

Abstract
Objective
To compare the relative rates and risk factors associated with stroke in adults vs. children with sickle cell disease (SCD) in the U.S. over the last decade.
Methods
We identified incident strokes in patients with SCD using ICD-9 codes for acute stroke and SCD and the California Patient Discharge Databases. We estimated SCD prevalence by using the incidence of SCD at birth with adjustment for early mortality from SCD.
Results
We identified 255 acute strokes (70 primary hemorrhagic and 185 ischemic) among 69,586 hospitalizations for SCD-related complications from 1998–2007. The rate of stroke in children [<18 years old (310/100,000 person-years)] was similar to young adults [18 to 34 years old (360/100,000 person-years)], but much higher in middle-aged [35 to 64 years old (1160/100,000 person-years)] and elderly adults [≥65 years old (4700/100,000 person-years)]. Stroke was associated with hypertension in children and hypertension, diabetes mellitus, hyperlipidemia, atrial fibrillation, and renal disease in adults. Most acute strokes (75%) and in-hospital deaths from stroke (91%) occurred in adults.
Interpretation
Our results suggest that the rate of stroke in SCD peaks in older adults and is 3-fold higher than rates previously reported in African-Americans of similar age (35 to 64 years) without SCD. Stroke in SCD is associated with several known adult risk factors for ischemic and hemorrhagic stroke. Studies for the primary and secondary prevention of stroke in adults with SCD are urgently needed.
Keywords: Stroke, sickle cell disease, epidemiology, transfusion
Introduction
Sickle cell disease (SCD) confers a greatly increased risk of ischemic and primary hemorrhagic stroke. The association with stroke was first described by Sydentricker in a three-year-old child with left hemiparesis and presumably sickle cell anemia (HbSS) (1). The predisposition to stroke in early childhood has been confirmed in multiple case series (2) and cohort studies, with an incidence of first stroke of 500 to 1000 per 100,000 person-years in those with HbSS (3–5) and 400 to 800 for all patients with SCD (3, 5). The high rate of first and recurrent stroke (3) have been the impetus for multiple studies of primary (6–8) and secondary (9–12) prevention of stroke in children with SCD. However, there have not been rigorous studies of stroke prevention in adults with SCD. The few studies describing the epidemiology of stroke in adults with HbSS have been limited by small size and provide imprecise and likely biased estimates of the incidence and prevalence of stroke, and associated outcomes (3, 5, 13, 14). We hypothesize that both the number and incidence rate of stroke in adults with SCD has increased from earlier reports since the widespread use of interventions to prevent early mortality from SCD (15).
RESULTS
Stroke Incidence
From 1998 to 2007, there were 255 discharges for acute stroke (70 primary hemorrhagic and 185 ischemic) among 69,586 discharges (including in-hospital deaths) from acute care hospitals for patients with SCD. Discharged patients with stroke had the diagnosis of homozygous sickle cell (51.8%), SCD, not otherwise specified (40.4%), other SCD (2.8%), sickle-hemoglobin C disease (2.4%), or sickle-thalassemia (2.8%). Information on age was masked in 12% of the records of patients with SCD. We estimated that there were 43,542 person-years of observation, including 19,616 for children and 23,926 for adults. Rates of stroke were similar for children <18 years old (310/100,000 person-years, 95% CI 230–390) and young adults 18 to 34 years old (360/100,000 person-years, 95% CI 270–480); however, they increased significantly in middle-aged adults 35 to 64 years old (1160/100,000 person-years, 95% CI 950–1,390, p<0.001) and older adults ≥65 years old (4700/100,000 person-years, 95% CI 2,700–7,600, p<0.001) compared to children. Adults 35 to 64 years old had the greatest absolute number of ischemic strokes and the highest incidence rates of ischemic stroke were in these middle-aged (740/100,000 person-years) and older adults [3500/100,000 person-years (Table 1)]. The variation in rates and associated confidence intervals for incident stroke were greater for incidence by 5 year-age groups than for other sub-analyses, since there were relatively few events within each age group (Table 2, Figure 1).
Table 1.
Stroke Type and Mortality by Age Group in Sickle Cell Disease
| Age Group | Hemorrhagic | Case Fatality | Ischemic | Case Fatality |
|---|---|---|---|---|
| Children <18 years | 8 | 13% | 52 | 4% |
| Young Adults 18 to 34 years | 16 | 31% | 35 | 9% |
| Older Adults 35 to 64 years | 40 | 38% | 71 | 7% |
| Elderly Adults ≥65 years | 4 | 0% | 12 | 8% |
| Unknown | 2 | 0% | 15 | 13% |
| Total | 70 | 30% | 185 | 7% |
Table 2.
Rate of Acute Stroke and Person-Years of Observation by Age Group (online publication only)
| Age group | Stroke per 100,000 person-years (95% CI) | Person-Years of Observation |
|---|---|---|
| <5 years | 78 (21 – 201) | 5101 |
| 5 – 9 years | 454 (299 – 660) | 5950 |
| 10 – 14 years | 283(162 – 460) | 5650 |
| 15 – 19 years | 205 (98 – 377) | 4875 |
| 20 – 24 years | 499 (309 – 762) | 4211 |
| 25 – 29 years | 329 (175 – 563) | 3951 |
| 30 – 34 years | 387 (217 – 638) | 3875 |
| 35 – 39 years | 594 (368 – 908) | 3534 |
| 40 – 44 years | 456 (236 –797) | 2631 |
| 45 – 49 years | 1680 (1130 – 2430) | 1717 |
| 50 – 54 years | 2750 (1780 – 4060) | 910 |
| 55 – 59 years | 2760 (1510-4630) | 507 |
| 60 – 64 years | 2330 (936 – 4800) | 301 |
Figure 1.
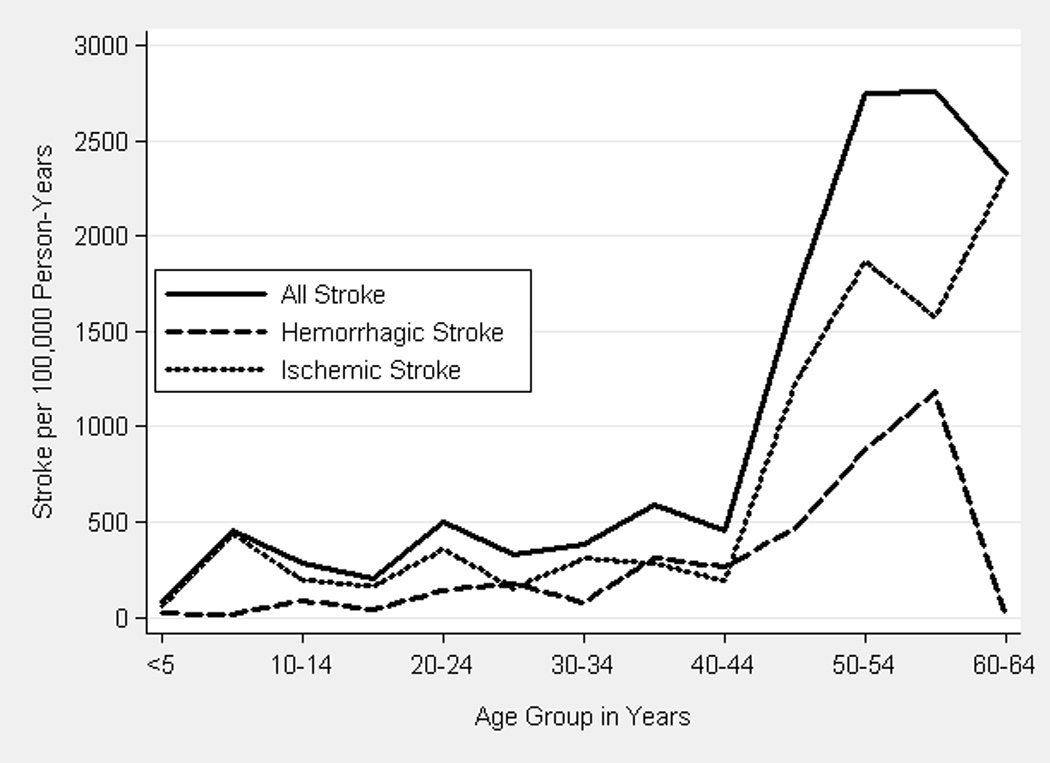
Rates of Acute Hemorrhagic and Ischemic Stroke by 5 Year Age Group
Primary hemorrhagic stroke accounted for 13% of pediatric (32/100,000 person-years) and 34% of adult acute stroke, with a peak in both absolute numbers and incidence in adults 35 to 64 (330/100,000 person-years) and ≥65 years old [1100/100,000 person-years (Table 1, Figure 1)]. Rates of hemorrhagic stroke were similar by calendar year for children and adults <65 years of age, but, on average, decreased 19% (95% CI 1–38%, p<0.05) per year for adults ≥65 years old.
Case Fatality Rates
Thirty-four (7%, 95% CI 5.1–10.0) of the 467 deaths in hospitalized patients with SCD were associated with acute stroke. Deaths were seen in all age groups, with an overall case fatality rate of 13% for acute stroke. The case fatality rate per SCD admission (other than for acute stroke) was significantly lower (0.7%, 95% CI 0.6–0.72) than that of ischemic (7%, 95% CI 3.8–11.7) or hemorrhagic stroke (30%, 95% CI 19.6–42.1, p<0.0001 for pair wise comparisons). Case fatality rates were similar across age groups (Table 1).
Length of Stay and Hospital Costs
Mean length of stay was similar for ischemic (9 days, median 6, IQR 3–10) and hemorrhagic stroke (10 days, median 7, IQR 2–15, p >0.5) but on average significantly longer than for other diagnoses in patients with SCD (6 days, median 4, IQR 2–7, p<0.001 for ischemic and hemorrhagic stroke). Hospital charges were significantly higher for hemorrhagic stroke (median $79,307, IQR 24,301–153,405) compared to ischemic stroke (median $32,216, IQR 19,214–63,704, P<0.005) or other diagnoses ($16,005, IQR 8,519–30,815, p<0.0001)
Comorbid Conditions and Treatments
Acute stroke in SCD was strongly associated with ICD-9 codes for a number of comorbid conditions that are known risk factors for stroke. In adults, we identified a significantly increased odds ratio for hypertension, hyperlipidemia, renal disease, and atrial fibrillation for ischemic stroke and for hypertension, renal disease and coagulopathy for hemorrhagic stroke (Table 3). Hypertension was associated with hemorrhagic and ischemic stroke in children and adults. The association with hypertension was significantly stronger for hemorrhagic than ischemic stroke (Table 3). A diagnosis of renal failure (OR 3.6, 95% CI 1.3–9.4 p<0.005) or coagulopathy [OR 7.2 (1.2–40, p<0.005] was associated with an increased risk of death from acute stroke. There were no significant differences in mortality for patients with other risk factors for stroke. A diagnosis of asthma was not significantly associated with all acute stroke, but was less frequent in patients with hemorrhagic stroke. A diagnosis of sickle cell crisis was significantly less frequent (37%) in patients with stroke compared to those without (72%). Transfusion was more frequent in patients with ischemic (48%) vs. hemorrhagic stroke (26%) and in children (53%) vs. adults (39%, P<0.05). Only 26% of transfusions for ischemic stroke and 6% for hemorrhagic stroke were coded as exchange transfusions.
Table 3.
Odds Ratio (and 95% CI) of Stroke Compared to No Stroke for Co-morbid Diagnoses and Procedures
| Diagnosis | Prevalence | Ischemic | P-Value | Hemorrhagic | P-Value |
|---|---|---|---|---|---|
| Hypertension | 9.5% | 4.1 (2.9 – 5.7) | <0.0001 | 7.7 (4.7 – 12.7) | <0.0001 |
| Obesity | 1.7% | 1.0 (0.2 – 3.0) | NS | 0 (0 – 3.3) | NS |
| Tobacco use | 7.6% | 1.2 (0.7 – 2.0) | NS | 1.2 (0.4 – 2.7) | NS |
| Diabetes mellitus | 3.4% | 2.2 (1.2 – 3.9) | <0.05 | 0.9 (0.1 – 3.2) | NS |
| Hyperlipidemia | 0.7% | 6.9 (2.9 – 14) | <0.0001 | 0 (0 – 8.4) | NS |
| Renal disease | 2.6% | 4.2 (2.4 – 6.8) | <0.0001 | 7.2 (3.4 – 13.9) | <0.0001 |
| Myocardial Infarction (old) | 0.7% | 3.1 (0.8 – 8.1) | <0.05 | 0 (0 – 7) | NS |
| Atrial Fibrillation | 1.1% | 4.9 (2.2 – 9.5) | <0.0005 | 4.3 (0.9 – 13) | <0.05 |
| Coagulopathy | 0.8% | 1.9 (0.4 – 5.8) | NS | 9.1(2.8 – 22.4) | <0.0005 |
| Asthma | 10.0% | 0.9 (0.5 – 1.4) | NS | 0.3 (0.03 – 0.99) | <0.05 |
| Crisis | 70.6% | 0.3 (0.2 – 0.34) | <0.0001 | 0.2 (0.1 – 0.3) | <0.0001 |
| Transfusion | 27.5% | 2.4 (1.8 – 3.2) | <0.0001 | 0.9 (0.5 – 1.6) | NS |
NS indicated not statistically significant at P<0.05.
Discussion
We provide, to our knowledge, the first population-based incidence of stroke in adults with SCD. By using a large database of discharge diagnoses and a stringent administrative definition of acute stroke, we were able to estimate the incidence of stroke in all patients in California with a known diagnosis of SCD. These results suggest that older adults have the greatest absolute number and rate of both hemorrhagic and ischemic stroke in patients with SCD. We found that the rate of stroke in patients with SCD was 3 fold higher than that of stroke in African-Americans of similar age (35 to 64 years). We conclude that SCD is an important risk factor for stroke in older adults with SCD as it is in young adults (approximately 20 fold increased risk) (24).
Our estimates of the incidence of stroke in children with SCD (310/100,000 person-years) were substantially lower than those reported from a large cohort study in southern California (800/100,000 person-years for initial stroke) (3), but similar to those reported in the Cooperative Study of Sickle Cell Disease (CSSCD, 400/100,000) and the Baltimore-Washington Cooperative Study of Stroke (285/100,000) (5, 25). They are also lower than those reported by Fullerton et al. in a study of first stroke in children with HbSS that also used the California Patient Discharge Databases for 1991 to 1998 (880 per 100,000 person years) (22). This may reflect their exclusion of other genotypes of SCD with lower stroke incidence than HbSS or our exclusion of ICD-9 codes with poor specificity for acute stroke, including codes for transient ischemic attacks, late effects of stroke, and occlusion of the precerebral arteries. An alternative explanation is that the incidence of stroke has truly decreased during the late 1990’s. Fullerton et. al reported much lower rates of stroke for 1999 and 2000 (500 and 170/100,000 person-years) and postulated that this was the result of widespread screening for elevated cerebral blood flow velocities by transcranial Doppler ultrasound and treatment of those at high risk with regular blood transfusions (22).
Our estimates of stroke incidence are higher than those reported from the CSSCD for older adults (26), but a four-decade cohort study of patients with SCD from a single institution also demonstrated a higher rate of hemorrhagic stroke in patients 35 to 54 years old (13). The CSSCD may have underestimated stroke rates in older adults because of survival or enrolment bias, since patients with more severe SCD were less likely to survive to the 5th and 6th decade of life. In addition, both cohorts had fewer person-years of observation for adults ≥35 years old, resulting in much less precise estimates of stroke rates than this study (5, 13).
We also found differences in case fatality rates. We found similar overall hospital mortality to the CSSCD [13% (34/255) vs. 8% (11/133, P=NS) after acute stroke with higher hospital mortality for ischemic [7% (13/185) vs. 0% (0/96, P<0.01)] but not hemorrhagic stroke [30% (21/70) vs. 26% (9/35, P=NS). We propose that the case fatality rate for ischemic stroke in SCD may be higher outside formal cohort studies or dedicated centers that provide comprehensive care for SCD. We also identified potentially higher overall stroke mortality in children (5%) than an earlier study using the same California discharge data and limited to first stroke in children with SCD (1.1%, p=0.14) (22). This may reflect an increased risk of death with recurrent stroke or our more stringent definition of stroke. Case fatality rates for primary hemorrhagic stroke in SCD are similar to the general population (20–45%) (27), but significantly lower than those reported for ischemic stroke in children (15%) (28) or adults (10–20%) in the general population (27) .
We evaluated several previously identified risk factors for stroke, including hypertension (29) obesity, tobacco use, diabetes mellitus, hyperlipidemia, renal disease, history of myocardial infarction, atrial fibrillation, and coagulopathy (23, 30), as well as asthma, and concomitant crisis. We found a strong relationship between stroke and hypertension and also identified new risk factors for stroke in SCD, including diabetes mellitus, hyperlipidemia, renal disease, and atrial fibrillation. Hypertension and coagulopathy were more strongly associated with hemorrhagic than ischemic stroke; however, the temporal relationship between hypertension and stroke cannot be established from this study, since hypertension may be secondary to increased intracranial pressure from stroke. High blood pressure has been associated with an increased volume of hemorrhage (31) and increased mortality (32) in stroke, but we identified an increased risk of death only in those with concomitant renal failure or coagulopathy. End-stage renal failure is associated with a greatly increased risk of both ischemic and hemorrhagic stroke in the general population. This risk has been attributed to both the accelerated atherosclerotic vascular disease and the bleeding diathesis associated with renal failure. In addition, patients on dialysis for end stage renal failure have an increased risk of death after both intracranial hemorrhage (56% vs. 16%) and ischemic stroke (28% vs. 17%) than the population overall (33). One potential strategy to decrease stroke incidence and mortality in patients with SCD might be to aggressively treat known risk factors for stroke.
It is surprising that only about half of children with acute ischemic stroke had a transfusion documented during their hospital admission. This is higher than the 33% of children that received transfusion in an earlier study which used the California discharge database(22), and may reflect incomplete ascertainment of transfusion, errors in the coding, or lower rates of transfusion at certain facilities. Immediate transfusion remains the standard treatment for acute ischemic stroke in children with SCD (34). The relative benefit of an immediate simple transfusion versus the delay often required to coordinate an exchange transfusion remains an area of controversy; however, a retrospective study from 14 pediatric sickle cell centers demonstrated a substantial decrease in risk of recurrent stroke in children initially treated with exchange transfusion when compared to those receiving simple transfusion (35). For adults with SCD, treatment recommendations for acute ischemic stroke include urgent consultation with a hematologist about the utility of transfusion, aspirin, and thrombolytics, if strict eligibility criteria are met (34, 36).
This study had several limitations. We used an administrative dataset to identify stroke and other diagnoses, based on ICD-9 coding that may not have accurately reflected the actual diagnoses. However, we limited the ICD-9 codes for stroke to those most reliably associated with acute stroke in adults (17, 18). Similar strategies have only been evaluated for all (acute and prevalent) stroke in children (37). In addition, we only included hospitalized patients and some patients (with severe hemorrhagic stroke) may die before admission while patients with mild stroke may not seek medical attention or be treated as outpatients. Our conservative assumptions likely resulted in an underestimate of the number of strokes and co-morbid conditions and may have decreased the power to detect associations between stroke and other conditions.
Our estimates of the number of patients alive with SCD depends on several assumptions, such as the prevalence of SCD at birth, the population of California by age and ethnicity, and the age-specific survival of people with SCD and African-Americans overall. The first two assumptions were based on high-quality data sources (the 2000 Census and the published newborn screening results for California), but survival was calculated based on the age of death for all patients with a diagnosis of SCD who died in non-federal hospitals in California. We assumed that the age of death was similar for people with SCD who died outside of a hospital setting and that the age of death had not changed over time. Our estimates of survival were similar to those obtained for sickle cell anemia from the CSSCD (38) and a large Californian cohort study (13), but about 10 years less than a Jamaican cohort study that adjusted for excess mortality before registration in the cohort (39). An increase in estimated survival would result in a decrease in our estimates of stroke incidence particularly in the older age groups. This is unlikely, because our population-based estimates likely underestimate mortality in the patients born before newborn screening and other advances in care for SCD and reflect a broad range of patients with SCD (not just those enrolled at referral centers).
In this study of SCD, the burden of acute stroke was significantly greater in adults than in children, with 75% of total strokes and 91% of deaths in adults with SCD. This disparity may become even more pronounced with the widespread adoption of screening with transcranial Doppler ultrasound and regularly scheduled transfusion to maintain sickle hemoglobin less than 30% for the primary prevention of stroke in children with SCD (6, 22). Primary hemorrhagic stroke continues to account for most of the mortality from stroke, and the role of emergency or scheduled transfusion or other treatments for this complication in children and adults is poorly defined (23). Likewise, the recommendations for the treatment of ischemic stroke in adults with SCD are based almost entirely on expert opinion and extrapolation from children with SCD and the general population (34), since there have been few reports of treatment of stroke (40, 41) and no studies of prevention of stroke in adults with SCD (36). Prospective studies of both treatment (acute transfusion, aspirin or other platelet antagonists, or thrombolytics) and secondary prevention (hydroxyurea, platelet antagonists, or regularly scheduled transfusion) of stroke are warranted and should be feasible, given the ease of identifying those at increased risk (known SCD) and the frequency of events in adults with SCD.
Methods
We obtained the public dataset of California Patient Discharge Databases (1998–2007) from the California Office of Statewide Planning and Development. This dataset includes up to 25 discharge diagnoses and up to 21 procedure codes per patient, and other variables that include age, sex, ethnicity, length of stay, hospital charges, source of admission, type of insurance, and disposition for over three million discharges (and deaths) per year from non-federal hospitals in California. To protect patient confidentiality, individual records with a unique combination of selected demographic variables have one of more of these variables masked to prevent identification. We extracted discharges related to SCD by International Classification of Disease, 9th revision (ICD-9) code (16). Use of this administrative dataset without personal identifiers was exempt from IRB review.
Inclusion/Exclusion Criteria
We included discharges and deaths from acute care hospitals with any diagnostic code for SCD [sickle-thalassemia (282.41-2), SCD, unspecified (282.60), HbSS (282.61-2), sickle-hemoglobin C disease (282.63-4), and other SCD (282.68-9)] but excluded sickle cell trait (282.5) and screening for SCD (V78.2). We identified patients with acute stroke by the ICD-9 codes for primary hemorrhagic [subarachnoid hemorrhage (SAH, 430) or intracerebral hemorrhage (ICH, 431) or ischemic stroke [(occlusion of cerebral arteries, (434) or acute, but ill defined cerebrovascular disease, (436)] in the first three discharge diagnoses (16). ICD-9 codes more commonly associated with prior stroke or cerebrovascular disease without acute stroke were not included [occlusion and stenosis of precerebral arteries (433), transient cerebral ischemia (435), other and ill-defined cerebrovascular disease (437), or late effects of cerebrovascular disease (438)]. We also excluded ICD-9 codes for cerebral sinus thrombosis (325) and other and unspecified intracranial hemorrhage. This approach has a high sensitivity and specificity for the identification and classification of acute stroke from administrative databases (17, 18) and permits the inclusion of recurrent acute strokes.
Population at Risk
We estimated the number of persons at risk by using the prevalence of SCD by race identified by the California newborn screening program from 1990 to 1996 (19), and the population of California by race and 5 year age groups from the 2000 United States Census (20). To estimate the number of people with SCD by 5 year age group, we multiplied the birth prevalence of SCD for each racial group by the number of people in each age group (assuming no increase in mortality for SCD). Adjustment for early mortality was based on proportion of deaths among patients with SCD from the California Discharge Databases by age group divided by the survival reported for African- Americans in the United States for 2000 for the midpoint of each 5 year age group (21). We assumed that the prevalence of SCD at birth remained constant for each group (22) and that survival was constant for both those with SCD and African-Americans for the period of the study.
Validation
We compared the performance of this ICD-9 based strategy to identify strokes in patients with SCD to those included in an existing research database of strokes in patients with SCD at Johns Hopkins Hospital from 1994–2008 with confirmation by review of medical records (23). We identified 31 of 42 acute strokes using ICD-9 codes and 8 false positives. The ICD-9 based strategy had a sensitivity of 74% (95% CI 58–86) and a specificity of 99.9% (95% CI 99.8–99.96) for the identification of discharges related to acute stroke in patients with SCD.
Incidence and Case Fatality Rates
We estimated the incidence rates of all acute strokes, as well as hemorrhagic and ischemic strokes, as the number of events divided by the number of person-years at risk for each age group. We calculated person-years at risk as the population for each age group multiplied by the 10 years of observation. We calculated case fatality rates as the number of hospital deaths divided by the number of events for each stroke subtype.
Comorbid Conditions and Treatments
To examine the association between comorbid conditions, treatments, and stroke, we identified patients with ICD-9 codes for hypertension, obesity, tobacco use, diabetes mellitus, hyperlipidemia, renal disease, history of myocardial infarction, atrial fibrillation, coagulopathy, asthma, sickle cell crisis, and procedure codes for transfusion.
Statistical Analysis
We calculated 95% confidence intervals for incidence rates and odd ratios using exact methods and compared incidence rates between and among age groups and years by Poisson regression. We compared charges and length of stay among groups with the Wilcoxon rank sum test and the Kruskal-Wallis test for equality of populations, since these variables were not normally distributed. We compared case fatality rates by Fisher’s exact test and used Intercooled Stata 10.1 (Stata Corporation, College Station, TX) for all analyses.
Acknowledgement
Dr. Strouse was supported by the NIH (K23HL078819), the American Society of Hematology and the Doris Duke Charitable Foundation. Dr. Jordan was supported by the NIH (K23NS062110). Dr. Casella was supported by the NIH Basic and Translation Research Program 5U54HL090515 and NIH grants 1RO1HL091759 and 5K12HL087169.
Footnotes
California Office of Statewide Planning and Development provided the public-use data without charge for academic use.
None of the authors have financial conflicts of interest related to this manuscript.
REFERENCES
- 1.Sydentricker V, Mulherin W, Houseal R. Sickle Cell Anemia: Report of Two Cases in Children with Necropsy in One Case. American Journal of Disease in Children. 1923;26:132–154. [Google Scholar]
- 2.Obama MT, Dongmo L, Nkemayim C, Mbede J, Hagbe P. Stroke in children in Yaounde, Cameroon. Indian Pediatr. 1994;31:791–795. [PubMed] [Google Scholar]
- 3.Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65:461–471. doi: 10.1016/0002-9343(78)90772-6. [DOI] [PubMed] [Google Scholar]
- 4.Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr. 1992;120:360–366. doi: 10.1016/s0022-3476(05)80897-2. [DOI] [PubMed] [Google Scholar]
- 5.Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294. [PubMed] [Google Scholar]
- 6.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 7.Riddington C, Wang W. Blood transfusion for preventing stroke in people with sickle cell disease. Cochrane Database Syst Rev. 2002:CD003146. doi: 10.1002/14651858.CD003146. [DOI] [PubMed] [Google Scholar]
- 8.The Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP II) Trial Investigators. Discontinuing Prophylactic Transfusions Used to Prevent Stroke in Sickle Cell Disease. N Engl J Med. 2005;353:2769–2778. doi: 10.1056/NEJMoa050460. [DOI] [PubMed] [Google Scholar]
- 9.Cohen AR, Martin MB, Silber JH, Kim HC, Ohene-Frempong K, Schwartz E. A modified transfusion program for prevention of stroke in sickle cell disease. Blood. 1992;79:1657–1661. [PubMed] [Google Scholar]
- 10.Sarnaik S, Soorya D, Kim J, Ravindranath Y, Lusher J. Periodic transfusions for sickle cell anemia and CNS infarction. Am J Dis Child. 1979;133:1254–1257. doi: 10.1001/archpedi.1979.02130120046009. [DOI] [PubMed] [Google Scholar]
- 11.Ware RE, Zimmerman SA, Sylvestre PB, Mortier NA, Davis JS, Treem WR, Schultz WH. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. J Pediatr. 2004;145:346–352. doi: 10.1016/j.jpeds.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 12.Wang WC, Kovnar EH, Tonkin IL, Mulhern RK, Langston JW, Day SW, Schell MJ, Wilimas JA. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J Pediatr. 1991;118:377–382. doi: 10.1016/s0022-3476(05)82150-x. [DOI] [PubMed] [Google Scholar]
- 13.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 14.Njamnshi AK, Mbong EN, Wonkam A, Ongolo-Zogo P, Djientcheu Vd-P, Sunjoh FL, Wiysonge CS, Sztajzel R, Mbanya D, Blackett KN, Dongmo L, Muna WFT. The epidemiology of stroke in sickle cell patients in Yaounde, Cameroon. Journal of the Neurological Sciences. 2006;250:79–84. doi: 10.1016/j.jns.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Sickle Cell Research for Treatment and Cure. National Institutes of Health, National Heart, Lung, and Blood Institute; 2002. [Google Scholar]
- 16.Physician ICD-9-CM. Clinical Modification. 9th Revision. Dover: American Medical Association; 2007. International Classification of Disease. [Google Scholar]
- 17.Goldstein LB. Accuracy of ICD-9-CM coding for the identification of patients with acute ischemic stroke: effect of modifier codes. Stroke. 1998;29:1602–1604. doi: 10.1161/01.str.29.8.1602. [DOI] [PubMed] [Google Scholar]
- 18.Tirschwell DL, Longstreth WT., Jr Validating administrative data in stroke research. Stroke. 2002;33:2465–2470. doi: 10.1161/01.str.0000032240.28636.bd. [DOI] [PubMed] [Google Scholar]
- 19.Lorey FW, Arnopp J, Cunningham GC. Distribution of hemoglobinopathy variants by ethnicity in a multiethnic state. Genet Epidemiol. 1996;13:501–512. doi: 10.1002/(SICI)1098-2272(1996)13:5<501::AID-GEPI6>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.US Census Bureau; American FactFinder. 2006 US Census Bureau: http://factfinder.census.gov/.
- 21.Arias E. National vital statistics reports. Hyattsville, Maryland: National Center for Health Statistics; 2002. United States life table, 2000. [Google Scholar]
- 22.Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood. 2004;104:336–339. doi: 10.1182/blood-2004-02-0636. [DOI] [PubMed] [Google Scholar]
- 23.Strouse JJ, Hulbert ML, DeBaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics. 2006;118:1916–1924. doi: 10.1542/peds.2006-1241. [DOI] [PubMed] [Google Scholar]
- 24.Kissela B, Schneider A, Kleindorfer D, Khoury J, Miller R, Alwell K, Woo D, Szaflarski J, Gebel J, Moomaw C, Pancioli A, Jauch E, Shukla R, Broderick J. Stroke in a Biracial Population: The Excess Burden of Stroke Among Blacks. Stroke. 2004;35:426–431. doi: 10.1161/01.STR.0000110982.74967.39. [DOI] [PubMed] [Google Scholar]
- 25.Earley CJ, Kittner SJ, Feeser BR, Gardner J, Epstein A, Wozniak MA, Wityk R, Stern BJ, Price TR, Macko RF, Johnson C, Sloan MA, Buchholz D. Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative Young Stroke Study. Neurology. 1998;51:169–176. doi: 10.1212/wnl.51.1.169. [DOI] [PubMed] [Google Scholar]
- 26.Ohene-Frempong K. Stroke in sickle cell disease: demographic, clinical, and therapeutic considerations. Semin Hematol. 1991;28:213–219. [PubMed] [Google Scholar]
- 27.Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. The Lancet Neurology. 2003;2:43–53. doi: 10.1016/s1474-4422(03)00266-7. [DOI] [PubMed] [Google Scholar]
- 28.Fullerton HJ, Wu YW, Zhao S, Johnston SC. Risk of stroke in children: ethnic and gender disparities. Neurology. 2003;61:189–194. doi: 10.1212/01.wnl.0000078894.79866.95. [DOI] [PubMed] [Google Scholar]
- 29.Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G, Vichinsky E. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. 1997;102:171–177. doi: 10.1016/s0002-9343(96)00407-x. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein LB, Adams R, Alberts MJ, Appel LJ, Brass LM, Bushnell CD, Culebras A, DeGraba TJ, Gorelick PB, Guyton JR, Hart RG, Howard G, Kelly-Hayes M, Nixon JV, Sacco RL. Primary Prevention of Ischemic Stroke: A Guideline From the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline. Stroke. 2006;37:1583–1633. doi: 10.1161/01.STR.0000223048.70103.F1. [DOI] [PubMed] [Google Scholar]
- 31.Broderick J, Connolly S, Feldmann E, Hanley D, Kase C, Krieger D, Mayberg M, Morgenstern L, Ogilvy CS, Vespa P, Zuccarello M. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage in Adults: 2007 Update: A Guideline From the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke. 2007;38:2001–2023. doi: 10.1161/STROKEAHA.107.183689. [DOI] [PubMed] [Google Scholar]
- 32.Willmot M, Leonardi-Bee J, Bath PMW. High Blood Pressure in Acute Stroke and Subsequent Outcome: A Systematic Review. Hypertension. 2004;43:18–24. doi: 10.1161/01.HYP.0000105052.65787.35. [DOI] [PubMed] [Google Scholar]
- 33.Iseki K, Fukiyama K. Clinical demographics and long-term prognosis after stroke in patients on chronic haemodialysis. Nephrol Dial Transplant. 2000;15:1808–1813. doi: 10.1093/ndt/15.11.1808. [DOI] [PubMed] [Google Scholar]
- 34.Stroke and Central Nervous System Disease. Bethesda, MD: National Institutes of Health, National Heart, Lung, and Blood Institute, Division of Blood Diseases and Resources; The Management of Sickle Cell Disease: NIH Publication No 02-2117. 2002
- 35.Hulbert ML, Scothorn DJ, Panepinto JA, Scott JP, Buchanan GR, Sarnaik S, Fallon R, Chu JY, Wang W, Casella JF, Resar L, Berman B, Adamkiewicz T, Hsu LL, Smith-Whitley K, Mahoney D, Woods G, Watanabe M, DeBaun MR. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr. 2006;149:710–712. doi: 10.1016/j.jpeds.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 36.Lottenberg R, Hassell KL. An Evidence-Based Approach to the Treatment of Adults with Sickle Cell Disease. Hematology. 2005;2005:58–65. doi: 10.1182/asheducation-2005.1.58. [DOI] [PubMed] [Google Scholar]
- 37.Golomb MR, Garg BP, Saha C, Williams LS. Accuracy and yield of ICD-9 codes for identifying children with ischemic stroke. Neurology. 2006;67:2053–2055. doi: 10.1212/01.wnl.0000247281.98094.e2. [DOI] [PubMed] [Google Scholar]
- 38.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 39.Wierenga KJ, Serjeant BE, Serjeant GR. Cerebrovascular complications and parvovirus infection in homozygous sickle cell disease. J Pediatr. 2001;139:438–442. doi: 10.1067/mpd.2001.117070. [DOI] [PubMed] [Google Scholar]
- 40.Oyesiku NM, Barrow DL, Eckman JR, Tindall SC, Colohan AR. Intracranial aneurysms in sickle-cell anemia: clinical features and pathogenesis. J Neurosurg. 1991;75:356–363. doi: 10.3171/jns.1991.75.3.0356. [DOI] [PubMed] [Google Scholar]
- 41.Vicari P, Sampaio Silva G, de Cassia Rosario Cavalheiro R, Massaro AR, Figueiredo MS. Fulminant stroke in an adult patient with sickle cell anemia. Acta Haematol. 2006;116:67–69. doi: 10.1159/000092351. [DOI] [PubMed] [Google Scholar]