

Abstract
The mechanisms responsible for polysaccharide-induced immunological paralysis have remained unexplained almost a century after this phenomenon was first described. Cryptococcus neoformans capsular polysaccharides glucuronoxylomannan (GXM) and galactoxylomannan (GalXM) elicit little or no antibody responses. This study investigates the immunological and biological effects of GalXM in mice. GalXM immunization elicits a state of immunological paralysis in mice characterized by the disappearance of antibody-producing cells in the spleen. Immunological paralysis and lack of immunogenicity could not be overcome by immunization with GalXM conjugated to a protein carrier, Bacillus anthracis protective antigen. Additionally, immunization with GalXM in either complete or incomplete Freund's adjuvant was associated with spleen enlargement in Balb/c mice. Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) and flow cytometry revealed widespread apoptosis in the spleen after GalXM administration. Administration of a cocktail of Caspase-3 Inhibitor Z-DEVD-FMK and General Caspase Inhibitor Z-VAD-FMK or Fas-deficient mice abrogated the complete disappearance of antibody producing cells. Analysis of spleen cytokine expression in response to GalXM systemic injection revealed that GalXM down-regulated the production of inflammatory cytokines. Hence, we conclude that GalXM-induced immune paralysis is a result of specific B-cell depletion mediated by its pro-apoptotic properties in the context of widespread dysregulation of immune function.
Keywords: Fungal, Tolerance, Vaccination, B-cells
Introduction
The phrase immunological paralysis was first coined by Lloyd Felton in the early 20th century in reference to the inability of pneumococcal polysaccharide to elicit antibody responses in mice (1). The phenomenon was studied extensively with pneumococcal polysaccharide in the early to mid-20th century (1). The term paralysis was broadly defined as a condition produced in the host by an antigen given in such doses, that a subsequent immunization fails to elicit an immune response (1). Unfortunately, no complete satisfactory mechanism was identified, but two important explanations for the phenomenon were proposed, immunological paralysis and immunological unresponsiveness. Immunological paralysis was thought to be a consequence of the suppression of antibody formation at its source while immunological unresponsiveness was believed to result from neutralization of formed antibody by excess antigen (1). Later studies provided evidence suggesting that T-cells were implicated, despite the dogma that polysaccharides were T-cell-independent antigens (2). Given that antibody responses to polysaccharide antigens are often protective against encapsulated pathogens the phenomenon of immunological paralysis has great medical importance. In recent years, the development of polysaccharide-protein conjugate vaccines has emerged as an effective means to bypass the lack of responsiveness often associated with capsular antigens.
Cryptococcus neoformans is an encapsulated fungal pathogen that causes life-threatening infections in humans that usually manifests clinically as meningoencephalitis (3). Prior to the 1980's, cryptococcosis was relatively uncommon and was associated with the immunosuppression resulting from corticosteroid therapy or with lymphomas (4–6). However, as a result of the AIDS pandemic, the medical importance of this microbe has increased dramatically (7, 8). In addition to those with advanced HIV infection, other groups at risk are patients receiving immunosuppressive therapies for cancer and following transplants. One of the major virulence factors of C. neoformans is its capsule. The capsular polysaccharide is known to be composed of Glucuronoxylomannan (GXM), Galactoxylomannan (GalXM) andMannoproteins (MP) (7, 9, 10). The major capsular component on a mass basis is GXM and this polysaccharide has been shown to have numerous deleterious effects on host immunity including inhibiting leukocyte migration, inhibiting phagocytosis, inducing the expression of Fas Ligand (Fas L) in macrophages and producing a state of immunological unresponsiveness (8, 11–13). GalXM is composed of an α-(1,6)-galactan backbone with several branches of α-Man-(1–3) - αMan (1–4)-β-Gal- trisaccharide with variable amounts of β-(1–2) or β -(1–3)-xylose side groups (14). Light scattering measurements revealed that GalXM was on average approximately 20-fold smaller than GXM with an average mass of 1 × 105 Daltons. The radius of gyration (Rg) is the average distance between the center of the GalXM molecule to the outer edges and was determined as 95 nm in comparison to GXM which has a radius of gyration that depending on the strain can range from 68–208 nm (10). Since GalXM has a smaller molecular mass, the molar concentration of GalXM in polysaccharide that is shed could exceed that for GXM in C. neoformans exopolysaccharides (10).
Percolini et al., reported that GalXM inhibited peripheral blood mononuclear cells and T-cell proliferation, increased interferon-γ and interleukin-10 production and induced apoptosis of T-lymphocytes by DNA fragmentation through the activation of caspase 8. GalXM is able to induce Fas directly, making this a unique property of GalXM and not of GXM (15). GalXM, is also able to induce T-cell apoptosis through the induction of FasL on antigen-presenting cells such as monocytes/macrophages (15, 16). GalXM-induced apoptosis is mediated by the interaction of GalXM with the glyco-receptors such as CD7, CD43 and CD45, which are expressed on the T cell surface and are normally involved in galectin-induced apoptosis. It was found that CD45 activation correlated with the induction of apoptosis in Jurkat cells, while CD7 and CD43 activation correlated with apoptosis of T cells (17). These results suggest that GalXM has a functional role in virulence by targeting human T-cells, impairing specific T-cell responses and that its mechanism may potentially be different from GXM (18). Recently, Villena et al. described that GalXM was more potent than GXM at induction of Fas/FasL expression and apoptosis on macrophages (16). Their findings revealed the induction of FasL dependent macrophage apoptosis. Although GalXM and GXM induced Fas and FasL and upregulated Fas expression on the macrophage surface, lower doses of GalXM were sufficient to block proliferation in RAW 264.7 cells and induce FasL expression. Neutralizing mAbs specific for FasL were used to restore a proliferative response in vitro. The study also looked at FasL-deficient gld mice. The results revealed that GalXM and GXM failed to induce apoptosis in macrophages in vivo.
Early studies using total polysaccharide from C. neoformans revealed this antigen had a propensity for inducing immunological paralysis (19, 20). Murphy et al., showed that polysaccharide had a dose-dependent primary immunological response in mice using the hemolytic plaque assay as indicated by the increase in antibody producing cells. However, after a subsequent challenge with capsular polysaccharide a state of immunological unresponsiveness was induced. There was a diminution of antibody response and the abolition of spleen plaque forming cells. Here we investigate the antibody response to GalXM in mice and show that GalXM is a potent immunomodulatory molecule. Like GXM, GalXM, causes immunological paralysis in mice. However, unlike GXM, the state of immunological unresponsiveness to GalXM is accompanied by pathological changes and the induction of apoptosis in splenic tissue.
Materials and Methods
Animals
Six to eight week old Balb/c female mice were obtained from the National Cancer Institute (NCI). Three to five week Fas-deficient mice MRL/MpJ-Faslpr/J and the genetically matched controls MRL/MpJ were obtained from the Jackson Laboratory (Bar Harbor, Maine).
Immunizations
Mice were used for the intraperitoneal (i.p.) immunization with 0.5. 5, 50 and 500 μg of galactoxylomannan (GalXM) in PBS or Galactoxylomannan-Protective Antigen (GalXM-PA) conjugate in complete Freund's adjuvant. The first group of mice was immunized with the above mentioned doses and sera were collected for ELISA four days after immunization. Mice were sacrificed and the spleens were harvested for ELISA spot assay. The second group of mice was also immunized with the above mentioned doses and subsequently challenged with 0.1 μg of GalXM or conjugate in incomplete Freund's adjuvant (IFA) at day 14. The mice were bled and sacrificed at day 28. MRL/MpJ-Faslpr/J and the genetically matched controls MRL/MpJ were initially injected with 50 μg of GalXM and boosting immunization was done at day 7 with 5 μg of GalXM in IFA, these were then followed to day 14. All animal experiments were conducted in accordance with institutional guidelines.
GalXM isolation
GalXM was isolated from the culture supernatant using a modified method from previously described protocols (10, 14). Briefly, a 400 ml culture of Cryptococcus neoformans acapsular mutant of strain cap67 was grown in peptone supplemented with 2% galactose for 7 days. The culture supernatant was separated from the cells by centrifugation at 1,500 g for 15 minutes at room temperature, and then concentrated using a 10,000 MW cut-off Amicon centrifugal filter (Millipore, Bedford, MA). The material was then dialyzed for one week against distilled water, and the 10 KDa retentate, containing GalXM and mannoproteins, was passed through a 0.2 um filter. The retentate was lyophilized and stored at room temperature. To separate GalXM from the mannoproteins, the freeze-dried mixture was dissolved in 25 ml start buffer (0.01 M Tris base and 0.5 M NaCl solution, pH 7.2, to which CaCl2 and Mn(II)Cl2 was sequentially added at final concentrations of 1 mM each). The GalXM and mannoprotein solution was then continuously passed through a Concanavalin A-Sepharose 4B column (2.5 × 10 cm) (Sigma) for 16 h at 4 °C using a peristaltic pump with a flow rate of 16 ml/h. The material was eluted with 5 column washes of start buffer and collected as 20-ml fractions. GalXM-containing fractions were identified by the phenol-sulfuric acid assay (21). The fractions were combined, ultra-concentrated as described before, and dialyzed against water for 7 days. GalXM was then recovered by lyophilization. Compositional analysis of GalXM was confirmed by combined gas chromatography/mass spectrometry of the per-O-trimethylsilyl derivatives of the monosaccharide methyl glycosides produced from the sample by acidic methanolysis. Composition analysis revealed that the mole percent for Xylose 17 %, Mannose 28% and Galactose 55 %. These numbers closely approximate the mass composition described by (14): Xylose 22%, Mannose 29% and Galactose 50%. The possibility of Concanavalin- A contamination of GalXM preparations was evaluated by immunoblot analysis of purified GalXM with an anti-Con A antibody (Vector labs, Burlingame, CA). No concanavalin A was detected in the GalXM preparations by Western blot analysis (data not shown). Lyophilized GalXM was reconstituted in 1X PBS and dialyzed in Endosafe LPS-free water for 3 weeks until the dialysate was negative by the Limulus amoebocyte assay (Cambrex, Pittsburg PA).
Synthesis of GalXM-Bacillus anthracis protective antigen Conjugate
GalXM hydroxyl groups were activated using cyanogen bromide (CNBr) (Sigma, St. Louis, MO). The activated GalXM was then derivatized with the bifunctional linker adipic acid dihydrazide (ADH) (Sigma, St. Louis, MO). GalXM (5 mg/ml in 0.2 M NaCl) was activated with 5 mg/ml of CNBr with continuous pH monitoring such that the pH was maintained between 10.5–11.0 for 6 min at 4°C. Subsequently, an equal volume of 0.5 M NaHCO3 at pH 8.5 containing 0.5 M ADH was added. The reaction mixture was tumbled at 4°C for 18 hrs and dialyzed against 0.2 M NaCl. The reaction mixture containing GalXM-AH was then mixed with 5.0 mg/ml of Protective Antigen (PA) from Bacillus anthracis (Wadsworth Laboratories, New York) and brought to pH 5.6 with 0.05N HCl and 0.05M of the water soluble carbodiimidine, 1-ethyl -3-(3 dimethylaminopropyl)-carboiimide (EDAC) (Sigma, St.Louis, MO). The pH was continuously maintained at 5.6 for 1 hr at 4°C. The reaction mixture was dialyzed against 0.2 M NaCl for 24 hrs at 4°C (22). Biorad Quick Start Protein Assay was used according to manufactures instructions to detect protein in the conjugate. In some experiments the conjugate was further purified by capture with the PA-binding mAb 10F4 (23) conjugated to agarose beads by AminoLink plus coupling resin kit according to manufacturers instructions (Pierce, Rockford, IL).
High performance liquid chromatography (HPLC)
To establish that the GalXM-PA conjugate contained both carbohydrate and protein, we tested for the presence of carbohydrates and protein using the phenol-sulfuric assay and the Bradford protein colorimetric assay, respectively. The analysis revealed that the conjugate was positive for both protein and carbohydrate (not shown). Additionally, we used HPLC to detect the conjugate by comparing differences in elution time between GalXM and PA alone and the conjugate. GalXM-PA, GalXM and PA were analyzed in an Alltech Prevail Carbohydrate Es Su column (250 mm × 4.6 mm) (Alltech, Deerfield, IL) using a Waters 600 liquid chromatography system (New York, NY). The mobile phase consisted of 75% acetonitrile in water. Sample volumes were 20 μl at a concentration of 1mg/ml and were detected at 280 nm with a flow rate of 1.0 ml/min with a Waters 486 detector. The column was maintained at room temperature and the samples were stored at 4°C after injection. The major elution peak for PA is 11.44 min, GalXM 5.50 min and 6.57 min for the GalXM-PA conjugate. The conjugate preparation did not contain any material eluting at 11.44 min and 5.50 min. Capture ELISA using anti-PA antibody 7.5 G and hyperimmune sera tested positive for both GalXM and PA (data not shown) (23).
Serum Antibodies
Blood was collected from pre-immune and immunized mice for ELISA. Costar plates were coated with 50 μg of GalXM, the plates were blocked with 2% BSA and a 1:100 dilution of serum was serially diluted in a 1 to 3 ratio along the plate. A (1 μg/ml) cocktail of IgM, IgA, IgG (H+L)-alkaline phosphatase conjugated (Southern Biotechnology, Birmingham, AL), was used as the secondary antibody. P-Nitrophenyl Phosphate (PNPP) was used to reveal the presence of the alkaline phosphatase. Absorbance was measured at 405 nm using a microplate reader (Labsystems, Multiskan). BSA coated plates were used as a negative control.
Immunofluorescence
C. neoformans strains cap67, B3501, 24067 and H99 were grown in Sabouraud dextrose broth (Difco Laboratories, Detroit, Mich.) for 3 days at 30°C. The cells were washed three times with sterile phosphate-buffered saline (PBS; pH 7.4) and then counted using a hemocytometer. Strains were normalized to a suspension of 2 × 106 cells/ml and incubated with a 1:100 dilution of GalXM-PA pre-immune serum or serum not stimulated by the antigen as a control in 2% BSA and 0.05% Goat Serum. Cells were washed three times with 2% BSA and 0.05% Goat Serum and incubated with 4 μg /ml of goat anti mouse IgM-FITC.
ELISA Spot Assay
Costar plates were coated overnight with 50 μg of GalXM or 2% BSA as a control in coating buffer (20 mM K2HPO4, 10 mM KH2PO4, 1 mM Na-EDTA, 0.8% NaCl, 0.01% NaN3). Prior to plating spleen cells plates were blocked using 2% BSA for 2 hrs at 37°C. Plates were then washed with 2X with DMEM. Spleens were homogenized and passed through a 0.20 μm Falcon cell strainer into 2 ml of DMEM supplemented with 10% FCS, 10% NTC-109, 1 % nonessential amino acids and 1% Penicillin-Streptomycin. Red blood cells were lysed by suspending the cell pellet in 0.17 M, NH4Cl buffer for 2 min at room temperature. Cells were placed in a dish for 2 hrs to allow adherent cells to settle to the bottom. Non-adherent cells were collected by centrifugation and the pellet was re-suspended in DMEM media. Viable cells were counted with trypan blue. 1 × 106 cells of non adherent cells are added to the first well and one log dilutions are plated across. Cells were incubated overnight at 37°C in a 10% CO2 incubator. Cells were then washed away from the plate with washing buffer (10 mM Tris, 150 mM NaCl, 0.1% Tween 20. ph 7.2). A cocktail of IgM, IgA, IgG1, IgG2a, IgG2b, IgG3 biotin conjugated (Southern Biotechnology, Birmingham, AL) antibodies was used as the secondary antibody and incubated overnight at 4 °C. Thirty minutes prior to washing the plates a Vectastain ABC (Vector Labs, Burlingame, CA) streptavidin buffer was prepared in (1X PBS and 0.1% Tween 20). The plates were incubated with the Vectastain (Vector Labs, Burlingame, CA) for 30 min. at room temperature. Plates were then washed and incubated with 2-bromo-4 chloro-3indoyl phosphate (5-BCIP) (Sigma, St. Louis, MO) in AMP buffer (203 mg MgCl2-6H20, 0.1 ml Triton X-405, 95.8 ml of 2-amino-2 methyl-1-propanol (AMP), pH was adjusted to 9.8 with HCl in 1L of dH20) for 3 hrs at room temperature until a blue color developed. Plates were rapidly washed twice with water and blue spots counted using a Zeiss inverted light microscope.
In vivo caspase inhibition
Balb/c female mice were injected i.p. with 50 μg of Z-DEVD-FMK an irreversible and cell permeable peptide used to inhibit caspase 3 (BD Pharmigen, San Jose, CA). Mice were subsequently injected i.p. with 50 μg of a general pan caspase inhibitor, the peptide Z-VAD-FMK (BD Pharmigen, San Jose, CA). In preliminary experiments we used several controls such 50 μg Z-FA-FMK a negative control peptide for caspase inhibitors. Since these peptide inhibitors are only soluble in Dimethyl Sulfoxide (DMSO) (Sigma, St. Louis, MO), and DMSO may cause cellular toxicity, the peptides were then diluted in PBS to dilute the DMSO. As an additional negative control, mice injected with diluted DMSO in PBS at the same final concentration used for diluting the inhibitors. All inhibitors were injected on a daily basis and within the same time for 23 days. The mice were also injected i.p. with 2 μg of galactoxylomannan (GalXM) or saline in complete Freund's adjuvant at the second day of inhibitor injections. Mice were sacrificed at days 3, 7, 14 and 23 days. Mice sacrificed on day 23 were boosted with 0.1 μg of GalXM. These were then compared to mice that were not given the inhibitors. ELISA spot Assay was performed to determine the number of antibody-producing cells.
Capture ELISA
Microtiter plates were coated with 1 μg /ml goat Ab to mouse IgG2b (Southern Biotechnology, Birmingham, AL). MAb 7.5G to PA was then added in a solution of 2 μg /ml as the antigen capture antibody (Ab). The GalXM-PA conjugate was incubated in the microtiter plate. The presence of GalXM was then detected with hyperimmune sera generated from mice immunized with the GalXM-PA conjugate. These specific antibodies to GalXM in the hyperimmune sera were mostly IgM and IgA. Alkaline phosphatase conjugated mouse anti-IgM MAb was used as the secondary antibody. p-Nitrophenyl Phosphate (PNPP) was used as the substrate for the enzyme alkaline phosphatase. Absorbance was measured at 405 nm.
Histology
Spleens were harvested and fixed in 10 % buffered formalin. Sections were paraffin embedded and processed with Hemotoxylin and Eosin, Trichrome and Mucincarmine stains. Apoptosis determination was done on paraffin slides using ApopTag Plus fluorescein in situ apoptosis kit (Chemicon, Billerica, MA), following the manufacturer's instructions. Slides were viewed using an Olympus AX 70 microscope (Olympus America, Inc., Melville N.Y). Images were captured with a QImaging, Retiga 1300 digital camera using the QCapture Suite V2.46 software (QImaging, Burnaby BC, Canada). Brightness and contrast was adjusted using Adobe Photoshop 7.0 in the TUNEL image to show clarity.
SDS-Page
A 4.5 % stacking and 10 % resolving SDS-polyacrylamide gel was made to resolve the conjugate. Protective antigen, GalXM and the Conjugate were analyzed under non-denaturing conditions.
Cytokine measurements
Balb/c, 6–8 week-old female mice were treated with different concentrations of GalXM as described above. Spleens were harvested and cell count was normalized and these were homogenized with 1X cell lysis buffer (Raybiotech, Norcross, GA) and Complete® protease inhibitors (Roche, Indianapolis, IN). Raybiotech Mouse Inflammation Cytokine Antibody Array 1.1 Array membranes were incubated with spleen cell lysates, washed and developed according to the manufacturer's instructions (Raybiotech, Norcross, GA). Percent protein cytokine expression was evaluated by analyzing spot densities with Image J software (Bethesda, Maryland). Background was also subtracted from the spot density. Densities were reported as the percent from the positive control. The positive control is a set of spots on the array membrane that are colored when the assay is developed and also show that that they array is working properly.
B-cell isolation
Spleens from Balb/c or Nu/Nu female mice were harvested and splenocytes were isolated by homogenization in DMEM. Cells were spun at 1200 rpm for 5 minutes and red blood cells were lysed by re-suspending the pellet in 0.17 M, ice cold NH4Cl buffer for 5 min. B-cells were isolated using Dynal® (Invitrogen, Carlsbad,CA) B-cell negative isolation kit according to manufacturer instructions. Isolated B-cells were diluted in RPMI supplemented with 10% FCS, 1% nonessential amino acids and 1% Penicillin-Streptomycin and plated in 6 well plates with 5ug of LPS to stimulate the cells. Cells were treated with GalXM or PBS and incubated for 18 hours at 37°C in 10% CO2.
Flow Cytometry Analysis
Balb/c female mice were injected i.v. with 0.5, 5, 50 or 500 μg of galactoxylomannan (GalXM) or saline in complete Freund's adjuvant. Mice were subsequently challenged with 0.1 μg of GalXM in incomplete Freund's adjuvant at day 14. Splenic macrophages were isolated by homogenization in DMEM supplemented with 10% FCS, 10% NTC-109, 1% nonessential amino acids and 1% Penicillin-Streptomycin. Cells were spun at 1200 rpm for 5 minutes and red blood cells were lysed by re-suspending the pellet in 0.17 M, ice cold NH4Cl buffer for 5 min. Pellet was re-suspended in staining buffer (1% FCS in PBS) and cell viability was determined by trypan blue staining. Cellular Fc receptors were blocked with mouse Ab to Fc (FcyIII/II anti mouse CD16/CD32 (BD Pharmigen, San Jose, CA) for 30 min. in staining buffer (Fetal Bovine Serum in PBS) with gentle rocking at room temperature. Approximately 5 × 107 cells were incubated for 30 minutes at room temperature with the following antibodies, F4/80+(Serotec, Raleigh, NC), CD4+(BD Pharmigen, San Jose, CA), CD8+(BD Pharmigen, San Jose, CA), Neutrophils (Serotec), CD11c+(BD Pharmigen, San Jose, CA), CD19+ (Serotec, Raleigh, NC) CD5+ (Ly1) (eBioscience, San Diego,CA) and CD21+(eBioscience, San Diego,CA). Cells were washed 3X after antibody incubation with staining buffer. Fifteen minutes prior to analysis apoptosis probes AnnexinV-Alexa 350 and 7AAD were added to samples in annexin binding buffer (10 mM HEPES, 140 mM NaCl and 2.5mM CaCl2 pH 7.4). Flow cytometry was performed in a BD FACSaria cytometer and analyzed with FloJo version 8.7.
Recombinant scFv using GalXM hybridomas
We extracted the mRNA from three hybridoma cultures before they became unstable and cloned their cDNA in bacterial vectors. We obtained 18 valid sequences for the variable heavy (VH) and variable light regions (VL) from these three cultures. Despite not being clonal, we were able to isolate a common kappa light chain and two common heavy chain sequences, one of the gamma isotype and the other from the mu isotype, from them. The variable gene usage was very similar to previously described anti-carbohydrate antibodies (24). The heavy chain variable region of the gamma isotype used VH5.1 and JH2 gene elements linked by an unknown D gene the same that were found in the anti-GXM mAb 18B7(24). The mu chain used VH3, DH2.1 and JH4 genes. The gamma heavy chain had several mutations when compared to the germline, indicating considerable affinity maturation. The sequences were deposited in Genbank (Accession numbers: FJ233886-FJ233904). Using genetic engineering, we produced two single-chain fraction variable (scFv) antibodies using either the IgG or the IgM variable region linked to the variable light chain. These recombinant antibodies were able to bind GalXM (data not shown).
Statistical Analysis
Statistical analysis was done by the Kruskal Wallis test (Primer; McGraw Hill, New York, NY) and Microsoft Excel.
Results
Serum Antibody Responses to GalXM and GalXM-PA Conjugate
This study began with an attempt to generate monoclonal antibodies to GalXM for capsule analysis. However, the problems encountered with making antibodies to GalXM led us to investigate the immunologic mechanisms responsible for the poor immunogenicity of this polysaccharide. To identify mice suitable for spleen harvest mice were injected with GalXM or the GalXM-PA conjugate and serum titers to GalXM were examined. GalXM immunization elicited modest amounts (titer 1/900) of serum antibodies reactive to GalXM regardless of immunization dose. Older mice had a slightly better response with serum titers of 1/2700. Additionally, mice were also immunized with LPS-free GalXM and the results were identical with both young and older mice (data not shown). We also consistently noted that after subsequent challenge at day 14 with GalXM, antibody titers where reduced to levels that were no longer measurable (Table I). To confirm that these effects were different from GXM, mice were injected with the GXM polysaccharide and a modest dose-dependent response was observed with the highest titer (1/2700) at 0.5 μg and the lowest response (1/100) at 500 μg (data not shown). The GXM results confirm earlier work done by Murphy et al. (12). To evaluate whether the poor immunogenicity of GalXM could be overcome by protein conjugation, the polysaccharide was conjugated with the 83,000 Da protective antigen (PA) protein from B. anthracis. Immunization studies with the conjugate revealed that although the primary antibody response to the conjugate was better than with GalXM alone it was still modest (1/900), but a booster immunization at day 14 elicited a significantly increased antibody response (1/8100) (Table I). Hybridomas with both GalXM and GalXM-PA yielded unstable clones that ceased to produce or grow during soft agar cloning, but were suitable donors for immunoglobulin genes that recognized GalXM (see below). However a screen done in parallel for PA yielded several positive antibodies in which one was protective in mice (data not shown), suggesting that the B-cell unresponsiveness was limited to cells producing mAb to GalXM.
Table I.
Effect of GalXM and GalXM-PA Conjugate on Antibody Titers 1
| GalXM (ug) | Titers Day 14 | Titers Day 28 | GalXM-PA Conjugate (ul) | Titers Day 14 | Titers Day 28 | ||
|---|---|---|---|---|---|---|---|
| n=3 | 6–8 weeks | 52 weeks | 6–8 weeks | 52 weeks | 6–8 weeks | 6–8 weeks | |
| 0.5 | 1/300 | 1/900 | 1/100 | 1/100 | 50 | 1/900 | 1/8100 |
| 5.0 | 1/900 | 1/2700 | 1/100 | 1/300 | 50 | 1/900 | 1/2700 |
| 50 | 1/900 | 1/900 | 1/100 | 1/100 | 100 | 1/2700 | 1/8100 |
| 500 | 1/900 | 1/2700 | 1/100 | 1/100 | CFA | 1/100 | 1/100 |
| CFA | 1/100 | 1/300 | 1/100 | 1/100 | PBS | 1/100 | 1/100 |
| PBS | 1/100 | 1/300 | 1/100 | 1/100 | BSA | 1/100 | 1/100 |
| BSA | 1/100 | 1/300 | 1/100 | 1/100 | |||
Fourteen days after initial immunization antibody response was measured using ELISA. After subsequent challenge with 0.1 μg of GalXM or the same initial dose of the conjugate, titers were evaluated at day 28. n = 3 mice per group for GalXM injected mice.
Hyperimmune serum from mice immunized with the conjugate was used for immunoflourescence on C. neoformans strains cap67, B3501, 24067 (serotype D) and H99 (serotype A). The polyclonal antibody bound near the cell wall in acapsular strains (data not shown), a pattern consistent with prior reports that GalXM is mostly cell wall-associated (25). In encapsulated serotype D strains the serum bound on all around outer edge in a punctuate pattern. In H99 cells the fluorescence pattern was scattered and punctuate (26). We also generated a recombinant single chain fragment variable region (scFv) antibody fragments derived from the mRNA of GalXM-PA hybridoma cell lines that secreted anti-GalXM antibodies prior to becoming unstable. Preliminary results with these scFv's showed a punctuate fluorescence pattern like the GalXM-PA serum (data not shown) (14, 25). The recovery of immunoglobulin genes from unstable hybridomas producing antibody to GalXM that reacted with GalXM when assembled into scFv suggested that the problem in recovering hybridomas lied with the cells and not the antibody molecules.
Spleen plasma cell response to GalXM immunization
Given our inability to recover stable hybridomas we proceeded to investigate the B cell response to GalXM immunization, and enumerated the antibody producing cells in the spleen following GalXM immunization by ELISA spot (Figure 1). The lowest immunizing dose, 0.5 μg of GalXM yielded the highest number (130 cells) of antibody producing cells. The effect of GalXM in suppressing antibody-producing cells was dose dependent, such that immunization with the larger amounts significantly reduced the number of cells. However, after a subsequent GalXM immunization at day 14, no antibody producing cells were detectable in the spleens of immunized mice by day 23. Repeating the experiments with LPS-free GalXM also yielded the same results (data not shown). As a control, we also examined the number of antibody producing cells in spleen in response to GXM. Although the number of antibody producing cells to GXM decreased in a dose dependent manner (300 cells for 0.5 μg of GXM and 10 cells for 500 μg of GXM), antibody-producing cells to GXM did not disappear completely as was observed with GalXM immunization (Figure 1). We also examined spleens that were injected with 50 μl of the conjugate in CFA followed by a boost of 10 μl at day 14. The results revealed the presence of 200 antibody producing cells however these results were similar to the CFA control (data not shown). We examined whether the loss of antibody producing cells was limited to B cells producing GalXM specific antibodies. Mice treated with two immunizations of GalXM continued have some antibody producing cells secreting IgM and IgG antibodies despite the dearth of cells producing antibodies to GalXM (Figure 2).
Figure 1.
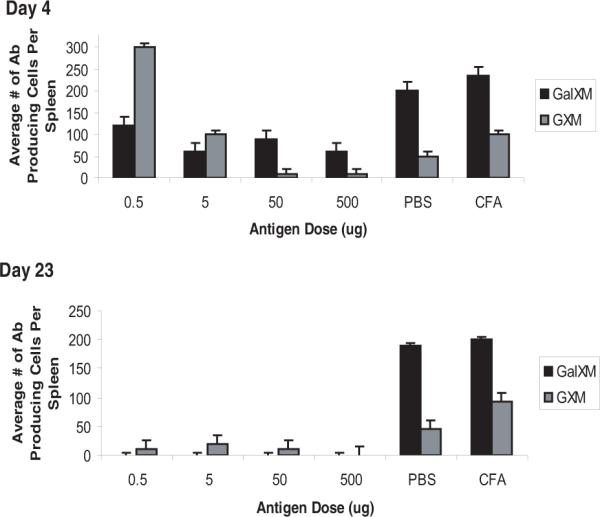
ELISA Spot Assay for spleen antibody producing cells to GalXM. Antibody producing cells were detected as blue spots on ELISA plates. Black bars are spots detected for GalXM, grey bars are spots detected for GXM. (Top panel) The number of antibody producing cells decrease in a dose-dependent manner for both antigens at day 4. (Lower panel) After an immune challenge, antibody producing cells are completely abolished for GalXM treated mice by day 23. Although there is a reduction for antibody producing cells against GXM, these are still detectable. n = 5 mice per group
Figure 2.
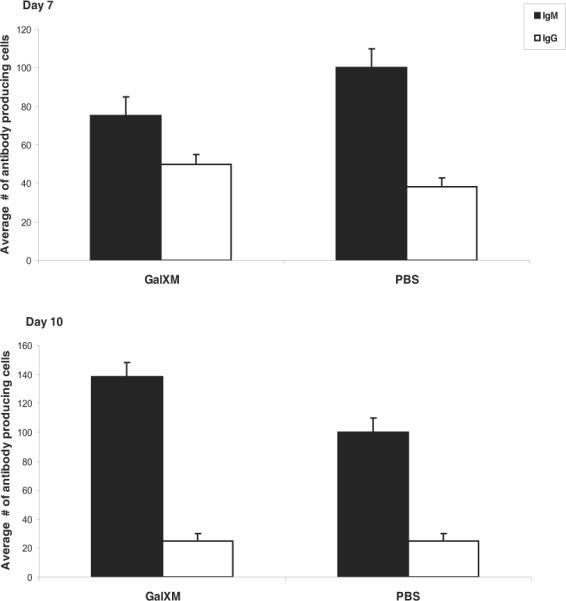
Splenocytes in Balb/c mice are able to produce non-specific antibodies after GalXM immunization. ELISA spot assay shows the average number of antibody producing cells against IgM or IgG in Balb/c after injection of 20 μg of GalXM, and PBS. The results are after 10 days of treatment. The mice were boosted with 0.5 μg of GalXM at day 8. n = 5 mice per group.
GalXM immunization causes spleen enlargement
During spleen harvesting we observed that mice injected with GalXM, and in particular those receiving the 500 μg dose had a significantly enlarged spleen. The average weight of the spleen (0.320 g) was three times higher than the average of spleen from mice receiving a mock injection with PBS alone (0.111 g) (Figure 3). At other GalXM immunization doses splenic enlargement was also observed, but the increase was not as dramatic as with 500 μg. Splenic sections using hematoxylin and eosin reveal that at doses of 500 μg splenic follicle definition is lost (Figure 4). Pathological analysis revealed moderate to marked extramedullary hematopoiesis (EMH) with increased myeloid and megakaryocyte precursors (data not shown). There was also mild lymphoid depletion as compared to PBS control. All the spleens samples had splenic capsulitis and peritonitis and early fibrosis due to IP injection of CFA. A TUNEL assay was used to detect if apoptosis was occurring in the spleen and if the effect was dose dependent. The results revealed that GalXM induced apoptosis in the spleen at all doses administered compared to the PBS control (Figure 5).
Figure 3.
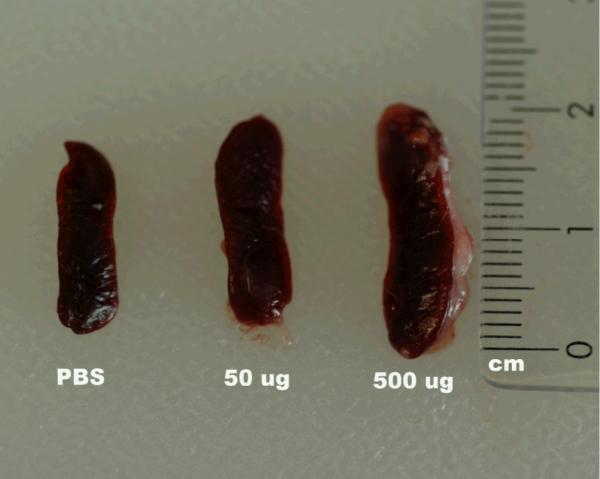
GalXM Causes Splenic Enlargement. After two challenges with GalXM spleen weights triple in size at a dose of 500 μg (2.0 cm, 0.320 g) as compared to 50 μg (1.5cm, 0.150 g) of GalXM and the PBS control (1.4 cm, 0.144 g). Spleen sizes are measured in cm and spleen weights measured in grams.
Figure 4.

Spleen histology after GalXM immunization. Normal splenic architecture with defined red pulp, white pulp and marginal zones can be seen in most sections. At 500 μg of GalXM, however, follicle definition is lost due to intense extramedullary hematopoiesis. Objective is 40X.
Figure 5.

TUNEL reveals apoptosis in the spleen of mice immunized with GalXM. Apoptotic bodies (green dots) are present at low doses 5 ug and high doses 500ug of GalXM as compared to the PBS control. Scale bar is 110 μm.
GalXM affects cells in the spleen
Given the observation that GalXM ablated the specific B cell response we investigated the effect of this polysaccharide on other major cell types in the spleen. Since GalXM is reported to cause apoptosis in T-cells and macrophages in-vitro (15) we enumerated CD19+ B cells, F4/80+ macrophages, CD4+ and CD8+ T-lymphocytes in the spleen after GalXM immunization. Eight days following the first initial injection of GalXM (10 μg) there was a decrease in the percentage of CD19+ B cells. In parallel, we looked at the percentage of the CD19+ cells that were annexin V positive, a marker of apoptosis. The results also revealed that at 8 days post GalXM injection there was an increase in the percentage of cells that are annexin V positive. After a second injection of GalXM there was a recovery of CD19+ B-cells as seen on day 23. However ELISA spot revealed a complete disappearance of GalXM specific B-cells after a second injection. We also tested if CD5+ and CD21+ specific B-cells were affected by GalXM but found no differences between spleens from GalXM treated mice and control mice (data not shown). For F4/80+ macrophages the initial injection of GalXM caused an increase in the percentage of macrophages by day 8 and a second injection caused an additional modest increase in the percentage of these cells by day 23. Annexin V stain could not be evaluated since it non-specifically binds to monocytes. For both CD4+ and CD8+ T-lymphocytes there was a decrease in the percentage of cells after the first injection of GalXM through day 14. Annexin V staining shows an increase in the percentage of cells undergoing apoptosis with the first injection (Figure 6). The effect of GalXM on isolated splenic B-cells was also tested. Isolated B-cells were treated with a low dose (0.5 μg) and a high dose (50 μg) of GalXM for 18 hours. Flow cytometry analysis for CD19+ and Fas or FasL double positive showed a modest increase in Fas at a dose of 50 μg but the differences were not statistically significant p = 0.806 (Figure 7). Hence, GalXM injection had a qualitative effect in causing a disappearance of GalXM-binding B-cells and significant quantitative effects on the numbers and proportions of total B and T-lymphocytes and macrophages.
Figure 6.

GalXM immunization negatively affects several types of splenocytes (Top Panel): Percentage of CD19+ B-cells, CD4+ and CD8+ T-cells and F4/80+ macrophages during 3, 8, 14, and 28 days after 10 μg of GalXM injection. Balb/c mice that were followed to day 28 received a GalXM boosting immunization of 0.5 μg of GalXM. For CD19+ B-cells injected with GalXM there is a statistically significant decrease p = 0.006, denoted by (*) when compared to the PBS control. (Lower Panel): Percentage of cells that are double positive for the marker and Annexin V. Representative absolute number of cells in the spleen range as follows: CD19+ B-cells; GalXM: (3.9 × 106 −1.69 × 106 cells), PBS (9.7 × 107 −2.2 × 107cells), CFA (8.9 × 106 −1.7 × 107cells). CD4+ T-cells; GalXM: (1.4 × 107 −3.4 × 107cells), PBS (3.0 × 107 −4.8 × 107cells),CFA (2.5 × 107 −3.5 × 107cells) and CD8+ T-cells; GalXM: (6.7 × 106 −1.8 × 107cells), PBS (9.0 × 106 −2.0 × 107cells), CFA (1.0 × 107 −1.6 × 107cells) and F4/80+; GalXM: (3.9 × 106 −7.6 × 107cells), PBS (3.3 × 106 −6.9 × 106 cells), CFA (6.8 × 106 −9.8 × 106 cells). n = 4 mice per group.
Figure 7.
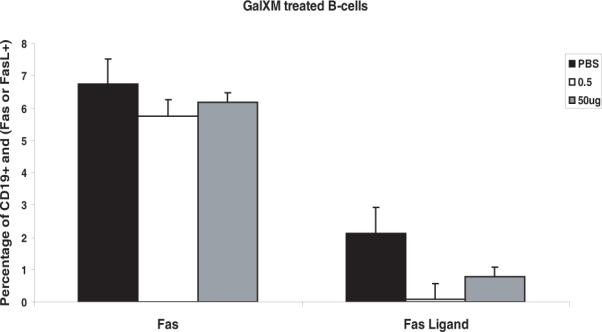
Low dose or high dose of GalXM does not increase splenic Fas or FasL in isolated B- cells. The percentage of CD19+ and Fas or FasL double positive B-cells, after isolated B-cells were treated with 0.5 μg or 50 μg of GalXM. n = 5 mice per group.
Cytokine Expression in Spleen
Since splenic follicle morphology had been drastically altered and apoptosis was observed in the spleen of mice receiving GalXM. We investigated global cytokine expression at 5 μg and 500 μg of GalXM. Cytokine array revealed that the expression of the inflammatory cytokines decreased with prolonged exposure to GalXM. The cytokine levels of mice that received boosting immunization at day 14 decreased regardless of the GalXM dose (Figure 8). At day 7, there is a specific decrease in granulocyte colony stimulating factor (G-CSF), which was produced mostly by macrophages and activates neutrophils and granulocytes; Granulocyte macrophage colony stimulating factor (GM-CSF), which is produced mostly by Th2 T-cells and activates T-lymphocytes, monocytes neutrophils and precursors (27); Interferon-inducible T-cell alpha chemoattractant (I-TAC), which is produced by neutrophils and is a T-cell chemoattractant; Keratinocyte chemoattractant (KC), which is produced by keratinocytes and monocytes and is a potent neutrophil chemoattractant (28); Leptin (Lep), which is expressed in adipose tissue can modulate the immune response by regulating the proliferation of T-cells in monocytes. It can increase the production of TNF and IL-12 (29); Interleukin IL1β which is produced by many immune cells such as macrophages, B and T cells, NK and neutrophils activates T, B and NK cells (27); IL-2, which is produced by CD4+ T-lymphocytes promotes T - and B-cell growth and differentiation (27); Il-3, which is produced by T-cells and activated NK cells (27), IL-13 which is produced by T-cells and promotes B-cell growth and differentiation (27); IL-17 which is produced by activated T-cells and whose function is to further activate T-cells (27). At day 14 there is a specific decrease in Fas-L, a type-II transmembrane protein that plays an important role in immune regulation by binding to its receptor Fas (CD95) and inducing apoptosis (30). LPS induces chemokine (LIX) an inflammatory chemokine that is strongly induced by bacterial LPS and is able to interact with matrix metalloproteases (31), Eotaxin 2 that induces the chemotaxis of eosinophils and T-cell activation-3 (TCA-3), that is produced by T-cells. Day 21 a decrease is seen in all other cytokines that affect mostly B and T-lymphocytes. The cytokines that were markedly reduced at day 21 are TCA-3, which is produced by T-cells after cell activation. LIX; IL1-beta and IL-12p70 that is produced mainly by B-cells and to a lesser extent, by T-cells and stimulates the activation of lymphocytes. Hence, GalXM immunization was associated with global dysregulation of spleen cytokine production.
Figure 8.

Immunization with GalXM results in dose dependent global reduction of inflammatory cytokine expression. Inflammatory cytokine arrays show the percent signal intensity of cytokine expression of Balb/c mice injected with PBS (black bars), CFA (red) and 5μg (green) and 500 μg (yellow) of GalXM in IFA.
Caspase Inhibition rescues GalXM-binding B cells
Prior in-vitro studies had shown that GalXM caused apoptosis in T-cells through the activation of caspase 8. Since our results also indicated apoptosis in the spleen, we carried out a pilot study using several caspase inhibitors, including the General Caspase inhibitor (Z-VAD-FMK), Caspase 8 inhibitor (Z-IETD-FMK), Caspase 3 inhibitor (Z-DEVD-FMK) and Negative Control inhibitor (Z-FA-FMK). The negative control inhibitor had no inhibitory effect on apoptosis mediated by caspases and only inhibited cysteine proteases when mice were given 0.5, 5, 50, and 500 μg of GalXM. In the first experiment, administration of either the general pan caspase inhibitor or the caspase 8 inhibitor. The general pan caspase inhibitor maintained the number of antibody producing cells above 250 cells regardless of GalXM challenge (data not shown). However, the caspase 8 inhibitor alone did not overcome the effects of GalXM since we observed a GalXM dose-dependent decrease in the number of antibody producing cells enumerated by ELISA spot. In a second experiment we repeated the experiment using a caspase 3 inhibitor instead of caspase 8, since caspase 3 is a downstream effector caspase in the extrinsic apoptotic pathway. The caspase 3 inhibitor maintained the number of antibody producing cells above 400 cells regardless of GalXM challenge (data not shown). In a subsequent experiment we injected mice with a combination of the caspase 3 and general pan caspase inhibitor. Mice were injected with 5 μg of GalXM twenty four hours after the inhibitor cocktail, then the inhibitors were given every other day and these were followed at days 3,7,14 and 23. The mice were also challenged with GalXM at day 14. These were then compared to a second set of mice that were not given the inhibitor cocktail and were also challenged at day 14. At days, 3 and 7 both groups had approximately 300–500 cells. At day 14, there was a slight decrease in the number of cells for the group with no inhibitor. However, by day 23 the mice not receiving the inhibitor had only an average of 50–100 whereas those given the inhibitor cocktail had an average of 450 cells, a difference that is statistically significant (p = 0.008) as determined by Kruskal Wallis test. We also observed that the spleens of mice not receiving the inhibitors were twice as large at 0.2 grams (Figure 9). We attempted to generate antibodies from mice treated with the cocktail of inhibitors but none were recovered because the hybridomas were not stable producers
Figure 9.
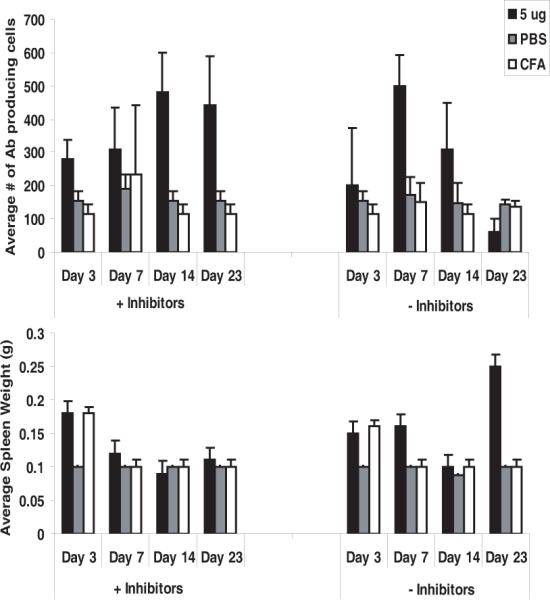
Capsase inhibitors prolong the presence of anti-GalXM antibody producing cells. (Top Panel) ELISA spot assay shows the average number of antibody producing cells in Balb/c mice with or without caspase inhibitor cocktail after a GalXM injection of 5 μg. Mice followed to day 21 received two doses of GalXM. (Lower Panel) The average spleen weights per treatment groups. n = 5 mice per group
Fas deficiency increases spleen plasma cell response to GalXM
Since GalXM is reported to up-regulate the Fas receptor in T-cells, we tested the effects of GalXM in vivo by enumerating the number of antibody producing cells. The results revealed that the number of anti-GalXM antibody producing cells was significantly larger in Fas-deficient mice (1600 cells) compared to MRL/MpJ control mice (350 cells). The results also reveal that there was no significant difference in spleen size between Fas receptor-deficient mice and the MRL/MPJ control mice (Figure 10).
Figure 10.
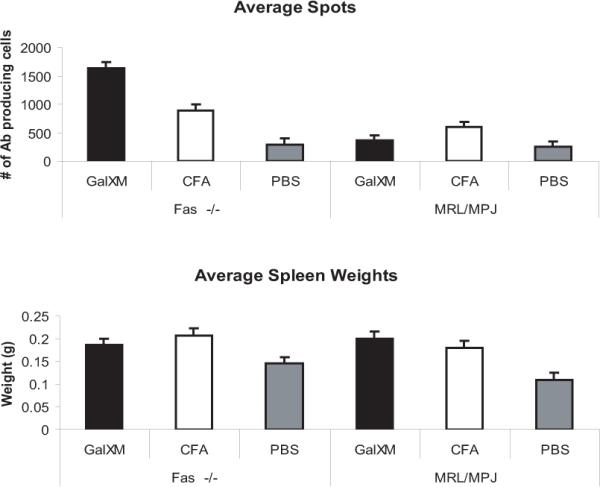
Fas deficient mice sustain GalXM antibody producing cells. (Top Panel) ELISA spot assay shows the average number of antibody producing cells in MRL/MpJ-Faslpr/J and MRL/MPJ control mice after injection of GalXM, CFA and PBS. The results are after 14 days of treatment. The mice were boosted with 5 μg of GalXM at day 7. (Lower Panel) The average spleen weights per treatment groups. n = 5 per group
Discussion
Many attempts have been made to render the cryptococcal capsular polysaccharides, namely GXM, immunogenic in animals by coupling it to BSA (32), bovine gamma globulin (33) and sheep erythrocytes (34). However, a major success in the field was the conjugation of GXM to tetanus toxoid which yielded a protective vaccine that was used to generate many of the monoclonal antibodies (mAb) used to study C.neoformans (22, 35, 36). While attempting to make a monoclonal antibody against GalXM we observed that GalXM alone was a poor immunogen since initial titers were modest and the titers dropped to non-immune levels after boosting with additional GalXM. We attempted to circumvent this problem in several ways. To make GalXM more immunogenic, we immunized mice with a GalXM in a CFA emulsion, but this approach yielded only low and transient titers of antibodies to GalXM. The second approach was by generating a GalXM-protein conjugate using the protective antigen from B. anthracis. Although initial immunization yielded modest titers of GalXM-binding antibodies, boosting immunization was also associated with a decrease in GalXM-specific titers. This result suggested that the conjugate helped to overcome initial immunological tolerance in the spleen but that its effect was short lived, which was seen by the instability of the hybridomas that we generated. Our ability to generate GalXM-binding scFv's from mRNA recovered from dying GalXM-binding hybridomas implies that the problem with unstable hybridomas is likely to reflect a host cell problem rather than loss of antibody production or specificity from ongoing somatic mutation.
The reduction in serum titers to non-immune levels following GalXM immunization, and literature reports showing that GalXM-induced apoptosis through the extrinsic apoptotic pathway by promoting the cleavage of caspase 8 in T-cells (15, 17), led us to hypothesize that GalXM was producing a state of immunological paralysis (19, 20) through direct induction of apoptosis. Consistent with this notion, GalXM administration was associated with a reduction in the number of specific antibody producing cells in the spleen, a dose-dependent effect. After subsequent GalXM challenge, antibody-producing cells recognizing GalXM completely disappeared from the spleen. These results are in contrast to GXM in which not all antibody producing cells disappear from the spleen (12). We tested whether the GalXM affected antibody production of all B-cells. Mice treated with two immunizations of GalXM were still able to make antibody producing cells that secreted IgM and IgG suggesting that the GalXM effect was specific to that polysaccharide. Further evidence that the effect was specific was that the immunization with GalXM-PA produced high serum titers to PA and we recovered several PA-binding hybridomas from the spleens of immunized mice (data not shown). The instability of hybridomas producing antibodies against GalXM was observed in four fusions and remains a,perplexing observation that raises the possibility that B-cell encounters with GalXM in the spleen induce cell damage that lasts through fusion with myeloma cells to produce short-lived hybrid cells.
We were able to avoid the complete depletion of B cells producing antibody to GalXM by administering a cocktail of general pan caspase and caspase 3-inhibitors. During a pilot study using the inhibitors separately we found that the caspase 8 inhibitor was unable to block the reduction of antibody producing cells while the caspase 3 and general pan caspase inhibitors are able to do so. In a subsequent experiment, our results showed that the cocktail of general pan caspase and caspase 3 inhibitors maintained the number of antibody producing cells despite increased GalXM concentrations or challenge. However, hybridoma instability was observed in these caspase inhibitor treated mice. We also show that Fas receptor-deficient mice also maintained the number of GalXM antibody producing cells. This result suggests that Fas is responsible for GalXM-induced apoptosis. However, when we isolated B-cells and treated these with GalXM, only a high dose of the polysaccharide modestly increased the presence of the Fas receptor but the results were not statistically significant. These results argue against a direct pro-apoptotic toxicity of GalXM on B cells and suggest that other cell types may be involved in the upregulation of the Fas receptor in B-cells. A preliminary experiment in nude mice showed that GalXM did not affect the upregulation of Fas in CD19+ B-cells suggesting that T-cells may not be the primary cells involved in B-cell apoptosis (data not shown).
Like GXM, GalXM causes immunological paralysis in the spleen as seen in the ELISA spot assay. However, GalXM induces more dramatic changes on immune function than GXM. For example, massive enlargement of the spleen followed GalXM immunization characterized by loss of splenic follicle definition, localized inflammation and cellular apoptosis. Although CFA caused some splenic enlargement on its own, remarkable splenic enlargement was observed only in mice that received GalXM and CFA. The inflammatory cytokine profile shows that GalXM causes a global reduction in cytokine expression. These changes in cytokine expression are paralleled by dramatic changes in the cellular numbers of B and T lymphocytes and macrophages. Flow cytometry revealed that the percentage of CD19+ B cells decreased after the initial injection of GalXM, but these cells recovered to initial levels after GalXM boosting. However, GalXM specific B-cells were completely abrogated by a second immunization with GalXM. This observation suggests that the first dose of GalXM affects all general CD19+ B cells while the second dose affects primarily GalXM specific B-cells. The specific subset of CD5+ - CD21+ B-cells were not affected by GalXM. CD4+ and CD8+ T-cells are affected by single doses of GalXM and these do not recover to initial levels. In contrast, the percentage of F4/80+ macrophages increased modestly in response to GalXM injections, consistent with macrophage infiltration to replenish cells undergoing apoptosis. We observed that after GalXM injection there was a modest increase in annexin V staining. Our in-vivo results correlated with those reported by Percolini et al. (15) in T-cells and most recently in macrophages by Villena et al. in-vivo. The large increase in spleen mass associated with GalXM immunization appears to be the result of profuse extra medullary hematopoetic activity that disrupted spleen architecture such that the distinction between marginal zones and pulp was lost. Splenomegaly has been observed during systemic infection by encapsulated pathogens such as Streptococcus pneumoniae this is due to increased B and T-cells and macrophages (37).
In summary, our results show that the mechanism of immunological paralysis after GalXM immunization is the result of apoptotic ablation of B cells producing antibodies to GalXM in the context of global spleen immune dysregulation, characterized by a global diminution in cytokine production and derangements in lymphocyte and macrophage prevalence. To our knowledge, this is the first mechanism explaining the classical observation that polysaccharides induce immune paralysis. Although GalXM was shown to induce T cell apoptosis in vitro (15), this mechanism did not extend to B cells and the specificity of paralysis for the antibody response to GalXM remains unexplained. A comparison of the spleen cytotoxic effects of GalXM and GXM indicate different effects suggesting that the mechanisms of immunological paralysis are likely to differ depending on the specific polysaccharide. GalXM is a powerful immunomodulatory molecule that could conceivably find applications for the treatment of autoimmune diseases as was recently reported for GXM in rheumatoid arthritis model (38). However, GalXM appears to have greater effects on host immune function than GXM and effects reported here provide additional mechanisms for the profound immune suppression that often accompanies cryptococcal infections.
Acknowledgements
We thank the Complex Carbohydrate Research Center at The University of Georgia for GalXM composition analysis. We also thank Wadsworth laboratories and Dr. Johanna Rivera for the production and providing the protective antigen protein. We thank Susan Buhl for sharing her expertise during the generation of the hybridoma cell lines. We thank Dr. Rani Sellers Comparative Pathologist at the AECOM Histopathology facility for her expert insight in the GalXM treated sections. We thank Matthew Marks for invaluable discussions on the caspase inhibitors. We thank Dr. Richard Kitsis for sharing his expertise on the Fas-deficient mice experiments and providing very helpful advice on which mouse strains to study. We thank Patrícia Andrade for helping us with the GalXM-PA scFv's protein purifications. We thank the AECOM Flow Cytometry Core Facility, P30CA013330. The data in this paper are from a thesis to be submitted by Magdia De Jesus in partial fulfillment of the requirements for the degree of doctor of philosophy in the Sue Golding Graduate Division of Medical Science, Albert Einstein College of Medicine, Yeshiva University, Bronx, N.Y.
This work was supported by NIH grants AI33774, AI33142, and HL59842-01 to A.C. M.D. was supported by NCI/NIH training grant 2T32CA009173-31. The Complex Carbohydrate Research Center is supported by the Department of Energy Center for Plant and Microbial Complex Carbohydrates, DE-FG09-93ER-20097. The Wadsworth Laboratories is supported by Northeast Biodefense Center under grant 2U54AI057158-06.
References
- 1.Halliday WJ. Immunological paralysis of mice with pneumococcal polysaccharide antigens. Bacteriol Rev. 1971;35:267–289. doi: 10.1128/br.35.3.267-289.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker PJ. Regulation of magnitude of antibody response to bacterial polysaccharide antigens by thymus-derived lymphocytes. Infect Immun. 1990;58:3465–3468. doi: 10.1128/iai.58.11.3465-3468.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grinsell M, Weinhold LC, Cutler JE, Han Y, Kozel TR. In vivo clearance of glucuronoxylomannan, the major capsular polysaccharide of Cryptococcus neoformans: a critical role for tissue macrophages. J Infect Dis. 2001;184:479–487. doi: 10.1086/322787. [DOI] [PubMed] [Google Scholar]
- 4.Rivera J, Mukherjee J, Weiss LM, Casadevall A. Antibody efficacy in murine pulmonary Cryptococcus neoformans infection: a role for nitric oxide. J Immunol. 2002;168:3419–3427. doi: 10.4049/jimmunol.168.7.3419. [DOI] [PubMed] [Google Scholar]
- 5.Lendvai N, Casadevall A, Liang Z, Goldman DL, Mukherjee J, Zuckier L. Effect of immune mechanisms on the pharmacokinetics and organ distribution of cryptococcal polysaccharide. J Infect Dis. 1998;177:1647–1659. doi: 10.1086/515329. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee J, Casadevall A, Scharff MD. Molecular characterization of the humoral responses to Cryptococcus neoformans infection and glucuronoxylomannan-tetanus toxoid conjugate immunization. J Exp Med. 1993;177:1105–1116. doi: 10.1084/jem.177.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherniak R, Sundstrom JB. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect Immun. 1994;62:1507–1512. doi: 10.1128/iai.62.5.1507-1512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker SC, Casadevall A. Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc Natl Acad Sci U S A. 2002;99:3165–3170. doi: 10.1073/pnas.052702799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherniak R, Reiss R, Turner SH. A Galactoxylomannan Antigen of Cryptococcus neoformans Serotype A. Carbohydrate Reserach. 1982;103:239–250. [Google Scholar]
- 10.McFadden DC, De Jesus M, Casadevall A. The physical properties of the capsular polysaccharides from Cryptococcus neoformans suggest features for capsule construction. J Biol Chem. 2006;281:1868–1875. doi: 10.1074/jbc.M509465200. [DOI] [PubMed] [Google Scholar]
- 11.Monari C, Pericolini E, Bistoni G, Casadevall A, Kozel TR, Vecchiarelli A. Cryptococcus neoformans capsular glucuronoxylomannan induces expression of fas ligand in macrophages. J Immunol. 2005;174:3461–3468. doi: 10.4049/jimmunol.174.6.3461. [DOI] [PubMed] [Google Scholar]
- 12.Murphy JW, Cozad GC. Immunological unresponsiveness induced by cryptococcal capsular polysaccharide assayed by the hemolytic plaque technique. Infect Immun. 1972;5:896–901. doi: 10.1128/iai.5.6.896-901.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vecchiarelli A, Retini C, Monari C, Casadevall A. Specific antibody to Cryptococcus neoformans alters human leukocyte cytokine synthesis and promotes T-cell proliferation. Infect Immun. 1998;66:1244–1247. doi: 10.1128/iai.66.3.1244-1247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaishnav VV, Bacon BE, O'Neill M, Cherniak R. Structural characterization of the galactoxylomannan of Cryptococcus neoformans Cap67. Carbohydr Res. 1998;306:315–330. doi: 10.1016/s0008-6215(97)10058-1. [DOI] [PubMed] [Google Scholar]
- 15.Pericolini E, Cenci E, Monari C, De Jesus M, Bistoni F, Casadevall A, Vecchiarelli A. Cryptococcus neoformans capsular polysaccharide component galactoxylomannan induces apoptosis of human T-cells through activation of caspase-8. Cell Microbiol. 2006;8:267–275. doi: 10.1111/j.1462-5822.2005.00619.x. [DOI] [PubMed] [Google Scholar]
- 16.Villena SN, Pinheiro RO, Pinheiro CS, Nunes MP, Takiya CM, DosReis GA, Previato JO, Mendonca-Previato L, Freire-de-Lima CG. Capsular polysaccharides galactoxylomannan and glucuronoxylomannan from Cryptococcus neoformans induce macrophage apoptosis mediated by Fas ligand. Cell Microbiol. 2008;10:1274–1285. doi: 10.1111/j.1462-5822.2008.01125.x. [DOI] [PubMed] [Google Scholar]
- 17.Pericolini E, Gabrielli E, Cenci E, De Jesus M, Bistoni F, Casadevall A, Vecchiarelli A. Involvement of glycoreceptors in galactoxylomannan-induced T cell death. J Immunol. 2009;182:6003–6010. doi: 10.4049/jimmunol.0803833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vecchiarelli A, Casadevall A. Antibody-mediated effects against Cryptococcus neoformans: evidence for interdependency and collaboration between humoral and cellular immunity. Res Immunol. 1998;149:321–333. doi: 10.1016/s0923-2494(98)80756-6. discussion 500–323. [DOI] [PubMed] [Google Scholar]
- 19.Gadebusch HH. Passive immunization against Cryptococcus neoformans. Proc Soc Exp Biol Med. 1958;98:611–614. doi: 10.3181/00379727-98-24123. [DOI] [PubMed] [Google Scholar]
- 20.Gadebusch HH. Active immunization against Cryptococcus neoformans. J Infect Dis. 1958;102:219–226. doi: 10.1093/infdis/102.3.219. [DOI] [PubMed] [Google Scholar]
- 21.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28:350–356. [Google Scholar]
- 22.Devi SJ, Schneerson R, Egan W, Ulrich TJ, Bryla D, Robbins JB, Bennett JE. Cryptococcus neoformans serotype A glucuronoxylomannan-protein conjugate vaccines: synthesis, characterization, and immunogenicity. Infect Immun. 1991;59:3700–3707. doi: 10.1128/iai.59.10.3700-3707.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera J, Nakouzi A, Abboud N, Revskaya E, Goldman D, Collier RJ, Dadachova E, Casadevall A. A monoclonal antibody to Bacillus anthracis protective antigen defines a neutralizing epitope in domain 1. Infect Immun. 2006;74:4149–4156. doi: 10.1128/IAI.00150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casadevall A, Cleare W, Feldmesser M, Glatman-Freedman A, Goldman DL, Kozel TR, Lendvai N, Mukherjee J, Pirofski LA, Rivera J, Rosas AL, Scharff MD, Valadon P, Westin K, Zhong Z. Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies. Antimicrob Agents Chemother. 1998;42:1437–1446. doi: 10.1128/aac.42.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Moer A, Salhi SL, Cherniak R, Pau B, Garrigues ML, Bastide JM. An anti-Cryptococcus neoformans monoclonal antibody directed against galactoxylomannan. Res Immunol. 1990;141:33–42. doi: 10.1016/0923-2494(90)90099-k. [DOI] [PubMed] [Google Scholar]
- 26.De Jesus M, Nicola AM, Rodrigues ML, Janbon G, Casadevall A. Capsular localization of the Cryptococcus neoformans polysaccharide component galactoxylomannan. Eukaryot Cell. 2009;8:96–103. doi: 10.1128/EC.00331-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curfs JH, Meis JF, Hoogkamp-Korstanje JA. A primer on cytokines: sources, receptors, effects, and inducers. Clin Microbiol Rev. 1997;10:742–780. doi: 10.1128/cmr.10.4.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bozic CR, Kolakowski LF, Jr., Gerard NP, Garcia-Rodriguez C, von Uexkull- Guldenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol. 1995;154:6048–6057. [PubMed] [Google Scholar]
- 29.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 30.Dockrell DH. The multiple roles of Fas ligand in the pathogenesis of infectious diseases. Clin Microbiol Infect. 2003;9:766–779. doi: 10.1046/j.1469-0691.2003.00669.x. [DOI] [PubMed] [Google Scholar]
- 31.Smith JB, Herschman HR. Targeted identification of glucocorticoid- attenuated response genes: in vitro and in vivo models. Proc Am Thorac Soc. 2004;1:275–281. doi: 10.1513/pats.200402-017MS. [DOI] [PubMed] [Google Scholar]
- 32.Dromer F, Salamero J, Contrepois A, Carbon C, Yeni P. Production, characterization, and antibody specificity of a mouse monoclonal antibody reactive with Cryptococcus neoformans capsular polysaccharide. Infect Immun. 1987;55:742–748. doi: 10.1128/iai.55.3.742-748.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckert TF, Kozel TR. Production and characterization of monoclonal antibodies specific for Cryptococcus neoformans capsular polysaccharide. Infect Immun. 1987;55:1895–1899. doi: 10.1128/iai.55.8.1895-1899.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozel TR, Cazin J., Jr. Induction of humoral antibody response by soluble polysaccharide of Cryptococcus neoformans. Mycopathol Mycol Appl. 1974;54:21–30. doi: 10.1007/BF02055969. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee J, Scharff MD, Casadevall A. Protective murine monoclonal antibodies to Cryptococcus neoformans. Infect Immun. 1992;60:4534–4541. doi: 10.1128/iai.60.11.4534-4541.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casadevall A, Mukherjee J, Scharff MD. Monoclonal antibody based ELISAs for cryptococcal polysaccharide. J Immunol Methods. 1992;154:27–35. doi: 10.1016/0022-1759(92)90209-c. [DOI] [PubMed] [Google Scholar]
- 37.Brendolan A, Rosado MM, Carsetti R, Selleri L, Dear TN. Development and function of the mammalian spleen. Bioessays. 2007;29:166–177. doi: 10.1002/bies.20528. [DOI] [PubMed] [Google Scholar]
- 38.Monari C, Bevilacqua S, Piccioni M, Pericolini E, Perito S, Calvitti M, Bistoni F, Kozel TR, Vecchiarelli A. A microbial polysaccharide reduces the severity of rheumatoid arthritis by influencing th17 differentiation and proinflammatory cytokines production. J Immunol. 2009;183:191–200. doi: 10.4049/jimmunol.0804144. [DOI] [PubMed] [Google Scholar]