elongation step was performed at 68°C for 5 min. The resultant PCR product was purified, digested with BamHI and EcoRI, and ligated into BamHI/EcoRI-digested pRSET B to generate pJTU942, which was sequenced to validate the inserted DNA sequence. A 0.6-kb NdeI/EcoRI fragment from pJTU942 harboring the N-terminal half of valD was ligated to the NdeI/EcoRI digested pJTU968 to give pJTU944. Subsequently, the 0.9-kb MfeI/EcoRI fragment was cleaved from pJTU944 and ligated into the EcoRI-digested pPM927 to give pJTU946. The plasmid was introduced into ZYR-4 through conjugation.
Construction of Recombinant C-terminal Half of ValD and Complementation of ZYR-4
The C-terminal half of valD encoding aa 220-451 was amplified by PCR with KOD-plus DNA polymerase with pJTU712 as template and primers ValD-C-F and ValD-C-R. The PCR amplification was carried under similar conditions for that of the N-terminal half of valD. The PCR products were digested with BamHI and EcoRI, and subsequently ligated into BamHI/EcoRI-digested pRSET B to generate pJTU943. The plasmid was sequenced to confirm the inserted DNA sequence. A 0.72-kb NdeI/EcoRI fragment from pJTU943 harboring the C-terminal half of valD was ligated to the NdeI/EcoRI-digested vector pJTU968 to give pJTU945. Then the 1.02-kb MfeI/EcoRI fragment was cleaved from pJTU945 and ligated into the EcoRI-digested pPM927 to give pJTU947, which was transferred into ZYR-4 through conjugation.
Site-directed Mutagenesis of ValD
The desired mutations were introduced by using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. The plasmid pValD (Table S2) was used as template and mutated primers (Table S3) were used in the PCR to give individual mutants H44N, E107A, H130N, E183A, H229N, E291A, H315N, and E366A. In order to generate H44N/H229N ValD, A 0.93-kb SacI/EcoRI fragment from pJTU3252 with the H44N mutation was ligated to the SacI/EcoRI-digested plasmid pJTU3250 with H229N mutation to give pJTU3262. Similarly, the E183A/E366A ValD was constructed. A 0.78-kb NcoI/EcoRI fragment from pJTU3253 with E366A mutation was ligated to the NcoI/EcoRI-digested plasmid pJTU3251 with E183A mutation to give pJTU3263. All single and double mutant forms of valD were confirmed by DNA sequencing.
Complementation of ZYR-4 with Single Site-directed Mutants of valD (H44N, E183A, H229N, or E366A)
The site-directed mutants of valD were cloned into the vector pPM927 in the same way as the full-length valD and were introduced into ZYR-4 by conjugation. The exconjugants were selected with thiostrepton and confirmed by PCR using the ValD-N-F and ValD-C-R as primers.
HPLC Analysis of Validamycin A
For HPLC analysis, the strains were cultured in 50 ml of YMG liquid medium in 250-ml baffled flasks at 37°C and 220 rpm for 6 days. The fermentation broth was acidified with oxalic acid and applied to a Dowex 50W column (25 ml) as previously reported (Dong et al., 2001). The extracts were loaded onto Agilent ZORBAX SB-C18 column (5 μm, 4.6 × 250 mm) for HPLC analysis (Waters 2690). The mobile phase (5 mM sodium phosphate buffer-methanol, 98:2, v/v) was applied with the flow rate of 0.6 ml/min at room temperature. The eluate was monitored at 210 nm with Waters 996 photodiode array detector and the data was analyzed with a Waters Millennium Chromatography Manager.
Cloning and Heterologous Expression of Recombinant His6-tagged ValD, N-terminal Half, and C-terminal Half of ValD
The valD gene was amplified with Platinum Pfx DNA polymerase (Invitrogen) using the cosmid clone 17F2 as template and primers valD-F and valD-R. The natural start codon (GTG) was replaced with ATG to create an NdeI site at the beginning of the gene. The PCR amplification was carried out in a thermocycler (Eppendorf) under the following conditions: 30 cycles of 45 s at 94°C, 45 s at 58°C, and 90 s at 72°C. The PCR products were digested with BamHI and EcoRI, and subsequently ligated into BamHI/EcoRI-digested pRSET B to generate pValD, which was verified by sequencing and introduced by heat-pulse transformation into E. coli BL21 Gold(DE3)pLysS (Stratagene). The transformants were grown in LBBS medium (50 ml) containing ampicillin (100 mg/ml) and chloramphenicol (25 mg/ml) at 37°C to an OD600 of 0.6. Isopropyl-β-D-thio-galactopyranoside (IPTG) was then added to a final concentration of 0.4 mM and the incubation was continued at 25°C for 24 h. The cells were harvested by centrifugation at 5,000 rpm for 10 min and stored frozen at -80°C until further use.
Preparation and Purification of His6-tagged ValD, N-terminal Half and C-terminal Half of ValD
Cells were thawed and resuspended in disruption buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0), the suspension was sonicated three times for 15 s each and cell debris was removed by centrifugation at 10,000 rpm for 7 min. The protein solution was applied to a BD TALONspin™ column (BD Biosciences) and centrifuged at 3,000 rpm for 1 min. The column was washed with washing buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0), twice, and washing buffer with 50 mM imidazole. The His6-tagged protein was eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 500 mM imidazole, pH 8.0) and dialyzed for 12 h against 1 liter of dialysis buffer (31 mM K2HPO4/KH2PO4 (pH 7.4) and 0.5 mM DTT). Protein concentration was measured by the Bradford protein micro-assay with bovine serum albumin as standard.
Enzymatic Assays
Coupled-enzyme assays were carried out using protein solutions of ValA (Yu et al., 2005) and ValD or mutated ValD (45 ml, 0.5 mg/ml) with sedoheptulose 7-phosphate (5 mM) as substrate in a 50 ml total volume containing 25 mM potassium phosphate buffer (pH 7.4), CoCl2 (1 mM), NaF (2 mM), and NAD+ (1 mM). The reactions were carried out at 30°C for 2 hours. For preliminary metal ion analysis, ValD was pretreated with 0.1 mM 1,10-phenanthroline and incubated with 2-epi-5-epi-valiolone (5 mM). After 2-h incubation, the protein was precipitated by methanol (50 μl) and centrifuged at 12,000 rpm for 10 min. The supernatant was collected and dried in vacuo. The product was analyzed by TLC (SiO2, nBuOH/EtOH/H2O 9:7:4) and stained with cerium- and molybdate-containing reagent.
Metal ion Analysis of ValD Using Inductively-coupled Plasma Optical Emission Spectrometry (ICP-OES)
Freshly purified His6-tagged ValD (4 mg) was digested with thrombin (4U) during dialysis against 20 mM Tris-HCl buffer (pH 7.5) overnight. The thrombin was removed with Thrombin CleanCleaveTM Kit (Sigma), and the Histag was then removed by passing the protein solution through nickel column. The purified Histag-free protein solution was split into two tubes, and one of them was treated with 0.5 mM EDTA (pH=8.0) for 20 min. Both samples were subsequently dialyzed against 20mM metal-ion-free Tris-HCl buffer (pH=7.5) prior to the metal ion analysis by ICP-OES (Iris Advangtage 1000, Thermo Electron) with a charge injector device (CID) detector. The dialysis buffer was used as negative control. Metal-ion-free buffers were prepared by extraction with a CHCl3 solution of dithizone (0.02%, w/v).
Synthesis of 2-epi-5-epi-valiolone and 5-epi-valiolone
2-epi-5-epi-Valiolone was synthesized enzymatically in a coupled reaction using transketolase (EC 2.2.1.1, Sigma) (Zhang et al., 2002) and ValA with ribose 5-phosphate and hydroxypyruvate as the initial substrates. The enzyme reaction was performed at 30°C in a total volume of 30 ml containing 0.5 units of transketolase (Sigma), 9 mg purified ValA, 10 mM hydroxypyruvate (Sigma), 10 mM ribose 5-phosphate (Sigma), 0.5 mM thiamine pyrophosphate (Sigma), 1 mM NAD+, 1 mM MgCl2, 0.025 mM CoCl2, 2 mM NaF, pH 7.6. The reaction was monitored by TLC. The mixture was then applied to Vivaspin 15R concentrator (VIVASCIENCE) with a membrane of 10,000 MWCO HY to remove the protein. The flow-through was lyophilized, dissolved in 1 ml of ddH2O, and subjected to a Dowex 1×8 (Cl- form, Sigma) column chromatography. The column was eluted with water and the fractions containing 2-epi-5-epi-valiolone were pooled and lyophilized. 5-epi-Valiolone was synthesized according to procedures reported previously (Mahmud et al., 2001).
Light Scattering Analysis of ValD
The protein was purified and analyzed by a DynaPro™ dynamic light scattering instrument with Dynamics Version 4.0 software at a concentration of 4 mg/ml. The detection was run at 4°C for 30 scans.
1H NMR Analysis of ValD Reaction
A solution of 2-epi-5-epi-valiolone (1 mg) in D2O (0.4 ml) was placed in a 5 mm NMR tube and the reaction was started by adding potassium phosphate buffer (pH 7.4, 25 mM) and purified ValD solution. The reaction was carried out at 25°C and 1H NMR measurements were performed under a water suppression condition in ten-minute intervals.
Supplementary Material
Acknowledgments
The authors thank Drs. Andrew P. Karplus, Patricia M. Flatt and Xiumei Wu for technical assistance and William Austin for metal ion analysis. Work at Oregon State University was supported by a grant from the National Institutes of Health (R01 AI061528). Work at Shanghai Jiao Tong University was supported by grants from the 973 and 863 Programs of the Ministry of Science and Technology, the Natural Science Foundation of China, and Shanghai Leading Academic Discipline Project B203. HX was in part supported by the Exchange Program from China Scholarship Council.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai L, Li L, Xu H, Minagawa K, Yu Y, Zhang Y, Zhou X, Floss HG, Mahmud T, Deng Z. Functional analysis of the validamycin biosynthetic gene cluster and engineered production of validoxylamine A. Chem Biol. 2006;13:387–397. doi: 10.1016/j.chembiol.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergdoll M, Eltis LD, Cameron AD, Dumas P, Bolin JT. All in the family: structural and evolutionary relationships among three modular proteins with diverse functions and variable assembly. Protein Sci. 1998;7:1661–1670. doi: 10.1002/pro.5560070801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AD, Olin B, Ridderstrom M, Mannervik B, Jones TA. Crystal structure of human glyoxalase I--evidence for gene duplication and 3D domain swapping. EMBO J. 1997;16:3386–3395. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Wu X, Choeng YH, Mahmud T, Jeong BC, Lee SH, Chang YK, Kim CJ, Hong SK. Genetic organization of the putative salbostatin biosynthetic gene cluster including the 2-epi-5-epi-valiolone synthase gene in Streptomyces albus ATCC 21838. Appl Microbiol Biotechnol. 2008;80:637–645. doi: 10.1007/s00253-008-1591-2. [DOI] [PubMed] [Google Scholar]
- Clugston SL, Yajima R, Honek JF. Investigation of metal binding and activation of Escherichia coli glyoxalase I: kinetic, thermodynamic and mutagenesis studies. Biochem J. 2004;377:309–316. doi: 10.1042/BJ20030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Mahmud T, Tornus I, Lee S, Floss HG. Biosynthesis of the validamycins: identification of intermediates in the biosynthesis of validamycin A by Streptomyces hygroscopicus var. limoneus. J Am Chem Soc. 2001;123:2733–2742. doi: 10.1021/ja003643n. [DOI] [PubMed] [Google Scholar]
- Dumas P, Bergdoll M, Cagnon C, Masson JM. Crystal structure and site-directed mutagenesis of a bleomycin resistance protein and their significance for drug sequestering. EMBO J. 1994;13:2483–2492. doi: 10.1002/j.1460-2075.1994.tb06535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltis LD, Bolin JT. Evolutionary relationships among extradiol dioxygenases. J Bacteriol. 1996;178:5930–5937. doi: 10.1128/jb.178.20.5930-5937.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa T, Yamamoto H, Shibata M. Studies on validamycins, new antibiotics. I. Streptomyces hygroscopicus var. limoneus nov. var., validamycin-producing organism. Jpn J Antibiot. 1970;23:595–602. [PubMed] [Google Scholar]
- Leadlay PF. Purification and characterization of methylmalonyl-CoA epimerase from Propionibacterium shermanii. Biochem J. 1981;197:413–419. doi: 10.1042/bj1970413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud T. The C7N aminocyclitol family of natural products. Nat Prod Rep. 2003;20:137–166. doi: 10.1039/b205561a. [DOI] [PubMed] [Google Scholar]
- Mahmud T, Tornus I, Egelkrout E, Wolf E, Uy C, Floss HG, Lee S. Biosynthetic studies on the alpha-glucosidase inhibitor acarbose in Actinoplanes sp.: 2-epi-5-epi-valiolone is the direct precursor of the valienamine moiety. J Am Chem Soc. 1999;121:6793–6983. [Google Scholar]
- Mahmud T, Xu J, Choi YU. Synthesis of 5-epi-[6-2H2]valiolone and stereospecifically monodeuterated 5-epi-valiolones: exploring the steric course of 5-epi-valiolone dehydratase in validamycin A biosynthesis. J Org Chem. 2001;66:5066–5073. doi: 10.1021/jo0101003. [DOI] [PubMed] [Google Scholar]
- Minagawa K, Zhang Y, Ito T, Bai L, Deng Z, Mahmud T. ValC, a new type of C7-Cyclitol kinase involved in the biosynthesis of the antifungal agent validamycin A. Chembiochem. 2007;8:632–641. doi: 10.1002/cbic.200600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa H, Hashizume H, Kubota Y, Sawa R, Takahashi Y, Arakawa K, Bowers SG, Mahmud T. Biosynthesis of the cyclitol moiety of pyralomicin 1a in Nonomuraea spiralis MI178-34F18. J Antibiot (Tokyo) 2002;55:578–584. doi: 10.7164/antibiotics.55.578. [DOI] [PubMed] [Google Scholar]
- Pakhomova S, Rife CL, Armstrong RN, Newcomer ME. Structure of fosfomycin resistance protein FosA from transposon Tn2921. Protein Sci. 2004;13:1260–1265. doi: 10.1110/ps.03585004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2nd. Cold Spring Harbor, NY: Cold Spring Harbor; 1989. [Google Scholar]
- Singh D, Seo MJ, Kwon HJ, Rajkarnikar A, Kim KR, Kim SO, Suh JW. Genetic localization and heterologous expression of validamycin biosynthetic gene cluster isolated from Streptomyces hygroscopicus var. limoneus KCCM 11405 (IFO 12704) Gene. 2006;376:13–23. doi: 10.1016/j.gene.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Smokvina T, Mazodier P, Boccard F, Thompson CJ, Guerineau M. Construction of a series of pSAM2-based integrative vectors for use in actinomycetes. Gene. 1990;94:53–59. doi: 10.1016/0378-1119(90)90467-6. [DOI] [PubMed] [Google Scholar]
- Stratmann A, Mahmud T, Lee S, Distler J, Floss HG, Piepersberg W. The AcbC protein from Actinoplanes species is a C7-cyclitol synthase related to 3-dehydroquinate synthases and is involved in the biosynthesis of the alpha-glucosidase inhibitor acarbose. J Biol Chem. 1999;274:10889–10896. doi: 10.1074/jbc.274.16.10889. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z. ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology. 2002;148:361–371. doi: 10.1099/00221287-148-2-361. [DOI] [PubMed] [Google Scholar]
- Wu X, Flatt PM, Schlorke O, Zeeck A, Dairi T, Mahmud T. A comparative analysis of the sugar phosphate cyclase superfamily involved in primary and secondary metabolism. Chembiochem. 2007;8:239–248. doi: 10.1002/cbic.200600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia TH, Jiao RS. Studies on glutamine synthetase from Streptomyces hygroscopicus var. jinggangensis. Sci Sin [B] 1986;29:379–388. [PubMed] [Google Scholar]
- Yu Y, Bai L, Minagawa K, Jian X, Li L, Li J, Chen S, Cao E, Mahmud T, Floss HG, et al. Gene cluster responsible for validamycin biosynthesis in Streptomyces hygroscopicus subsp. jinggangensis 5008. Appl Environ Microbiol. 2005;71:5066–5076. doi: 10.1128/AEM.71.9.5066-5076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Podeschwa M, Altenbach HJ, Piepersberg W, Wehmeier UF. The acarbose-biosynthetic enzyme AcbO from Actinoplanes sp. SE 50/110 is a 2-epi-5-epi-valiolone-7-phosphate 2-epimerase. FEBS Lett. 2003;540:47–52. doi: 10.1016/s0014-5793(03)00221-7. [DOI] [PubMed] [Google Scholar]
- Zhang CS, Stratmann A, Block O, Bruckner R, Podeschwa M, Altenbach HJ, Wehmeier UF, Piepersberg W. Biosynthesis of the C7-cyclitol moiety of acarbose in Actinoplanes species SE50/110. 7-O-phosphorylation of the initial cyclitol precursor leads to proposal of a new biosynthetic pathway. J Biol Chem. 2002;277:22853–22862. doi: 10.1074/jbc.M202375200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.