

Abstract
Insulin-like growth factor-1 (IGF-1) is neuroprotective and improves long-term function after brain injury. However, its clinical application to neurological disorders is limited by its large molecular size, poor central uptake and mitogenic potential. Glycine-proline-glutamate (GPE) is naturally cleaved from the IGF-1 N-terminal and it is also neuroprotective after ischemic injury, which provided a novel strategy of drug discovery for neurological disorders. GPE is not enzymatically stable, thus intravenous infusion of GPE becomes necessary for stable and potent neuroprotection. The broad effective dose range and treatment window of 3–7 h after the lesion suggest its potential for treating acute brain injuries. G-2meth-PE, a GPE analogue designed to be more enzymatic resistant, has a prolonged plasma half-life and is more potent in neuroprotection. Neuroprotection by GPE and its analogue may involve modulation of inflammation, promotion of astrocytosis, inhibition of apoptosis and vascular remodelling. Acute administration of GPE also prevents 6-OHDA-induced nigrostrial dopamine depletion. Delayed treatment with GPE does not prevent dopamine loss, but improves long-term function. Cyclo-glycyl-proline (cyclic Gly-Pro) is an endogenous DKP that may be derived from GPE. Cyclic Gly-Pro and its analogue cyclo-L-glycyl-L-2-allylproline (NNZ 2591) are both neuroprotective after ischaemic injury. NNZ2591 is highly enzymatic resistant and centrally accessible. Its peripheral administration improves somatosensory-motor function and long-term histological outcome after brain injury. Our research suggests that small neuropeptides have advantages over growth factors in the treatment of brain injury, and that modified neuropeptides designed to overcome the limitations of their endogenous counterparts represent a novel strategy of pharmaceutical discovery for neurological disorders.
Keywords: endogenous neuropeptides, diketopiperazine, GPE, Gm-2PE, NNZ 2591, ischemic brain injury, Parkinson disease and rat
Introduction
Endogenous response of insulin-like growth factor-1 (IGF-1) to brain injury suggests a role for IGF-1 in neuroprotection
In the days following hypoxia-ischaemia (HI) injury in the developing rat brain, the mRNAs for IGF-1, its binding protein and its receptor are expressed with different temporal and spatial characteristics depending on the area of infarction and IGF-1 protein can be immunohistochemically shown to be associated with reactive glia (Gluckman and Ambler, 1992; Beilharz et al., 1995). Similar findings have also been reported by Lee and Bondy in immature (Lee et al., 1993; 1996) and adult (Yamaguchi et al., 1991) rats. These data raised the possibility that IGF-1 may play an important role as a neurotrophic peptide following brain injury.
IGF-1 is neuroprotective after HI injury
Central administration of IGF-1 prevents neuronal and white matter injury after HI brain injury in the adult rat (Guan et al., 1993) as well as ischaemia in the fetal sheep (Johnston et al., 1996; Guan et al., 2001a). The treatment effects are dose dependent, with a clear bell-shaped dose dependency of neuronal survival following a 30 min period of cerebral ischaemia in fetal sheep (Guan et al., 1993; Johnston et al., 1996). However, a continuous infusion of IGF-1 (1 or 3 µg) for 24 h in addition to the initial short infusion of IGF-1 (3 µg) did not improve neuroprotection compared with a short infusion alone (Guan et al., 2001a). Treatment with IGF-1 improves long-term somatomotor-sensory function examined using the bilateral tactile test and long-term histological outcome examined 3 weeks after the injury (Guan et al., 2001b). Whereas 50 µg of IGF-1 is protective when given 2 h after HI injury, it is not protective when administered 6 h after HI (Guan et al., 2000b). This relatively short window of opportunity is now understood to be related to the rapid evolution of post-ischaemic neuronal injury. However, this window of opportunity can be significantly prolonged to 6 h if rats are in a cooled environment during the first 2 h of recovery. Thus, injury-induced brief cerebral cooling may prolong the window of opportunity for other therapies.
Limitations to the clinical application of IGF-1
Although the central uptake of IGF-1 has been reported to be poor (Pardridge, 1997), neuroprotection by IGF-1 after peripheral administration has been reported on several occasions (Saatman et al., 1997; Liu et al., 2001). The potential metabolic and mitogenic effects of IGF-1 are the major concerns for its use in the treatment of neurological disorders.
N-terminal tripeptide of IGF-1, GPE
Discovery of GPE
Following the discovery of des-(1-3)-IGF-1, a truncated form of IGF-1 isolated from brain tissue, most scientific interest focused on establishing the difference between the native form of IGF-1 and its truncated analogue, des-IGF-1. The other product of IGF-1 cleavage, its N-terminal tripeptide (GPE), was until the late 1980s generally believed to be a non-bioactive by-product of IGF-1 metabolism. The bioactivity of this tripeptide in cell culture was first reported in 1989 (Sara et al., 1989). Synthetic GPE stimulates both dopamine and acetylcholine release in brain slice culture without interacting with IGF-1 receptors (Nilsson-Hakansson et al., 1993). A novel ion-channel-associated receptor was suggested to be involved in the mode of action, since GPE shows only partial displacement by an NMDA receptor antagonist in cell culture (Nilsson-Hakansson et al., 1993). The discovery of an acid protease in both plasma and brain tissues provided further evidence for the existence of GPE as an endogenous neurobioactive peptide (Yamamoto and Murphy, 1995a,b; Yamamoto et al., 1999). This endogenous cleavage can be enhanced in an acidic environment.
GPE is neuroprotective in vitro
Given that GPE is neuroactive and possibly interacts with NMDA receptors, the neuroprotective effect of GPE in vitro was first examined after NMDA-induced neuronal injury (Saura et al., 1999). Using organotypic culture, Saura et al. showed that GPE dose-dependently protects hippocampal neurons from NMDA-induced neuronal toxicity. Neuroprotection by GPE was similar to that by MK801, a non-competitive NMDA receptor antagonist. Despite the consistent NMDA effects of GPE previously reported by Bourguignon's group (Bourguignon et al., 1993; Bourguignon and Gerard, 1999), the authors ruled out the possibility of NMDA-receptor mediated neuroprotection by GPE because of the weak binding of tritiated GPE in the dentate gyrus, where NMDA receptors are strongly expressed (Saura et al., 1999).
Neuroprotection of GPE after central administration
Following the demonstration that GPE is neuroprotective in cell culture, its possible neuroprotective effects in vivo were initially examined in the HI model, the same model used for examining the neuroprotective effects of IGF-1. Following central administration of GPE at 2 h after HI injury, at a dose equimolar to the effective dose of IGF-1, there was a reduction in the extent of cortical infarction (Guan et al., 1999). As with IGF-1, the treatment effects were seen in most brain regions examined, particularly in the lateral cortex, striatum and some sub-regions of the hippocampus. Interestingly, a profound treatment effect of GPE was found in the CA1-2 sub-regions of the hippocampus, where IGF-1 has never been seen to be neuroprotective (Guan et al., 1996; 1999). The most obvious explanation for the lack of effect in this particular brain region would be a relatively lower density of IGF-1 receptors. This spatial difference between IGF-1 and GPE also suggests a unique mode of action for GPE in neuroprotection.
The striatum consists of multiple neuronal phenotypes and each of them is involved in different forms of neurotransmission. The majority of neurons in the striatum are GABAergic projection neurons, which express calbindin, while others express several different neuronal peptides associated with either cholinergic or GABAergic interneurons (Kawaguchi, 1997). Inspired by its ability to promote acetylcholine release, the authors also examined the possibility of selective neuroprotection by GPE in different neuronal phenotypes (Guan et al., 1999).
HI brain injury resulted in a significant loss of several phenotypes of striatal neurons, including ChAT-containing cholinergic neurons and GABAergic projection neurons (calbindin+) and various subgroups of GABA interneurons, which can be distinguished by neuronal markers such as GAD, neuronal NOS, somatostatin and parvalbumin. A single dose of GPE (3 µg) given centrally 2 h after HI injury selectively prevented the loss of ChAT+ cholinergic neurons and GAD- and SMT-immunopositive GABA interneurons, but not of those containing parvalbumin and neuronal NOS, in the striatum compared with the vehicle-treated control.
As it appears that the activity of GPE is somewhat lower in the developing brain, its neuroprotective effects were examined in infant rats. Using a modified Levine rat model in 21-day-old animals, a 10-fold higher dose of GPE (30 µg) was required for infant rats to achieve neuroprotective effects compared to the dose used in adult rats (3 µg) after central administration (Sizonenko et al., 2001).
With no interactions with the IGF-1 receptor and a small molecular size, GPE appeared to overcome the disadvantages that have prevented the clinical application of IGF-1 for CNS disorders; however, as an endogenous peptide, GPE is not enzymatically stable.
Pharmacokinetics of GPE
As determined with a locally developed radioimmunoassay and by HPLC mass spectrometry, the half-life of GPE in plasma is extremely short after single-bolus i.v. (<2 min) or i.p. (<4 min) administration to normal Wistar rats (Batchelor et al., 2003; Baker et al., 2005). Endogenous proteases appear to have a role in GPE metabolism, as Bestatin, a protease inhibitor, can depress GPE metabolism in both plasma and brain tissues (Baker et al., 2005). Protease activity in the CNS ex vivo appears to be lower than in plasma, which may provide an explanation for the longer half-life of GPE (>30 min) in CSF and brain tissues (Batchelor et al., 2003; Baker et al., 2005). Thus, to maintain efficacious plasma levels of GPE, continuous intravenous administration would be necessary to achieve consistent neuroprotection after ischaemic brain injury.
To determine a possible role for the BBB in central uptake of GPE, an efficacious dose of GPE was injected intravenously either 2 h after HI injury or to normal control rats. Elevation of GPE was observed only in HI injured rats, suggesting that central penetration of GPE is injury-dependent despite its small molecular size (Guan et al., 2004). It is well known that the BBB can become functionally and morphologically more permeable as a result of brain injury (Preston et al., 1993; Betz et al., 1994). Ischaemia-induced disruption of the BBB can be as early as 2 h after the injury due to the loss of tight junctions of the endothelium. However, the hydrophilic nature of GPE has been hypothesized to be a barrier to its central penetration. Elevations of MMP-2 and -9 can disrupt the tight junction of the BBB because of digestion of the endothelial basal lamina (Fujimura et al., 1999; Planas et al., 2001). Therefore, the loss of basal lamina due to activation of MMP-2 and -9 (Fujimura et al., 1999) would be associated more with GPE central uptake. Compared to substances with free access to the CNS, injury-mediated central penetration allows specificity to injured brain regions while limiting unwanted effects outside these regions.
Using a micro-autoradiographic technique, tritiated GPE signals were found to co-localize with morphologically identified glial cells (Sizonenko et al., 2001) and GFAP-positive astrocytes (unpublished data) when tritiated GPE was given centrally to infant rats with mild HI injury. This glial-specific localization of GPE may support a different mode of action in neuroprotection by GPE, as the tritiated IGF-1 signal is more specific to neurons, particularly those with apoptotic morphology (Guan et al., 2000a).
Neuroprotection of GPE after peripheral administration
A neuroprotective effect of GPE has been demonstrated after either an i.p. bolus injection or continuous i.v. infusion. Neuroprotection by GPE following a single bolus injection was limited and not reliable (Guan et al., 2004; Baker et al., 2005), which is probably a result of its enzymatic instability in plasma (Batchelor et al., 2003; Baker et al., 2005). To overcome the extremely short half-life of GPE, continuous intravenous infusion appears to maintain efficacious blood levels and thus stable central uptake. Intravenous infusion of GPE consistently achieved robust neuroprotection in all the brain regions examined (Figure 1), with a broad effective dose range between 0.3 and 30 mg·kg·h−1 over a 4 h continuous infusion (Table 1). Although the rapid plasma clearance of GPE suggests only limited application for chronic neurological conditions, this could be favourable for treating acute neurological conditions, since adverse effects associated with drug accumulation can be minimized (Guan et al., 2004).
Figure 1.
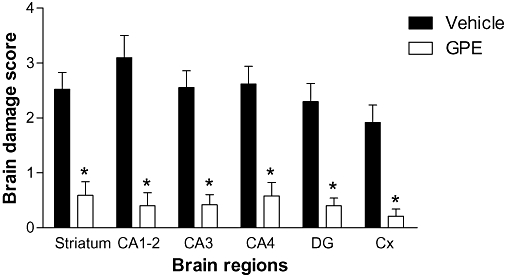
The protective effect of glycine-proline-glutamate (GPE) after hypoxia-ischemia injury in rats. A single i.v. bolus injection of GPE (3 mg·kg−1) followed by a continuous 4 h i.v. infusion of GPE (3 mg·kg·h−1) given 1–5 h after injury. Histology examined after 4 days. The group with GPE treatment significantly reduced the degree of brain damage compared to the group treated with the vehicle. Cx, lateral cortex, DG, dentate gyrus. *P < 0.05.
Table 1.
The dose range and the route of administration of IGF-1 and its related peptide after brain injuries
| i.c.v. | i.v. | i.p. | |
|---|---|---|---|
| IGF-1 | 5–50 µg in rats 1–3 µg in sheep | ||
| GPE | 3 µg in rats | 0.3–30 mg·kg·h−1 | 15 mg·kg−1 |
| G-2mPE | 1.2 mg·kg−1 | ||
| Cyclic GP | 0.2–2 µg | ||
| NNZ 2591 | 2–20 ng | 1 mg·kg−1 | |
| Cyclic HP | 1 mg·kg−1 | ||
| 35b | 1 mg·kg−1 |
GPE, glycine-proline-glutamate; G-2mPE, glycine-2methyl-proline-glutamate; IGF-1, insulin-like growth factor-1.
Another key practical issue in drug discovery for acute neurological conditions is that treatment of patients in a timely manner is a formidable problem (Fisher and Schaebitz, 2000). For example, despite the proven efficacy of anticoagulants after acute ischaemic stroke, the great majority of patients are not enrolled within the 3 h treatment window (Lees, 2002). The majority of compounds demonstrated to be neuroprotective in experimental models have a rather short window of opportunity of less than 3 h. Few compounds can be administered later than 6 h after injury, and such delayed treatment generally has lower efficacy than does early treatment. However, a delayed 4 h infusion of GPE initiated at either 3 or 7 h after HI injury showed a similar degree of neuroprotection compared with earlier administration initiated at 1 h after injury (Guan et al., 2004). Treatment was not effective when the 4 h infusion was initiated at 24 h after HI injury; no data exist for treatment between 7 and 24 h after injury. With a broad effective dose range, the extended window of opportunity that the GPE offers further promise to its clinical development for treating acute brain injury.
The secondary phase of neuronal loss after brain injury can continue in a progressive ‘tertiary’ phase over many months (Hsu et al., 1994; Coimbra et al., 1996; Dirnagl et al., 1999), as demonstrated in this particular rat model (Guan et al., 2001b; 2004). In reality, although most experimental studies report histological endpoints exclusively between 24 h and 1 week after initial injury, early protection by some compounds may not be maintained over the long term (Fisher and Schaebitz, 2000). Therefore, short-term neuronal outcome cannot be seen to reflect realistic efficacy of treatment and can be misleading in clinical development. Conversely, i.v. infusion of GPE initiated 1 h after HI injury improved long-term neuronal outcome examined 21 days later (Guan et al., 2004).
A well-recognized reason for failure in the transformation of results from animal models to humans is that preclinical development is evaluated more by histological outcomes, whereas clinical trials are generally evaluated by long-term functional and neurological outcomes (Fisher and Schaebitz, 2000). Our model of HI injury results in unilateral damage within the territory of the middle cerebral artery (Ginsberg and Busto, 1989; Guan et al., 2001b), which is associated with somatosensory function. This particular distribution of neuronal damage in the cerebral cortex results in loss of somatosensory-motor function, which can be assessed by measuring the time to contact a patch placed on the forelimb that has lost function. The rats that showed delayed contact to the patch also were often observed to miss the patch during the trial. This delayed response may suggest a deficiency of motor co-ordination, as loss of multiple phenotypic neurons in the striatum has been reported after HI injury (Guan et al., 1999). Like its parent peptide IGF-1, treatment with GPE improves recovery in somatosensory function (Guan et al., 2004).
Effect of GPE after 6-OHDA induced dopamine depletion
We used an animal model of 6-OHDA-induced nigrostriatal depletion to examine the specific treatment effect of GPE on dopamine neurons. Injection of 6-OHDA into the MFB leads to a massive loss of TH-immunopositive neurons and also severe loss of TH immunoreactivity in both the ipsilateral substantia nigra pars compacta (SNc) and the striatum 14 days later (Figure 2, Guan et al., 2000c). This animal model is considered to reflect some of the pathological aspects of human Parkinson's disease (Barbosa et al., 1997).
Figure 2.
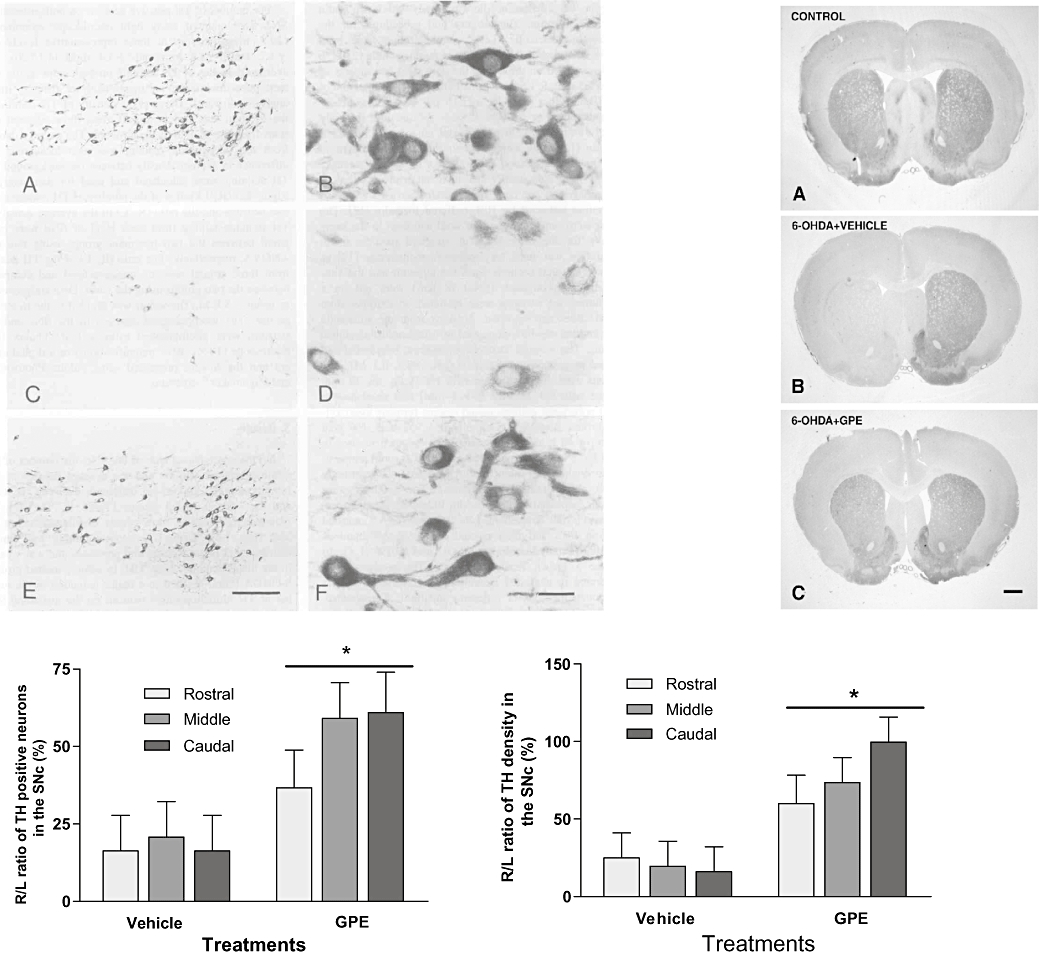
The photos show the distribution (A, C and E) and the morphology (B, D and F) of tyrosine-hydroxylase (TH) neurons of substantia nigra pars compacta (SNc) in normal control rat (A and B), the rat had 6-OHDA lesion and treated with the vehicle (C and D) and the rat with lesion and treated with GPE (E and F, left), as well as the effects of 6-OHDA lesion and GPE treatment on striatal TH terminals (right). In comparison with the vehicle-treated group, the treatment with GPE given 2 h after 6-OHDA lesion significantly prevented the loss of TH positive neurons (left) and the loss of TH terminals (right), particularly in the caudal level, *P < 0.05.
A single dose of GPE (3 µg) given intracerebroventricularly, a treatment regime known to be effective after ischaemic brain injury, at 2 h after the lesion prevented the loss of TH immunopositive neurons in the substantia nigra compared with the vehicle-treated group. The neuroprotection by GPE treatment was more pronounced at the caudal level and less pronounced at the rostral level. This difference is possibly due to the caudal neurons being more distal to the lesion, which may have resulted in a less severe and more reversible lesion (Figure 2, Guan et al., 2000c).
These results may also suggest a possible role for GPE in neuronal modulation in the dopaminergic system because of its ability to up-regulate TH immunoreactivity. This up-regulation in TH immunoreactivity appeared to involve both cytoplasm and the dendritic processes, perhaps related to enhanced transport of the enzyme. GPE treatment also may promote distal neurotransmission, as the TH immunoreactivity in the striatum was strongly increased compared with the vehicle-treated group (Guan et al., 2000c).
Although a single dose of GPE given 3 days after the lesion failed to prevent loss of dopamine neurons, the treatment significantly reduced the rotation, the result of unilateral dopamine oversensitivity and improved motor coordination at 12 weeks (Krishnamurthi et al., 2004). The delayed treatment was given after the onset of apomorphine-induced contralateral rotations, which normally occur after approximately 70–90% loss of nigrostriatal dopamine function (Schwarting and Huston, 1996). The effects of GPE on apomorphine-induced rotation appeared to be dose dependent, as either a lower or a higher dose failed to reduce the rotations. A similar bell-shaped dose-response effect of GPE has been reported after HI in infant rats (Sizonenko et al., 2001).
In addition to apomorphine-induced rotations, GPE treatment attenuated long-term forelimb akinesia as examined by Stepping Tests, a behavioural test for akinesia (Olsson et al., 1995; Krishnamurthi et al., 2004). In contrast to the effects of GPE on rotations, which tailed off at the end of experiment, the effects of GPE on step tests either remained or showed further improvement at the end of the experiment (Krishnamurthi et al., 2004). This difference in long-term recovery between rotation and step tests further suggests that apomorphine-induced rotations may be more indicative of nigrostriatal dopamine depletion, while stepping tests likely represent Parkinsonian motor dysfunction.
Mode of action of GPE
The mode of action of GPE is still not completely identified. However, a considerable amount of information for possible mode of action has been reported during the last two decades. Given that GPE does not interact with the IGF-1 receptor, hypotheses have mainly focused on NMDA receptors. With glycine and glutamate at the opposite ends of the molecule, GPE appears to have the perfect stereochemistry to activate NMDA receptors by binding to both the glutamate and glycine binding sites of the receptor. Using a model of NMDA-mediated secretion of GnRH, Bourguignon et al. examined the antagonistic effects of GPE on the NMDA receptor both in vivo and in vitro (Bourguignon et al., 1993; Bourguignon and Gerard, 1999). Potential NMDA antagonistic effects of GPE were examined by comparison with a classic NMDA receptor antagonist (AP-5), its parent peptide (IGF-1) and its sibling peptide (des-IGF-1), the product of removal of GPE from the N-terminus of IGF-1. The results suggested that both IGF-1 and GPE, but not des-IGF-1, have comparable antagonistic NMDA effects to that of AP-5. The most obvious explanation for lack of effects following des-IGF-1 treatment is the removal of GPE. These results suggested that the activity of GPE may be mediated by effects on the NMDA-mediated receptor, and that IGF-1 acts as a pro-hormone of GPE (Bourguignon et al., 1993; Bourguignon and Gerard, 1999). The NMDA-mediated effects of GPE and IGF-1 were also age-dependent, as immature brains appeared to be less responsive to the treatments (Bourguignon et al., 1993; Bourguignon and Gerard, 1999).
Ikeda et al. examined whether the two IGF-1 cleavage products, GPE and des-IGF-1, have different modes of action by measuring glial proliferation in vitro (Ikeda et al., 1995). They observed that MK801, an NMDA receptor antagonist, blocked the effects of GPE on glial proliferation and that there was an additive effect of des-IGF-1 and GPE on glial proliferation, suggesting that the action of des-IGF-1 is mediated via a different mechanism, presumably via IGF-1 receptors. The study provided further evidence that the bioactivity of IGF-1 is mediated via different modes of action through its two cleavage products (Ikeda et al., 1995). However, the neuroprotective effect of GPE appeared not to be comparable with that of MK801, as an effective dose of MK801 given immediately after ischaemic injury failed to be effective when given 2 h after HI injury, contrary to what is seen with GPE. It is possible that the NMDA effects observed can be a result of interfering with a number of signalling pathways that lead to the activation of NMDA receptors rather than directly interact with the receptors.
Possible mechanisms of GPE in neuroprotection
Guan et al. suggested that the inhibition of caspase-3-activated and non-caspase-3-activated apoptotic pathways may be involved in the neuroprotection by GPE after HI injury (Guan et al., 2004). HI injury can result both in necrosis, a morphologically recognized process of rapid neuronal death, and in apoptosis, in which neurons are committed to die via a more progressive process (Dirnagl et al., 1999). Neither form of neuronal death occurs immediately following the injury, which provides a window of opportunity for treatment. TUNEL and caspase-3 immunostaining have been widely used as markers of cells that undergo apoptosis. The degree of tissue damage assessed morphologically represents a mixture of neuronal necrosis and apoptosis, which appeared to be similar across the CA1-2, CA3 and CA4 sub-regions of the hippocampus in the group treated with the vehicle (Guan et al., 2004). Interestingly, while the increased TUNEL-positive cells were seen mainly in the CA3 sub-region of the hippocampus, the majority of caspase-3 positive cells were located in the CA4 sub-regions in the vehicle-treated group (Guan et al., 2004). Both TUNEL and caspase-3 positive cells were relatively low in the CA1-2 sub-regions. It is thought that, as an execution phase protease, capase-3 activation leads to fragmentation of DNA, which is detected by TUNEL labelling. This HI injury-induced spatial difference between caspase-3 activation and TUNEL labelling indicates that the caspase-3 pathway may not necessarily lead to positive TUNEL labelling. This disassociation between TUNEL and caspase-3 immunoreactivity has also been suggested outside of the CNS (Blomgren, 2007). Therefore, these spatial differences indicated that both caspase-3-dependent and -independent pathways were involved in neuronal injury in the hippocampus following HI injury. GPE treatment 1–5 h after HI injury significantly reduced the tissue damage, as well as TUNEL and caspase-3 positive cells, suggesting inhibition of both neuronal necrosis and apoptosis (Guan et al., 2004).
Furthermore, although controversial, a role for reactive glia in neuronal damage and recovery has been documented (Kraig et al., 1995; Maragakis and Rothstein, 2006). Treatment with GPE strongly suppresses microglial proliferation and completely prevents the HI-induced loss of astrocytes. A physiological role for reactive astrocytes has been suggested to be involved in BBB integrity, cell-to-cell communication, intracellular iron homeostasis, plasticity of neurons and neurotrophic actions by regulating growth factor metabolism (Kraig et al., 1995; Persidsky et al., 2006). Under physiological conditions, excitatory amino acid release from astrocytes is receptor mediated, whereas injury-induced excitatory amino acid leakage from astrocytes is due to astrocyte swelling (Kraig et al., 1995; Ridet et al., 1997; Panickar and Norenberg, 2005), which can lead to damaged homeostasis and contribute to further neuronal injury, particularly in neurodegenerative conditions (Maragakis and Rothstein, 2006). Loss of astrocytes following ischaemic injury has also been suggested to be an important part of the evolution of tissue infarction. Therefore, maintaining astrocyte integrity may be critical for neuroprotection by GPE.
Microglial cells are generally believed to have a role in brain inflammation, autoimmune responses and neuronal degeneration (Kraig et al., 1995; Panickar and Norenberg, 2005). In contrast to its parent peptide IGF-1 (Cao et al., 2003), GPE treatment reduced HI injury-induced isolectin B4 positive microglial cells. This may have involved the inhibition of cell proliferation, since PCNA-positive cells also were reduced by GPE treatment. Several neuroprotective agents have been identified to have anti-inflammatory properties, such as TGFβ-1 (McNeill et al., 1994), which could be involved in neuroprotection by GPE after HI injury.
Other possible mechanisms underlying neuroprotection by GPE also have been suggested by studies examining the treatment effects of GPE on striatal neuronal phenotypes (Guan et al., 1999). In this study, the authors showed that the loss of GAD-immunopositive neurons is strongly prevented by GPE treatment, whereas the loss of parvalbumin-positive neurons is not; both markers are co-expressed in some populations of GABA interneurons. Therefore, the complete restoration of GAD-immunopositive neurons at least partially may be due to an up-regulation of enzymatic immunoreactivity rather than due to increased survival of the cellular phenotype. Given that the accumulation of extracellular glutamate during brain injury plays a key role in the pathogenesis of ischaemic neuronal death (Tan et al., 1996; Dirnagl et al., 1999), up-regulation of GAD may also help protect neurons from glutamate-induced toxicity. Thus, alleviation of the excitotoxic effects of glutamate and elevation of GABA after ischaemia may be another potential mechanism of action of GPE on neuronal rescue.
GPE also appeared to up-regulate NOS, as there was an increase in neuronal NOS immunopositivity in the contralateral side of the striatum (Guan et al., 1999). As a neurotransmitter, nitric oxide (NO) plays an important role during the process of neuronal damage as well as neuronal recovery. Inhibiting NO synthesis can impair local cerebral blood flow and exacerbate brain injury (Tanaka et al., 1993). As GPE is an active moiety of IGF-1, up-regulation of NOS enzymatic activity and the consequent improvement in cerebral blood flow may be an important mechanism of the neuroprotective effects of GPE as well as IGF-1.
Neuroprotective effects of glycine 2-methyl proline glutamate (G-2mPE)
To overcome the instability of GPE in plasma, a GPE analogue was generated by adding a methyl group to the proline ring of GPE with the intention of increasing resistance to enzymatic degradation (Harris et al., 2005). The half-life of G-2mPE in the plasma was indeed significantly prolonged compared with that of GPE (Bickerdike et al., 2009). The neuroprotective effects of this analogue have been reported subsequently in both adult and neonatal rat models of ischaemic brain injury (Zhao et al., 2005; Svedin et al., 2007). Zhao et al. examined the treatment effect of GPE, G-2mPE and their combination with Caffeinol, a mixture of caffeine and ethanol, on neurological deficit after middle cerebral artery occlusion. Continuous i.v. infusion of either GPE (0.3 mg·kg·h−1 × 4 h) or G-2mPE at a 10-fold lower dose significantly reduced the neurological deficit compared with the vehicle-treated rats (Zhao et al., 2005). The authors also found an additive treatment effect between G-2mPE and Caffeinol, but not between GPE and Caffeinol (Zhao et al., 2005). More recently, Svedin et al. reported that daily treatment with G-2mPE given i.p. for 7 days improved neuronal outcome when examined 7 days after HI injury in post-natal day 7 rats (Figure 3). However, the treatment effect was less significant when a single dose was given 2 h after the injury.
Figure 3.
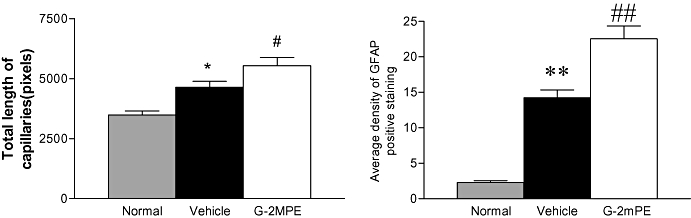
The hypoxia-ischemia injury-induced increase in vascular remodelling (left) is correlated to the up-regulation in glial fibrillar acidic protein (GFAP) positive cells (right *P < 0.05; **P < 0.01). The treatment with glycine-2methyl-proline-glutamate (G-2mPE) further increased the vascular remodelling and reactive astrocyte. #P < 0.05; ##P < 0.01.
Angiogenesis is a major pathway for injury-associated neovascularization involving endothelial sprouting and division of existing vessel lumens to form new vessels (Frontczak-Baniewicz and Walski, 2002). The formation of new cerebral vessels is fundamental for providing nutritional and trophic support to adjacent tissues, maintaining BBB integrity as well as tissue repair after injury. Astrocytes have an important role in vascularization (Salhia et al., 2000; Acker et al., 2001; Frontczak-Baniewicz and Walski, 2002). Ischaemic brain injury can cause neuro-endothelial damage and provoke astrocytosis (Brault et al., 2003). The injury-induced increase in GFAP-reactive astrocytosis has been found to be associated with neoangiogenesis (Salhia et al., 2000).
In this particular study, the authors also examined the effects of HI injury and treatment with G-2mPE on cerebral vascular vessels. The total length of blood vessels, measured in 2D profile for vascular density, was found to be reduced in the ipsilateral hemisphere as part of the pathology of HI injury. However, the hypoxic insult increased vascular density in the contralateral cortex and hippocampus, where histological damage was absent, compared with normal control rats. G-2mPE treatment increased the capillary density in selective brain regions. The study did not determine whether the treatment effects on capillary density are due to preservation of blood vessels following HI or to a higher degree of subsequent revascularization. However, the increased capillary density may be a contributing factor for neuroprotection, as the neuroprotection and increased capillary density were coherent in some brain regions (Svedin et al., 2007). In addition, it has been demonstrated that an increase in the density of blood vessels is associated with improved blood flow and contributes to the neuroprotective effects of hypoxic preconditioning in neonatal rats (Gustavsson et al., 2007).
The authors also reported clear association of GFAP-positive staining with vascular morphology (Figure 3). A role for GFAP-reactive astrocytes in angiogenesis has been well documented, showing induction of key vascular regulators, including vascular endothelial growth factor and angiopoietin-1 and its receptors (Acker et al., 2001). Treatment with G-2mPE also significantly increases GFAP density and revascularization, probably through promoting astrocytic angiogenesis. IGF-1, the parent peptide of GPE, has been reported to have a critical role in vascular remodelling by increasing vessel growth in the perilesional area after injury in adult mice brains (Lopez-Lopez et al., 2004). The role of G-2mPE in angiogenesis needs to be further investigated.
In addition, the neuroprotective effects of several other GPE analogues also have been tested in vitro (Alonso De Diego et al., 2006). However, the neuroprotective action of both GPE and its analogues recently has been suggested not to be associated with glutamate receptors (Alonso De Diego et al., 2006).
Diketopiperazine, DKP
The DKPs are a group of modified cyclic di-peptides that can be derived from endogenous neuropeptides. The most tested DKPs are cyclic His-Pro and its modified analogues. Cyclic His-Pro is naturally derived from the tripeptide thyrotropin-releasing hormone (TRH) (Faden et al., 1989; 1999). Both TRH and cyclic His-Pro are neuroprotective after traumatic brain injury (TBI) and spinal cord injury. Several bioactive analogues of cyclic His-Pro also prevent neuronal injury and improve motor and cognitive function (Faden et al., 1989; 1999; Takami et al., 1991). Inspired by their findings, we hypothesized that cyclic-Gly-Pro, another endogenous diketopiperazine with nootropic action (Samonina et al., 2002), may be an additional metabolite of GPE. Such cyclic structures may render a molecule resistant to enzymatic breakdown and more lipophilic for better central uptake. We have therefore examined the protective effects of cyclic Gly-Pro and its analogue, NNZ 2591, which has been modified to overcome potential disadvantages of the endogenous compound and to improve its potency.
The data suggest more potent neuroprotection by NNZ 2591 than by cyclic Gly-Pro after central administration, as the effective doses of NNZ 2591 are lower than those of cyclic Gly-Pro. The introduction of an allyl substituent at C-8a on the diketopiperazine skeleton may thus confer lipophilicity, thereby facilitating the ability of NNZ 2591 to cross the CSF-brain barrier (Guan et al., 2007) after the compound is delivered to the CSF.
The physiological role of the BBB is to selectively prevent large and/or hydrophilic substances from accessing the CNS (Pardridge, 2005). The central uptake of some molecules, regardless of their molecular size, is largely dependent on injury-induced BBB breakdown (Pardridge, 1991; Guan et al., 2004). However, central uptake of NNZ 2591 appears not to be injury dependent, as in normal control rats, the amounts of free NNZ 2591 detected in the CSF were similar to those found in the plasma (Figure 4, Guan et al., 2007).
Figure 4.
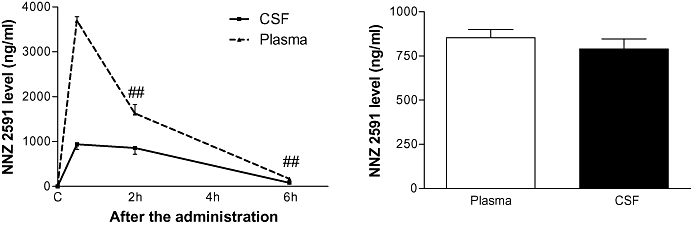
The left panel shows the half-life of NNZ 2591 in the blood and the cerebrospinal fluid (CSF) after single i.v. administration in hypoxia-ischemia injured adult rats. The right panel shows the level of NNZ 2591 in the blood and the CSF 2 h after single administration of NNZ 2591 in normal rats. ##P < 0.01.
Although the level of NNZ 2591 in blood fell to half of the initial value 2 h after a single intravenous dose, the CSF level remained the same between 0.5 and 2 h after a bolus injection, suggesting a threshold effect for central uptake (Figure 4; Guan et al., 2007). The window of CSF turnover, which is approximately 1 h in rats, is largely responsible for drug elimination from the CNS due to CSF absorption (Pardridge, 1991; Guan et al., 2004). The maintained CSF level to 2 h suggests a sustained central transfer of NNZ 2591 from plasma and that central uptake of NNZ 2591 compensates for the drug's elimination through CSF absorption. The compound is detectable 6 h after the administration.
Given that the BBB appears to be highly permeable to NNZ 2591, the effective dose range (0.6–3 mg·kg−1) used for peripheral administration is rather high compared with the dose range (2–20 ng rat-1) tested by central administration (Guan et al., 2007). An in vitro albumin binding assay suggests a potential protein binding-releasing of NNZ 2591 in the plasma, which may alter the pharmacokinetics of NNZ 2591 by influencing the level of free compound in the plasma (Guan et al., 2007). Furthermore, the neuroprotective effects of NNZ 2591 show complex dose dependency after central administration, with higher doses being ineffective (Guan et al., 2007). However, this dose dependency of NNZ 2591 is not as prominent after peripheral administration, as the three different doses tested were all effective. The possible binding-releasing process discussed previously may serve as a sustained release depot after the free compound is biotransformed or eliminated from plasma (Endrenyi, 1998).
Although a single treatment resulted in a modest histological improvement, repeated treatment with NNZ 2591 resulted in almost complete long-term protection when histology was examined 9 weeks later (Guan et al., 2007). Repeated treatment with NNZ 2591 also prevents HI injury-induced somatosensory-motor deficit (Guan et al., 2007). Faden et al. have also suggested that another diketopiperazine derived from thyrotropin-releasing hormone could improve recovery of motor and cognitive function following TBI in rats and mice (Faden et al., 2003; 2005). Given that the single treatment used by those authors presumably only prevented cell death within the first few hours after injury, repeated treatment may have targeted the more progressive brain injury which occurred several days after the initial insult. Long-lasting neuroprotective effects are critical for any treatment regimen, as the improved neuronal outcomes detected soon after the treatment may not always be maintained long term (Fisher and Schaebitz, 2000; Gladsrone et al., 2002). Delayed neuronal/glial injury can be mediated through necrotic, apoptotic and necroptotic pathways (Degterev et al., 2005); although the downstream cascades of these cell death pathways largely overlap (Yuan et al., 2003). Cleaved caspase-3 has been used as a marker for the caspase activation-dependent apoptotic pathway, whereas AIF is commonly used as a marker for apoptotic pathways that do not rely on activating caspases (Yuan et al., 2003). Treatment with NNZ 2591 inhibits the protein expression of caspase-3 but not AIF (Guan et al., 2007), suggesting that the inhibition of the caspase-mediated apoptotic pathway may mediate the neuroprotective effect of NNZ 2591.
Even after a broad receptor-binding screening, which included acetylcholine, GABA, glutamate, opoid, adrenergic and serotonergic receptors, the mode of action of DKP has not been identified (Faden et al., 2003). The neuroprotective mechanism of NNZ 2591 also needs further investigation. However, inhibition of reactive microglia and elevation of astrocytes may have a role in the protective effect of NNZ 2591 (Guan et al., 2007). Inhibiting both necrotic and apoptotic neuronal death pathways, without altering cerebral blood flow, has been suggested to be involved in neuroprotection of B35, another modified diketopiperazine, after TBI (Faden et al., 2003). Moreover, inhibiting glutamate-induced excitotoxicity, beta-amyloid induced cells death and promotion of survival factors may also mediate the neuroprotective activity of DKP in vitro (Faden et al., 2005).
The up-regulation of cell cycle proteins occurs in both mitotic and post-mitotic brain cells after brain injury during adulthood. The activation of cell cycle proteins induce cell proliferation in astrocyte and microglia, which are mitotic cells, and initiate caspase-related apoptosis in neurons, which are post-mitotic cells (Byrnes and Faden, 2007). The research from Faden's group also suggests that the effects of B35 on inhibiting neuronal apoptosis and microglial proliferation are mediated by inhibiting the expression of cell cycle proteins (Faden et al., 2005). Thus, inhibiting cell cycle protein may also be associated with the effect of NNZ 2591 on inhibiting caspase-3 activation-dependent apoptosis (Guan et al., 2007).
Conclusion
The induction of endogenous IGF-1 after brain injury suggests that it plays a role in preventing brain injury. Indeed, treatment with exogenous IGF-1 after brain injury is protective and leads to improved long-term neurological function. However, the poor central uptake of IGF-1 and its potential mitogenic nature prevent its clinical application. Intriguingly, the naturally cleaved N-terminal tripeptide of IGF-1, GPE, crosses the BBB and does not interact with IGF receptors. Intravenous infusion of GPE for 4 h prevents brain injury and improves long-term functional recovery. GPE has a broad effective dose range and a 3–7 h window of opportunity for treatment. Continuous administration is essential for neuroprotection by GPE because of its enzymatic instability. G-2-meth-PE, an analogue of GPE modified to improve enzymatic stability, has a prolonged plasma half-life and is neuroprotective after ischaemic brain injury in both adult and neonatal rats. Promotion of astrocytosis while inhibiting apoptosis and microglial activation may be the underlying mechanisms of neuroprotection by GPE and its analogue. Cyclic Gly-Pro (which may be a natural derivative of GPE) and its modified analogue NNZ 2591 are both neuroprotective after ischaemic brain injury; cyclization further improved enzymatic stability and conferred excellent central uptake. The pharmacology of the NNZ 2591 provides further advances in drug discovery for treating acute brain injury. Without knowing the mode of action, the beneficial effects of these IGF-1-related small peptides can be mediated via a panoply of direct and indirect actions against the diverse injury in neurones and glia.
Glossary
Abbreviations:
- AIF
apoptosis inducing factor
- BBB
blood brain barrier
- ChAT
choline acetyltransferase
- CNS
central nervous system
- CSF
cerebrospinal fluid
- cyclic Gly-Pro
cyclo-glycyl-proline
- Des-IGF-1
Des-N-(1-3) insulin-like growth factor-1
- DG
dentate gyrus of the hippocampus
- DKP
diketopiperazine
- GABA
γ-aminobutyric acid
- GAD
Glutamate acid decarboxylase
- GFAP
glial fibrillar acidic protein
- GPE
glycine-proline-glutamate
- G-2mPE
glycine-2methyl-proline-glutamate
- GnRH
gonadotrophin-releasing hormone
- HI
hypoxia-ischemia
- ICV
intracerebroventricular
- IV
intravenous
- IP
intraperitoneal
- IGF
insulin-like growth factor
- rhIGF-1
recombinant human IGF-1
- MMP
matrix metalloproteinase
- NMDA
N-methyl d-aspartate
- NNZ 2591
Cyclo-L-glycyl-L-2-allylproline
- NO
nitric oxide
- NOS
nitric oxide synthase
- NPY
neuropeptide-Y
- PD
Parkinson's disease
- PCNA
proliferating cell nuclear antigen
- PLP
proteolipid protein
- SMT
somatostatin
- TUNEL
terminal deoxynucleotidyl transferase mediated dUTP nick-end labelling
- TH
tyrosine-hydroxylase
- VEGF
vascular endothelial growth factor
Conflict of interest
Dr Jian Guan and Prof Peter Gluckman, the principal investigators, have been providing scientific consultancy to Neuren Pharmaceuticals Limited, Auckland through UniServices Limited, University of Auckland, New Zealand, and have equity interests in Neuren Pharmaceuticals Ltd. but have not been associated with Neuren Pharmaceuticals as employees.
References
- Acker T, Beck H, Plate KH. Cell type specific expression of vascular endothelial growth factor and angiopoietin-1 and -2 suggests an important role of astrocytes in cerebellar vascularization. Mech Dev. 2001;108:45–57. doi: 10.1016/s0925-4773(01)00471-3. [DOI] [PubMed] [Google Scholar]
- Alonso De Diego SA, Gutierrez-Rodriguez M, Perez de Vega MJ, Gonzalez-Muniz R, Herranz R, Martin-Martinez M, et al. The neuroprotective activity of GPE tripeptide analogues does not correlate with glutamate receptor binding affinity. Bioorg Med Chem Lett. 2006;16:3396–3400. doi: 10.1016/j.bmcl.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Baker AM, Batchelor DC, Thomas GB, Wen JY, Rafiee M, Lin H, et al. Central penetration and stability of N-terminal tripeptide of insulin-like growth factor-I, glycine-proline-glutamate in adult rat. Neuropeptides. 2005;39:81–87. doi: 10.1016/j.npep.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Barbosa ES, Limongi JCP, Cummings JL. Parkinson's disease. Neuropsych Basal Ganglia. 1997;20:769–790. [Google Scholar]
- Batchelor DC, Lin H, Wen J-Y, Keven C, van Zijl PL, Breier BH, et al. Pharmacokinetics of glycine-proline-glutamate, the N-terminal tripeptide of insulin-like growth factor-1, in rats. Anal Biochem. 2003;323:156–163. doi: 10.1016/j.ab.2003.08.032. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Bassett NS, Sirimanne ES, Williams CE, Gluckman PD. Insulin-like growth factor II is induced during wound repair following hypoxic-ischemic injury in the developing rat brain. Mol Brain Res. 1995;29:81–91. doi: 10.1016/0169-328x(94)00232-4. [DOI] [PubMed] [Google Scholar]
- Betz AL, Keep RF, Beer ME, Ren XD. Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J Cereb Blood Flow Metab. 1994;14:29–37. doi: 10.1038/jcbfm.1994.5. [DOI] [PubMed] [Google Scholar]
- Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. NNZ-2566: A Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke. J Neurol Sci. 2009;278:8–90. doi: 10.1016/j.jns.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Blomgren K. Pathological apoptosis in the developing brain. Apoptosis. 2007;12:993–1010. doi: 10.1007/s10495-007-0754-4. [DOI] [PubMed] [Google Scholar]
- Bourguignon JP, Gerard A. Role of insulin-like growth factor binding proteins in limitation of IGF-1 degradation into the N-methyl-D-aspartate receptor antagonist GPE: evidence from gonadotrophin-releasing hormone secretion in vitro at two developmental stages. Brain Res. 1999;847:247–252. doi: 10.1016/s0006-8993(99)02051-x. [DOI] [PubMed] [Google Scholar]
- Bourguignon JP, Gerard A, Alvarez Gonzalez ML, Franchimont P. Acute suppression of gonadotropin-releasing hormone secretion by insulin-like growth factor I and subproducts: an age-dependent endocrine effect. Neuroendocrinology. 1993;58:525–530. doi: 10.1159/000126586. [DOI] [PubMed] [Google Scholar]
- Brault S, Martinez-Bermudez AK, Marrache AM, Gobeil F, Jr, Hou X, Beauchamp M, et al. Selective neuromicrovascular endothelial cell death by 8-Iso-prostaglandin F2alpha: possible role in ischemic brain injury. Stroke. 2003;34:776–782. doi: 10.1161/01.STR.0000055763.76479.E6. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Faden AI. Role of cell cycle proteins in CNS injury. Neurochem Res. 2007;32:1799–1807. doi: 10.1007/s11064-007-9312-2. [DOI] [PubMed] [Google Scholar]
- Cao Y, Gunn AJ, Bennet L, Wu D, George S, Gluckman PD, et al. Insulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J Cereb Blood Flow Metab. 2003;23:739–747. doi: 10.1097/01.WCB.0000067720.12805.6F. [DOI] [PubMed] [Google Scholar]
- Coimbra C, Drake M, Boris-Moller F, Wieloch T. Long-lasting neuroprotective effect of postischemic hypothermia and treatment with an anti-inflammatory/antipyretic drug. Evidence for chronic encephalopathic processes following ischemia. Stroke. 1996;27:1578–1585. doi: 10.1161/01.str.27.9.1578. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Endrenyi L. Drug distribution. In: Kalant H, Roschlau WHE, editors. Principles of Medical Pharmacology. 6th edn. New York: Oxford Univrsity Press; 1998. pp. 28–37. [Google Scholar]
- Faden AI, Fox GB, Fan L, Araldi GL, Qiao L, Wang S, et al. TRH analog YM-14673 improves outcome following traumatic brain and spinal cord injury in rats: dose-response studies. Brain Res. 1989;486:228–235. doi: 10.1016/0006-8993(89)90509-x. [DOI] [PubMed] [Google Scholar]
- Faden AI, Fox GB, Fan L, Araldi GL, Qiao L, Wang S, et al. Novel TRH analog improves motor and cognitive recovery after traumatic brain injury in rodents. Am J Physiol. 1999;277:R1196–R1204. doi: 10.1152/ajpregu.1999.277.4.R1196. [DOI] [PubMed] [Google Scholar]
- Faden AI, Knoblach SM, Cernak I, Fan L, Vink R, Araldi GL, et al. Novel diketopiperazine enhances motor and cognitive recovery after traumatic brain injury in rats and shows neuroprotection in vitro and in vivo. J Cereb Blood Flow Metab. 2003;23:342–354. doi: 10.1097/01.WCB.0000046143.31247.FD. [DOI] [PubMed] [Google Scholar]
- Faden AI, Movsesyan VA, Knoblach SM, Ahmed F, Cernak I. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology. 2005;49:410–424. doi: 10.1016/j.neuropharm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Fisher M, Schaebitz W. An overview of acute stroke therapy. Arch Intern Med. 2000;160:3196–3206. doi: 10.1001/archinte.160.21.3196. [DOI] [PubMed] [Google Scholar]
- Frontczak-Baniewicz M, Walski M. Non-sprouting angiogenesis in neurohypophysis after traumatic injury of the cerebral cortex. Electron-microscopic studies. Neuro Endocrinol Lett. 2002;23:396–404. [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- Gladsrone DJ, Black SE, Hakim AM. Toward wisdom from failure- Lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Ambler GR. Therapeutic use of insulin-like growth factor I: lessons from in vivo animal studies. Acta Paediatr Suppl. 1992;383:134–136. [PubMed] [Google Scholar]
- Guan J, Williams C, Gunning M, Mallard C, Gluckman P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J Cereb Blood Flow Metab. 1993;13:609–616. doi: 10.1038/jcbfm.1993.79. [DOI] [PubMed] [Google Scholar]
- Guan J, Williams CE, Skinner SJ, Mallard EC, Gluckman PD. The effects of insulin-like growth factor (IGF)-1, IGF-2, and des-IGF-1 on neuronal loss after hypoxic-ischemic brain injury in adult rats: evidence for a role for IGF binding proteins. Endocrinology. 1996;137:893–898. doi: 10.1210/endo.137.3.8603600. [DOI] [PubMed] [Google Scholar]
- Guan J, Waldvogel HJ, Faull RL, Gluckman PD, Williams CE. The effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience. 1999;89:649–659. doi: 10.1016/s0306-4522(98)00338-8. [DOI] [PubMed] [Google Scholar]
- Guan J, Beilharz EJ, Skinner SJ, Williams CE, Gluckman PD. Intracerebral transportation and cellular localisation of insulin-like growth factor-1 following central administration to rats with hypoxic-ischemic brain injury. Brain Res. 2000a;853:163–173. doi: 10.1016/s0006-8993(99)02030-2. [DOI] [PubMed] [Google Scholar]
- Guan J, Gunn AJ, Sirimanne ES, Tuffin J, Gunning MI, Clark R, et al. The window of opportunity for neuronal rescue with insulin-like growth factor-1 after hypoxia-ischemia in rats is critically modulated by cerebral temperature during recovery. J Cereb Blood Flow Metab. 2000b;20:513–519. doi: 10.1097/00004647-200003000-00010. [DOI] [PubMed] [Google Scholar]
- Guan J, Krishnamurthi R, Waldvogel HJ, Faull RL, Clark R, Gluckman P. N-terminal tripeptide of IGF-1 (GPE) prevents the loss of TH positive neurons after 6-OHDA induced nigral lesion in rats. Brain Res. 2000c;859:286–292. doi: 10.1016/s0006-8993(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Guan J, Bennet L, George S, Wu D, Waldvogel HJ, Gluckman PD, et al. Insulin-like growth factor-1 reduces postischemic white matter injury in fetal sheep. J Cereb Blood Flow Metab. 2001a;21:493–502. doi: 10.1097/00004647-200105000-00003. [DOI] [PubMed] [Google Scholar]
- Guan J, Miller OT, Waugh KM, McCarthy D, Gluckman PD. Insulin-like growth factor-1 improves somatosensory function and reduces the extent of cortical infarction and ongoing neuronal loss after hypoxia-ischemia in rats. Neuroscience. 2001b;105:299–306. doi: 10.1016/s0306-4522(01)00145-2. [DOI] [PubMed] [Google Scholar]
- Guan J, Thomas GB, Lin H, Mathai S, Bachelor DC, George S, et al. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic-ischemic adult rats. Neuropharmacology. 2004;47:892–903. doi: 10.1016/j.neuropharm.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Guan J, Mathai S, Harris AP, Wen JY, Zhang R, Brimble MA, et al. Peripheral administration of a novel diketopiperazine, NNZ 2591, prevents brain injury and improves somatosensory-motor function following hypoxia-ischemia in adult rats. Neuropharmacology. 2007;53:749–762. doi: 10.1016/j.neuropharm.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Mallard C, Vannucci SJ, Wilson MA, Johnston MV, Hagberg H. Vascular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab. 2007;27:928–938. doi: 10.1038/sj.jcbfm.9600408. [DOI] [PubMed] [Google Scholar]
- Harris PWR, Brimble MA, Muir VJ, Lai M, Trotter NS, Callis DJ. Synthesis of proline-medified analogues of the neuroprotective agent glucyl-l-prolyl-glutamic acid (GPE) Tetrahedorn. 2005;61:10018–10035. [Google Scholar]
- Hsu M, Sik A, Gallyas F, Horvath Z, Buzsaki G. Short-term and long-term changes in the postischemic hippocampus. Ann NY Acad Sci. 1994;743:121–139. doi: 10.1111/j.1749-6632.1994.tb55790.x. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Waldbillig RJ, Puro DG. Truncation of IGF-I yields two mitogens for retinal Muller glial cells. Brain Res. 1995;686:87–92. doi: 10.1016/0006-8993(95)00473-4. [DOI] [PubMed] [Google Scholar]
- Johnston BM, Mallard EC, Williams CE, Gluckman PD. Insulin-like growth factor-1 is a potent neuronal rescue agent after hypoxic-ischemic injury in fetal lambs. J Clin Invest. 1996;97:300–308. doi: 10.1172/JCI118416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Neostriatal cell subtypes and their functional roles. Neurosci Res. 1997;27:1–8. doi: 10.1016/s0168-0102(96)01134-0. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Lascola CD, Cagiano A. Glial response to brain ischemia. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford University Press; 1995. pp. 964–976. [Google Scholar]
- Krishnamurthi R, Stott S, Maingay M, Faull RL, McCarthy D, Gluckman P, et al. N-terminal tripeptide of IGF-1 improves functional deficits after 6-OHDA lesion in rats. Neuroreport. 2004;15:1601–1604. doi: 10.1097/01.wnr.0000127461.15985.07. [DOI] [PubMed] [Google Scholar]
- Lee WH, Michels KM, Bondy CA. Localization of insulin-like growth factor binding protein-2 messenger RNA during postnatal brain development: correlation with insulin-like growth factors I and II. Neuroscience. 1993;53:251–265. doi: 10.1016/0306-4522(93)90303-w. [DOI] [PubMed] [Google Scholar]
- Lee WH, Wang GM, Seaman LB, Vannucci SJ. Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 1996;16:227–236. doi: 10.1097/00004647-199603000-00007. [DOI] [PubMed] [Google Scholar]
- Lees KR. Management of acute stroke. Lancet Neurol. 2002;1:41–50. doi: 10.1016/s1474-4422(02)00005-4. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Thorne RG, Frey WH., 2nd Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci Lett. 2001;308:91–94. doi: 10.1016/s0304-3940(01)01982-6. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez C, LeRoith D, Torres-Aleman I. Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc Natl Acad Sci USA. 2004;101:9833–9838. doi: 10.1073/pnas.0400337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- McNeill H, Williams C, Guan J, Dragunow M, Lawlor P, Sirimanne E, et al. Neuronal rescue with transforming growth factor-beta 1 after hypoxic-ischaemic brain injury. Neuroreport. 1994;5:901–904. doi: 10.1097/00001756-199404000-00012. [DOI] [PubMed] [Google Scholar]
- Nilsson-Hakansson L, Civalero I, Zhang X, Carlsson-Skwirut C, Sara VR, Nordberg A. Effects of IGF-1, truncated IGF-1 and the tripeptide Gly-Pro-Glu on acetylcholine release from parietal cortex of rat brain. Neuroreport. 1993;4:1111–1114. [PubMed] [Google Scholar]
- Olsson M, Nikkhah G, Bentlage C, Bjorklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci. 1995;15:3863–3875. doi: 10.1523/JNEUROSCI.15-05-03863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia. 2005;50:287–298. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. Neurorx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. Transnasal and intraventricular delivery of drugs. In: Pardridge WM, editor. Peptide Drug Delivery to the Brain. New York: Raven Press; 1991. pp. 99–122. [Google Scholar]
- Pardridge WM. Drug delivery to the brain. J Cereb Blood Flow Metab. 1997;17:713–731. doi: 10.1097/00004647-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Preston E, Sutherland G, Finsten A. Three openings of the blood-train barrier produced by forebrain ischemia in the rat. Neurosci Lett. 1993;149:75–78. doi: 10.1016/0304-3940(93)90351-k. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, et al. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- Salhia B, Angelov L, Roncari L, Wu X, Shannon P, Guha A. Expression of vascular endothelial growth factor by reactive astrocytes and associated neoangiogenesis. Brain Res. 2000;883:87–97. doi: 10.1016/s0006-8993(00)02825-0. [DOI] [PubMed] [Google Scholar]
- Samonina G, Ashmarin I, Lyapina L. Glyproline peptide family: review on bioactivity and possible origins. Pathophysiology. 2002;8:229–234. doi: 10.1016/s0928-4680(02)00018-4. [DOI] [PubMed] [Google Scholar]
- Sara VR, Carlsson-Sdwirut C, Bergman T, Jornvall H, Roberts PJ, Crawford M, et al. Indentification of Gly-Pre-Glu (GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncated in brain, as a novel neuroaction peptide. Bioch Bioph Res Com. 1989;165:766–771. doi: 10.1016/s0006-291x(89)80032-4. [DOI] [PubMed] [Google Scholar]
- Saura J, Curatolo L, Williams CE, Gatti S, Benatti L, Peeters C, et al. Neuroprotective effects of Gly-Pro-Glu, the N-terminal tripeptide of IGF-1, in the hippocampus in vitro. Neuroreport. 1999;10:161–164. doi: 10.1097/00001756-199901180-00031. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog in Neurobio. 1996;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Sizonenko SV, Sirimanne ES, Gluckman PD, Williams CE. Neuroprotective effects of the N-terminal tripeptide of IGF-1, glycine-proline-glutamate, in the immature rat brain after hypoxic-ischemic injury. Brain Res. 2001;922:42–50. doi: 10.1016/s0006-8993(01)03148-1. [DOI] [PubMed] [Google Scholar]
- Svedin P, Guan J, Mathai S, Zhang R, Wang X, Gustavsson M, et al. Delayed peripheral administration of a GPE analogue induces astrogliosis and angiogenesis and reduces inflammation and brain injury following hypoxia-ischemia in the neonatal rat. Develop Neurosci. 2007;29:393–402. doi: 10.1159/000105480. [DOI] [PubMed] [Google Scholar]
- Takami K, Hashimoto T, Shino A, Fukuda N. Effect of thyrotropin-releasing hormone (TRH) in experimental spinal cord injury: a quantitative histopathologic study. Jpn J Pharmacol. 1991;57:405–417. doi: 10.1254/jjp.57.405. [DOI] [PubMed] [Google Scholar]
- Tan WK, Williams CE, During MJ, Mallard CE, Gunning MI, Gunn AJ, et al. Accumulation of cytotoxins during the development of seizures and edema after hypoxic-ischemic injury in late gestation fetal sheep. Pediatr Res. 1996;39:791–797. doi: 10.1203/00006450-199605000-00008. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Fukuuchi Y, Gomi S, Mihara B, Shirai T, Nogawa S, et al. Inhibition of nitric oxide synthesis impairs autoregulation of local cerebral blood flow in the rat. Neuroreport. 1993;4:267–270. doi: 10.1097/00001756-199303000-00010. [DOI] [PubMed] [Google Scholar]
- Yamaguchi F, Itano T, Miyamoto O, Janjua NA, Ohmoto T, Hosokawa K, et al. Increase of extracellular insulin-like growth factor I (IGF-I) concentration following electrolytical lesion in rat hippocampus. Neurosci Lett. 1991;128:273–276. doi: 10.1016/0304-3940(91)90278-2. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Murphy LJ. Enzymatic conversion of IGF-I to des(1-3)IGF-I in rat serum and tissues: a further potential site of growth hormone regulation of IGF-I action. J Endocrinol. 1995a;146:141–148. doi: 10.1677/joe.0.1460141. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Murphy LJ. N-terminal truncated insulin-like growth factor-I in human urine. J Clin Endocrinol Metab. 1995b;80:1179–1183. doi: 10.1210/jcem.80.4.7536201. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Maake C, Murphy LJ. Enhanced proteolytic activity directed against the N-terminal of IGF-I in diabetic rats. J Endocrinol. 1999;162:243–250. doi: 10.1677/joe.0.1620243. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- Zhao X, Liu SJ, Zhang J, Strong R, Aronowski J, Grotta JC. Combining insulin-like growth factor derivatives plus caffeinol produces robust neuroprotection after stroke in rats. Stroke. 2005;36:129–134. doi: 10.1161/01.STR.0000149624.87661.18. [DOI] [PubMed] [Google Scholar]