

Abstract
The principles of inter-species dose extrapolation are poorly understood and applied. We provide an overview of the principles underlying dose scaling for size and dose adjustment for size-independent differences. Scaling of a dose is required in three main situations: the anticipation of first-in-human doses for clinical trials, dose extrapolation in veterinary practice and dose extrapolation for experimental purposes. Each of these situations is discussed. Allometric scaling of drug doses is commonly used for practical reasons, but can be more accurate when one takes into account species differences in pharmacokinetic parameters (clearance, volume of distribution). Simple scaling of drug doses can be misleading for some drugs; correction for protein binding, physicochemical properties of the drug or species differences in physiological time can improve scaling. However, differences in drug transport and metabolism, and in the dose–response relationship, can override the effect of size alone. For this reason, a range of modelling approaches have been developed, which combine in silico simulations with data obtained in vitro and/or in vivo. Drugs that are unlikely to be amenable to simple allometric scaling of their clearance or dose include drugs that are highly protein-bound, drugs that undergo extensive metabolism and active transport, drugs that undergo significant biliary excretion (MW > 500, ampiphilic, conjugated), drugs whose targets are subject to inter-species differences in expression, affinity and distribution and drugs that undergo extensive renal secretion. In addition to inter-species dose extrapolation, we provide an overview of dose extrapolation within species, discussing drug dosing in paediatrics and in the elderly.
Keywords: allometric scaling, body surface area, pharmacokinetics, pharmacodynamics, dose extrapolation, species difference, paediatric dosing, physiological time
Introduction
Healthcare professionals and researchers have access to a wide range of compounds that are approved for clinical use, under investigation as potential clinical therapies or used as tools to unravel physiological and/or pathophysiological mechanisms. Regardless of the purpose for which the drug is administered, the key to success is the selection of a safe and effective dose. If the safe and effective dose of a chosen drug is not known in a particular species, it must then be estimated based on information obtained from other species. The aim of dose extrapolation is to produce appropriate drug exposure to ensure efficacy and safety.
The principles that underlie the correct translation of doses among and within species are very poorly understood and applied by the healthcare, scientific and general communities. An often-quoted and tragic example is the case of Tusko, who was an Asiatic elephant at the Lincoln Park Zoo in Oklahoma City. In 1962, Tusko was given a dose of the psychotomimetic drug lysergic acid diethylamide (LSD), which had been estimated using a mg·kg−1 dose previously used in cats. Tusko went into status epilepticus 5 min after the first dose was administered and, despite efforts to control the seizures, died 1 h and 35 min later (West et al., 1962). A dose of LSD, which produced borderline effects in cats and primates, was toxic to the elephant; this difference arose because the rates of pharmacokinetic processes in elephants are slower, leading to an increase in the peak plasma concentration achieved by the dose and a prolongation of the effect. It is possible that differences in the sensitivity of elephants to LSD may have contributed to the remarkably fast onset of status epilepticus; however, the excessively high dose is the most likely reason for the fatal outcome.
A more recent example is given by the media coverage surrounding resveratrol, an antioxidant found to protect against metabolic dysfunction and aging-related disease (Baur et al., 2006; Lagouge et al., 2006). The media extrapolated the dose given to mice (22.4 mg·kg−1) on the basis of body weight and reported that the human dose would be 1344 mg·day−1, an unfeasibly high dose that created scepticism of the research. The correct extrapolation is 1.82 mg·kg−1·day−1, or 109 mg·day−1 for a 60 kg human, a dose that could feasibly be administered (Reagan-Shaw et al., 2008). Both of these cases illustrate that larger animals require smaller drug doses on a mg·kg−1 basis, a phenomenon that is often counterintuitive to the uninitiated.
However, scaling for size differences is not consistent among species. When a dose of aspirin was scaled for administration to cats, the cats experienced aspirin toxicity (Davis and Westfall, 1972). This problem arose because cats lack the capacity for glucuronidation, an important clearance mechanism of aspirin, and the aspirin dose must be reduced to account for the resulting decrease in aspirin clearance. The key to successful dose extrapolation therefore lies in understanding how doses should be scaled for size and adjusted for size-independent differences. The aim of this review is to provide an overview of the principles underlying both of these aspects.
An understanding of inter-species dose extrapolation is required in three main situations:
The selection of the maximum recommended starting dose in humans for Phase 1 clinical trials.
The selection of a safe and effective therapeutic dose in veterinary practice.
Dose selection for experimental purposes.
The journey of a drug from its absorption to its excretion, during which the drug interacts with its pharmacological targets (e.g. receptors, enzymes, ion channels), defines the points at which physiological and biochemical inter-species differences exert their influence. The concentration of the drug that reaches the site of action depends on its absorption, distribution, metabolism and excretion, the classic components of pharmacokinetics. The effect produced by the concentration at the site of action depends on the affinity of the drug for its target and the nature and magnitude of the response produced by the drug–target interaction. Some of these determinants are related to the size of the animal and some are not; the relative importance of each determines the utility of scaling on the basis of size.
The effect of animal size on metabolic rate
The origin of allometric scaling relationships in biology can be understood by considering the relationship between the metabolic rate of an animal and its size. As the size of animal species gets larger, the body surface area in relation to body weight decreases. Larger animals therefore have less capacity for losing heat than smaller animals. Larger animals could have adapted to this by evolving new metabolic machinery, which can function at higher temperatures, or by retaining the same machinery and decreasing the specific metabolic rate (the metabolic rate per unit mass). Evolution opted for the latter, simpler adaptation; the larger animal species retain similar anatomical features and biochemical machinery, but have a lower specific metabolic rate. Their heat production per unit weight is therefore decreased, and their body temperature is maintained (Huxley, 1932; Adolph, 1949). A mechanism for this adaptation is provided by the principle that, as animal species get larger, the transportation distances increase and the supply of nutrients to the distal tissues is constrained; the cells must therefore ‘slow down’ if they are to function in the face of these constraints (Brown et al., 2004; Duncan et al., 2007). Because evolution has chosen this route, it is valid to assume that there are many biochemical, physiological and anatomical similarities among animals. The decrease in the pace of these biochemical and physiological processes underlies many species differences in pharmacokinetics, and this assumption forms the basis for the use of allometric scaling to adjust for pharmacokinetic differences.
Allometric scaling
Scaling is a term borrowed from engineering, which, as its name suggests, refers to adaptations of a functional system to operate at different production scales. This is achieved by increasing the number of functional units, increasing the size of a functional unit, increasing flow through the functional unit (which may or may not involve alteration of the flow scheme) or by redesigning the system altogether. Biological systems take advantage of all four of these mechanisms when adapting to serve organisms of different sizes. Allometry is the study of size and its consequences, and the scaling of biological functional systems can be studied and described mathematically by using allometric equations. The term allometry is derived from the Greek alloios, meaning different, and is used to distinguish allometric scaling from isometric scaling. Isometric scaling applies to figures whose proportions remain the same at all sizes (i.e. geometrical figures), whereas allometric scaling applies to figures whose proportions change as a function of size (Schmidt-Nielsen, 1984).
The relationship between whole body metabolic rate and body size can be described mathematically by using the equation P=aWb, where P is the physiological parameter (metabolic rate) and W is the body weight in kilogram. The constant b is referred to as the exponent, which describes how the parameter scales over different values of body weight. It is important to realize that exponents are not constants and have no physiological meaning by themselves. They simply provide a means of describing, in mathematical language, the effect of size (W) on a given parameter (P). If P and W increased in direct proportion, the exponent would be 1. Blood volume, for example, increases in direct proportion to body mass and therefore scales with an exponent of 1; this is an isometric relationship. Haemoglobin concentration is constant across species and the exponent is therefore 0. Skeletal mass increases out of proportion with body mass in order to withstand the increased static load, locomotive stresses and impact forces generated and received by larger animals. In this case, the exponent is greater than 1 (see Schmidt-Nielsen, 1984 for overview).
The value of the exponent for whole body metabolic rate was originally calculated by Max Kleiber in 1932 to be 0.74 (Kleiber, 1932). A few years later, Brody et al. published their famous mouse to elephant curve and calculated the exponent to be 0.734 (Brody, 1945). A value of 0.75 is now accepted because it is easier to use, and the difference from 0.734 is considered to be statistically negligible (Schmidt-Nielsen, 1984). However, it should be noted that exponents in the range 0.6–0.8 have been reported for metabolic rate (Agutter and Wheatley, 2004). A value of 0.75 means that the whole body metabolic rate increases as body weight increases, but to a lesser extent than would be expected of a simple proportional relationship. It follows on from this that the specific metabolic rate (the metabolic rate per unit mass) decreases as animals get larger (the exponent is −0.25); the metabolic rate of 1 g blue whale tissue is 1000 times less than that of 1 g shrew tissue (Kirkwood, 1983).
Metabolic rate, physiological time and the fractal origin of scaling
The decrease in specific metabolic rate discussed above forms part of a wider phenomenon: how the meaning of time changes with animal size. Time can be regarded in one of two ways. The first is time as absolute, universal and above nature. This is the classical Newtonian view that we are most familiar with, represented by chronological time. The second view, first proposed by Meyen, is that time is related to variability and change, and that each self-contained system has its own time defined by specific events that occur within that system. Psychological time is an example of this, and we have an intuition of the concept in that time seems to flow faster when we are busy than when we are doing nothing in a waiting room, for example.
Both concepts of time are important to the study of biology. Of particular relevance to issues surrounding drug dosing is the fact that, as a result of their evolutionary adaptation to size, all the physiological processes of a larger animal are slower than those of a smaller animal (heart rate, respiration rate, movement, etc.). For example, the heart rate of an elephant is 30 beats per minute whereas that of a shrew is 1000 beats per minute. However, if one were to count the number of heartbeats or respirations a shrew and an elephant get through during their average lifespan, these turn out to be roughly the same (approximately 200 million breaths and 800 million heartbeats) (Schmidt-Nielsen, 1984).
The perspective of Meyen has given rise to the concept of physiological time in which time is defined by, for example, the number of heartbeats or the number of respirations an animal will have in its lifespan (Brody, 1945; Hill, 1950). Using the above example, we can see that the elephant and the shrew live an equivalent amount of physiological time. The difference in lifespan is seen only in chronological time; elephants live at a slower pace and get through their ‘allotted’ heartbeats and respirations later than a shrew. Their lifespan in chronological time is therefore longer than a shrew's. This example illustrates that physiological time relative to chronological time is unique for each animal species and increases as the size of the animal increases. As Boxenbaum put it, ‘physiological time may be defined as a species-dependent unit of chronological time required to complete a species-independent physiological event’ (Boxenbaum, 1982). This concept is reflected by the fact that the rates of a wide range of physiological processes are related to body size with exponents equal or close to the value −0.25 (Edwards, 1975; Schmidt-Nielsen, 1984). However, out of all of these, the specific metabolic rate is the most important index of the pace of an animal's life, and therefore of what the physiological time for that animal is relative to chronological time (Schmidt-Nielsen, 1984). This means that specific metabolic rate provides an index of how the rates of all the physiological processes involved in drug absorption, distribution, metabolism and excretion scale across species.
In 1997, West et al. proposed an elegant model that explains the origin of these scaling effects. All living things supply nutrients to the cells through a linear, space-filling branching network of vessels designed to limit the energy expended in transport (West et al., 1997). As organisms become larger, the transport distances and the number of cells increases, but the size of the individual cells remains the same. The structure of the transport network is found to conform to a fractal branching network. A fractal, as described by Mandelbrot, is ‘a rough or fragmented geometric shape that can be split into parts, each of which is (at least approximately) a reduced-size copy of the whole’ (Mandelbrot, 1982). Fractals can be observed throughout nature and are seen from the very small scale of atoms and molecules to the very large scale of galaxies. The property of self-similarity, such as exhibited by snowflakes or Russian dolls, is a key feature of fractals and is exhibited by biological transport systems such as the vascular tree. In the model proposed by West et al., the quarter power scaling emerges mathematically from the fractal and space-filling natures of the transport network and the invariant nature of the units supplied by that network. The implications of this are the following. The scaling relationships emerge from evolutionary adaptations that have enabled animals of all sizes to transport, utilize and disperse energy while conserving the same metabolic machinery and conforming to fundamental physical laws over which evolution has no power. These adaptations cause each animal species to have its own physiological time, and this is the effect that scaling accounts for.
Inter-species differences in the pharmacokinetic phase
The elimination of a drug from the bloodstream can exhibit first-order or zero-order kinetics. In first-order kinetics, a constant proportion of drug is eliminated per unit time, whereas in zero-order kinetics, a constant amount of drug is eliminated per unit time. The clearance of a drug is the volume of plasma from which the drug is completely eliminated per unit time; for first-order kinetics, the amount eliminated is proportional to the initial concentration. For zero-order kinetics, the amount eliminated is constant and is independent of the initial concentration. The volume of distribution (Vd) is the ratio of the amount of drug in the body and the concentration of the drug in the plasma; Vd is an index of drug distribution and is high for drugs that distribute extensively into the tissues. The elimination half-life of the drug is the time taken for the plasma concentration to reduce by 50%. Each of these parameters has been considered when accounting for inter-species pharmacokinetic differences, but, as discussed below, clearance has received the most attention.
Species differences in the pharmacokinetic phase have been extensively studied. The absorption of drugs is influenced by the physicochemical properties of the drug, which determine its membrane permeability (ionization constant, molecular size, solubility and lipophilicity) and physiological factors (pH, gastric and intestinal transit time, blood flow rate and first pass metabolism). The membrane permeability of a drug is a property unaffected by size and stays relatively constant across species. However, there are exceptions to this rule; the absorption of lipophilic compounds is greater in dogs than in other species for example (Clark and Smith, 1984). Inter-species differences in oral bioavailability typically arise as a result of differences in first pass metabolism in the gut and liver (Clark and Smith, 1984; Lin, 1998).
The rate of drug distribution is determined by blood flow and the rate of diffusion and/or transport to the target cells. Because circulation time and blood flow scale allometrically with increased body size, smaller animals would be expected to distribute drugs to their targets faster. Also, because of the increased blood flow to the liver and kidneys, smaller animals would be expected to eliminate the drugs faster. If the drug is a high-clearance drug, the effect of size will be accentuated. Based on these principles, one would expect that a high-clearance drug that is eliminated primarily by renal excretion and/or flow-limited hepatic elimination would be amenable to allometric scaling of its clearance and dose. Flow-limited metabolism refers to a situation in which the extraction and metabolism of a drug by the liver is both high and efficient, so that the major limiting factor on the rate of drug metabolism is the rate at which the drug is delivered to the liver.
Protein binding of drugs prevents their diffusion across barrier membranes and therefore restricts the distribution of drugs to their sites of action and excretion. It is well established that drug–protein binding varies considerably between species as a result of differences in the drug affinity and number of protein-binding sites (Callan and Sunderman, 1973). This variation in protein binding is unrelated to size and difficult to predict, so one would not expect the clearance of drugs that exhibit high protein binding to be amenable to straightforward scaling; however, correction for protein binding in these instances greatly increases the accuracy of the scaling. Diffusion of drugs across barrier membranes is also counteracted by the action of efflux transporters such as P-glycoprotein. Drug transporters such as P-glycoprotein contribute to the distribution of drugs into tissues and across barrier membranes and can therefore influence the extent and pattern of drug distribution. At present, very little information is available on inter-species differences in drug transporter activity, and so it is not possible to assess their impact on dose extrapolation. However, the volume of distribution, a measure of the overall extent of drug distribution, has been found to be amenable to allometric scaling; the exponents range from 0.8 to 1.10 (Mahmood, 2007).
One of the key factors to account for during inter-species dose extrapolation is drug metabolism. Despite the presence of the same fundamental biochemical machinery, different species have their own unique characteristics and differ in their ability to metabolize drugs. Some of the differences are marked. Cats lack glucuronidation whereas humans exhibit efficient glucuronidation. Dogs lack acetylation whereas rats are highly efficient acetylators and humans are intermediate between the two. Pigs lack sulphation (Van Miert, 1989; Riviere et al., 1997). Other differences are more complex. The most important group of drug metabolism enzymes is the cytochrome P450 system (CYPs) which mediates the hepatic metabolism of a diverse range of compounds. At least 57 CYP genes have been identified based on information from the human genome project (Guengerich, 2008), all of which have evolved from a single ancestral gene over the past 1.36 billion years (Nelson et al., 1996). There are considerable variations in the primary sequences of CYPs between species, and even a small change in the sequence can produce a profound change in substrate specificity. Similar differences are observed with other drug-metabolizing enzyme systems. The uridine diphosphate glycosyltransferases, which mediate glucuronidation, show complex differences in their profile and, as already mentioned, are absent in dogs (Resetar and Spector, 1989; Clarke and Burchell, 1994). In addition, species differences have emerged in the mechanisms by which drug metabolizing enzymes are induced and inhibited (Lin, 1998). Some of the differences in drug metabolism and drug transport can now be understood by the species differences in the drug activation of nuclear receptors, such as constitutive androstane receptor (CAR) and the pregnane X receptor (PXR), which regulate the expression of various drug-metabolizing enzymes and drug transporters (Stanley et al., 2006). This means that there are complex inter-species differences in drug metabolism and metabolite profiles that are extremely difficult to predict and account for. The situation is further complicated by the fact that metabolites can also be biologically active and must be accounted for when extrapolating the dose. The literature abounds with examples of these differences (Smith, 1997; Lin, 1998). Overall, inter-species differences in drug metabolism systems are not related to size, so one would not expect extensively metabolized drugs to be amenable to allometric scaling of their clearance or dose.
The elimination of drugs and their metabolites usually occurs as a result of biotransformation followed by renal and/or biliary excretion. Compounds with molecular weights exceeding 500, with amphipathic structures (structures containing polar and non-polar groups) or which undergo conjugation reactions tend to be excreted in the bile. Species differences exist in the biliary excretion rate; humans, primates, guinea pigs and rabbits are poor biliary excreters whereas mice, rats and dogs are good biliary excreters and cats are intermediate (Lin, 1998). These differences are not related to blood or biliary flow. It is now known that biliary excretion is mediated by several ATP-binding cassette transmembrane transporters including the multi-drug resistance proteins, P-glycoprotein, breast cancer-related protein and the canalicular multi-specific organic anion transporter (Muller and Jansen, 1997;Ishizuka et al., 1999; Leslie et al., 2005). Recent studies indicate that inter-species differences exist in the activity and regulation of these transporters, just as they do for the metabolizing enzymes, which accounts for species differences in biliary excretion (Bleasby et al., 2006; Nozaki et al., 2007). By contrast, renal excretion of drugs is related primarily to the glomerular filtration rate and the number of nephrons, both of which scale allometrically. The tubular secretion and reabsorption of the drug will also affect excretion. Renal secretion is dependent on renal blood flow but also on the activity of transporters in the proximal tubule; the latter is size-independent and may complicate attempts to scale clearance. Overall, one would expect that drugs that primarily undergo renal excretion would be amenable to allometric scaling whereas drugs that undergo biliary excretion, or a combination of biliary and renal excretion, would not be. Furthermore, drugs that undergo extensive renal secretion may also be less amenable to allometric scaling.
The impact of inter-species differences in pharmacokinetics is illustrated by the study of Rivere et al. (Riviere et al., 1997). In this study, pharmacokinetic data stored in the Food Animal Residue Avoidance Databank were used to examine how the plasma half-lives of 44 candidate drugs scaled as a function of body size. Only 11 of the drugs examined were amenable to allometric scaling. It should be noted that plasma half-life is known to scale less perfectly than volume of distribution and clearance, but this does not affect the key observations of this study. Most of the drugs that did scale allometrically were antibiotics. This is not surprising, as these drugs are usually not extensively metabolized and are renally excreted. Furthermore, their efficacy is determined by the concentration achieved in the extracellular fluid, so pharmacodynamic differences do not occur. Antibiotics are therefore the ‘ideal’ drugs for allometric scaling.
Inter-species differences in the pharmacodynamic phase
Just as the drug transport and metabolizing systems vary among species, so do the target cells and systems of the drugs and their active metabolites. These produce differences in the pharmacodynamic response by producing shifts in the dose–response relationship and also changes in the nature of the response. For example, the process of anaphylaxis is not uniform across species because of inter-species differences in the structure and function of the immune system: the mediators released by mast cells, and the tissue distribution of their receptors, vary between species. The clinical presentation is profoundly affected by these differences. Anaphylaxis targets the lung and vasculature in humans but targets the intestine and the liver in rats. Far from altering the dose of the drug, this even influences the choice of the drug. Antihistamines would be effective in humans, guinea pigs and dogs, but ineffective in cattle (Aitken, 1983; Haley, 2003). In another well-known example, differences in the CNS distribution of neurotransmitters cause differences in the response to analgesics and anaesthetics; opioid analgesics induce CNS depression in primates, dogs, rats and rabbits, but induce CNS excitation in horses, cats, ruminants and swine (Aitken, 1983). Indeed, species differences in the expression levels, tissue distribution and regulation of receptors exist for most of the receptor subtypes for which they have been investigated (Aitken, 1983; Crozatier et al., 1991; Smith 1997; LeCluyse and Rowlands, 2007).
Differences in receptor affinity also exist between species. A good example is provided by the Na+/K+ ATPase transporter, the target of cardiac glycosides. The sensitivity of the dog, sheep and human transporters is one thousand times greater than their counterparts in mice and rats, and these profound differences arise as a result of differences in two amino acids (Tobin and Brody, 1972; Price and Lingrel, 1988). This example illustrates that amino acid substitutions in drug targets can have more profound effects on the pharmacodynamic phase than on the pharmacokinetic phase. These differences in gene regulation, expression patterns and responsiveness reflect and create inter-species differences in feedback regulatory circuits from the level of gene transcription networks to entire systems (Woodman, 1997; Shi et al., 1999; Hollenberg, 2000). Again, species differences have been found in almost every system, at every level of organization, in which they have been looked for. The pharmacodynamic phase is therefore as important to account for as the pharmacokinetic phase, and inter-species differences in the pharmacodynamic phase are the result of the unique characteristics conferred by evolution to each species and therefore unrelated to size. Drugs subject to marked pharmacodynamic differences would not be expected to be amenable to scaling, and pharmacodynamic differences will, in some cases, influence the choice of drug as well as the dose. Scaling in this situation can be improved by introducing adjustment factors. For example, if receptor affinity differs between species, the dose–response curve can be adjusted to account for the difference.
Estimation of the first-in-human dose
There is no consensus on the best method for selecting a first dose in humans. In a recent review, Lowe et al. provided an overview of the approach used at Novartis to derive the first in human dose (Lowe et al., 2007); the pharmacodynamics of the drug in animals are worked out first, followed by the pharmacokinetics. Each is then extrapolated to humans and finally integrated to predict the exposure–response relationship and arrive at a dose. Because pharmacodynamic differences are size-independent, the major contribution of allometric scaling to this process is in the extrapolation of pharmacokinetic parameters from animals to humans.
A limited number of surveys have been conducted to determine which methods of dose estimation are currently used. These indicate that the ‘dose-by-factor’ approach and the ‘pharmacokinetically guided’ approach, which are described below, are the most commonly applied in the planning of Phase 1 studies (Blackwell and Martz, 1972; Reigner and Blesch, 2002). Both of these approaches rely on allometric scaling either of the dose itself or of drug clearance. For a detailed discussion of the application of allometry to pharmacokinetics, the reader is referred to several recent reviews (Lowe et al., 2007; Mahmood, 2007; Edginton et al., 2008). Several caveats apply to the use of allometry to predict pharmacokinetics (Lindstedt and Schaeffer, 2002). The allometric equations should be applied among species, not within species, and derived from species whose weight differs by at least three orders of magnitude (Calder, 1981). Better scaling is obtained when three or more species are used to derive the allometric equation. There are several sources of error in the derivation of allometric exponents. Measurement and analytical errors can be minimized by rigorous assessment of the assays and consistent sampling protocols; alternative assays may be considered to verify that the exponent is not unduly influenced by the choice of assay. Species differences that are size-independent will also influence the exponent. If this effect is pronounced, the dose extrapolation is best performed by using an alternative approach such as pharmacokinetic/pharmacodynamic (PKPD) modelling. Physicochemical properties of drugs determine the extent to which they are absorbed and the pattern of their distribution; neglecting these effects introduces error into the dose extrapolation, which prompted Wajima et al. to develop a prediction model to account for physicochemical properties (Wajima et al., 2002).
The dose-by-factor approach
In the ‘dose-by-factor’ approach, the ‘no-observed adverse effect level’ (NOAEL) of the drug is scaled by using simple allometry on the basis of body surface area to obtain the ‘human-equivalent dose’ (HED) (USFDA, 2005). The Food and Drug Administration of the United States (FDA) has formulated a table of conversion factors that allow the HED to be conveniently calculated by multiplying the NOAEL by the conversion factor (USFDA, 2005) (see Table 1). Note that the main application of this table is to calculate the HED. In order to account for the size-independent effects of pharmacokinetics and pharmacodynamics discussed above, the HED is divided by a safety factor. The default safety factor is 10, although this value does not have a firm scientific basis and several other values have been advocated for specific situations (Bokkers and Slob, 2007; USFDA, 2005). Recent attempts have been made to derive a data-based safety factor from toxicity data in animals, but this work is still ongoing (Bokkers and Slob, 2007). This approach has a good safety record because it is very conservative, and its simplicity makes it very practical and easy to apply. However, the use of this approach assumes that the drug shows similar pharmacokinetics and pharmacodynamics in both species. If these assumptions are not valid, this approach is prone to underestimate the effective dose, meaning that numerous dose escalations are required to find the therapeutic range and demonstrate efficacy (Reigner and Blesch, 2002).
Table 1.
Conversion of animal doses to human-equivalent doses (HEDs) by using the exponent 0.67 for body surface area
| Species | HED from mg·kg−1 dose in animal divide animal dose by | HED from mg·kg−1 dose in animal multiply animal dose by |
|---|---|---|
| Mouse | 12.3 | 0.081 |
| Hamster | 7.4 | 0.135 |
| Rat | 6.2 | 0.162 |
| Ferret | 5.3 | 0.189 |
| Guinea pig | 4.6 | 0.216 |
| Rabbit | 3.1 | 0.324 |
| Dog | 1.8 | 0.541 |
| Monkey | 3.1 | 0.324 |
| Marmoset | 6.2 | 0.162 |
| Squirrel monkey | 5.3 | 0.189 |
| Baboon | 1.8 | 0.541 |
| Micro-pig | 1.4 | 0.730 |
| Mini-pig | 1.1 | 0.946 |
Data obtained from FDA draft guidelines (USFDA, 2005).
The FDA approach uses the exponent for body surface area, 0.67, to scale doses between species. This practice was traditionally rationalized as a means of accounting for differences in metabolic rate. The link between body surface area and metabolic rate, as discussed above, is the evolutionary adaptation of animals to their size, so body surface area is an indirect and imperfect correlate of metabolic rate. There has been debate in the literature about whether the exponent 0.75 would be more appropriate to account for metabolic rate/physiological time (0.75 is the exponent used in veterinary practice). Indeed, better scaling of doses has been demonstrated with the use of the 0.75 exponent (Rennen et al., 2001). On a conceptual level it is more logical to use the exponent that directly accounts for differences in physiological time, rather than an imperfect correlate of metabolic rate, and the proposed fractal origin of the scaling relationship also supports 0.75 as the proper exponent to use (West and Brown, 2005). The justification for using 0.67 is safety related; it provides a more conservative dose estimate.
Example of the dose-by-factor approach
The most sensitive species for a particular drug is the rat, with a NOAEL of 15 mg·kg−1·day−1. To calculate the HED according to the FDA guidelines:
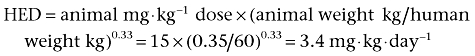 |
Assuming the human weight to be 60 kg, the HED is 206 mg·day−1. Applying a safety factor of 10, the starting dose in humans is 20.6 mg, so a dose of 20 mg·day−1 would be selected.
Pharmacokinetically guided dose extrapolation
The second approach, which is being increasingly used, is the ‘pharmacokinetically guided approach’. In this approach the NOAEL and its area under the curve are determined in several animal species, and the animal species that gives the lowest NOAEL is used as the index species for scaling. The area under the curve gives a measure of systemic exposure to the drug with a time dimension. Next, the clearance of the drug in the index species is scaled allometrically to obtain the estimated clearance in humans, and the starting dose is given by the product of the area under the curve in the index species and the estimated clearance in humans (Bonate and Howard, 2000; Reigner and Blesch, 2002). This method is also safe and has the advantage of accounting for preclinical pharmacokinetic data. Its use assumes that the pharmacokinetic parameters are linear, and that only the parent compound is active; the method must be adjusted if these assumptions are not valid. A limitation of this approach is the inability of the method to account for inter-species differences in drug potency. However, these can be controlled for by the introduction of a safety factor similar to that used in the ‘dose-by-factor’ approach. The key limitation of the method is the extent to which the pharmacokinetic parameters of the drug in humans can be accurately predicted. It is therefore important to consider the various methods available to estimate drug pharmacokinetics in humans.
Estimating clearance using the rule of exponents
Clearance is the most important pharmacokinetic parameter. It can be predicted by simple allometry alone, but it has been found that the ability of simple allometry to predict clearance in humans can be improved in certain situations by multiplying the clearance by the animal's maximum lifespan potential (MLP) or brain weight. It is unclear whether this improved predictive power has any physiological significance or is merely a convenient mathematical happenstance. To help choose the most appropriate of these methods, Mahmood and Balian developed the ‘Rule of Exponents’ (Mahmood and Balian, 1996). In this approach, the clearance of the drug is measured in a range of animal species, and an exponent can be derived by simple allometry. If the value of the exponent is in the range 0.55–0.70, the clearance in humans is best predicted by simple allometry. When the exponent is 0.71–0.99, the product of clearance and MLP best predicts clearance in humans. When the exponent is greater than 1, the product of clearance and brain weight best predicts clearance, although clearance is likely to be underestimated when the exponent rises above 1.3. Conversely, the clearance is likely to be overestimated when the exponent falls below 0.55. A disadvantage of the rule of exponents is that the correction factors depend on the species used, which can interfere with the accuracy of the prediction (Tang and Mayersohn, 2005a). However, the accuracy of the Rule of Exponents can be improved when it is combined with other approaches such as protein-binding correction.
In extreme cases, the clearance can be overestimated by many degrees of magnitude; diazepam, warfarin and valproic acid have predicted clearances (by simple allometry) that are many times greater than their actual clearance in humans. This phenomenon has been named ‘vertical allometry’ and highlights the extreme inaccuracies to which allometric scaling can be subject (Calder, 1984).
Estimating clearance by correction for protein binding
Because protein binding restricts the glomerular filtration of the drug, better scaling is obtained by considering the unbound fraction (fu). In the approach developed by Feng et al. (2000), better prediction of human clearance of 37 drugs was obtained by measuring the clearance of the unbound drug fraction and correcting the estimated clearance using brain weight. This result was confirmed by Sinha et al. in an examination of 24 compounds; the rule of exponents provided a more accurate prediction of clearance when combined with the fu correction method (Sinha et al., 2008). Tang and Mayersohn recently reported the ‘fu-corrected intecept method’, an alternative two-species scaling approach that uses the allometric exponent of clearance and the ratio of fu between rats and humans (Tang and Mayersohn, 2005b). The latter approach has the advantage of accounting for vertical allometry, but it is limited to scaling between rats and humans. The advantage of the fu correction method of Tang and Mayersohn is that it can be used to correct for protein binding between any species of interest, and that it obeys the general principle of using at least three species to scale clearance.
Estimating clearance by measurement of intrinsic clearance in vitro
In recent years, there has been increased interest in the use of in vitro data to improve prediction of in vivo clearance in humans. In this approach, the intrinsic clearance (the rate at which a drug can be removed from the blood by the liver when all confounding influences are absent) of a drug of interest is measured in vitro in human and animal hepatocytes. The intrinsic clearances of the drug in vitro are then integrated with in vivo animal data to predict in vivo clearance in humans (Lave et al., 1997; Lin, 1998). As with any extrapolation method, the use of in vitro metabolic data has failed to predict in vivo metabolic clearance in some cases (Lin, 1998; Mahmood, 2007). The reasons for this are unclear, but a number of factors could explain the discrepancies. Problems with the design and execution of the in vitro measurements are a major source of error, as is the choice of in vitro model (microsomes, recombinant enzymes and hepatocytes). In addition, this method estimates hepatic metabolic clearance and can only be applied to drugs for which this is the major route of elimination. Finally, even when accurate, the method would not be expected to be advantageous over other allometric approaches if the hepatic metabolism of the drug is purely flow-limited. A disadvantage of the method is that it is labour-intensive and time-consuming, because in vitro clearance must be measured in hepatocytes and/or microsomes from several species. However, there is the clear advantage of accounting for inter-species differences in hepatic metabolism. Further studies are required to establish the utility of this method, but it represents a promising new direction.
Estimating clearance by accounting for physicochemical drug properties
Wajima et al. developed an innovative new method of clearance extrapolation by taking a ‘molecule-centric’ approach to scaling. The underlying basis of this approach is the idea that the clearance of a drug is partly determined by its own properties (molecular weight, partition coefficient, number of hydrogen bond acceptors). Full validation of this method is awaited (Wajima et al., 2002).
Estimating clearance by accounting for physiological time: the Dedrick plot
It is possible to account for species differences in physiological time by transforming the time axis of the concentration time profile from Newtonian chronological time to a unit of physiological ‘species-invariant time’. If species differences in the concentration time profile are largely or solely due to differences in physiological time, correcting for physiological time should make the concentration time profiles from different species superimposable. This approach was first employed by Dedrick (Dedrick et al., 1970), who found that correcting for physiological time made the concentration–time profiles of methotrexate in five mammalian species superimposable. Several methods for carrying out this correction now exist. The simplest approach, referred to as the species invariant time method, is to adjust the time axis by using the allometric exponent 0.25. In the elementary Dedrick plot, the adjustment is made based on the allometric exponent of clearance derived from at least three species. In the complex Dedrick plot, the adjustment is made based on the allometric exponents of both clearance and volume of distribution. The most advanced approach is to generate a species invariant unit of time called the dienetichron, which involves adjusting for the allometric exponents of clearance and volume of distribution and for the MLP of the species of interest. The Dedrick plots are superior to the species invariant time approach because they use exponents derived from pharmacokinetic data. The advantage of Dedrick plots is that they can be used to scale concentration–time profiles, and are therefore useful when scaling of clearance or volume of distribution alone has produced inaccurate predictions.
Estimating clearance using physiologically based pharmacokinetic modelling
An alternative to allometric scaling is provided by physiologically based pharmacokinetic (PBPK) modelling. The advantage of this approach is that it allows doses to be scaled between species and within the same species based on real physiological data. The tissues of the organism of interest are represented as a series of compartments, and physiological data are used to create mathematical descriptions of the flow scheme for a particular drug in a particular species. The model is validated and refined by testing its predictions against experimental data. Once the model is able to make accurate predictions in the index species, the model can be used to predict the pharmacokinetics of the drug in another species by inputting the physiological data for that species. Studies comparing PBPK with allometric scaling are scarce. In a retrospective analysis of 19 compounds developed by Hoffmann-La Roche, better prediction of pharmacokinetic parameters was obtained when PBPK was used (Jones et al., 2006). However, the clear advantage of PBPK is that it provides a method of dose extrapolation with a mechanistic basis and a more comprehensive description of the pharmacokinetics; size-independent differences such as drug metabolism can be accounted for. PBPK can also be used to extrapolate between administration routes. A disadvantage of PBPK is its accessibility; it requires an advanced understanding of pharmacokinetics from the user. Furthermore, large amounts of experimental data are required to develop and refine the models. PBPK does not account for pharmacodynamic differences, but PBPK approaches can be integrated with pharmacodynamic modelling to improve the dose estimate (see Toutain, 2002 for review).
It is worth considering the range of applications to which PBPK can be put. In addition to its use as a tool to predict pharmacokinetic parameters between species, PBPK is also used within the same species to predict the effects of age, gender, obesity and various disease states (e.g. liver cirrhosis) on pharmacokinetic parameters (see Edginton et al., 2008 for review). PBPK can therefore be used to guide dose selection throughout the phases of clinical drug development. On a population level, PBPK is used to predict inter-individual variability and reduce the need for extensive population pharmacokinetic studies; these models vary in complexity, from those that consider clearance alone to those that include all of the underlying components of the system (Price et al., 2003; Yang et al., 2006; Willmann et al., 2007). PBPK can also be used to study the mechanism of pharmacokinetic patterns, an application that has received sparse attention (Peters, 2008a,b; Peters and Hultin, 2008); in this application, the parameters of the model are varied to see which combinations of parameters replicate the experimental data. Hypotheses can be generated by examining the compartment affected and the magnitude and direction of the change. For example, the observed pattern may be replicated by increasing gastric emptying time or by decreasing hepatic metabolism.
A more advanced approach, employed to predict the human dose–response relationship, is to combine pharmacokinetic and pharmacodynamic models to generate response–time profiles for proposed dosing regimens (PKPD modelling) (Lowe et al., 2007). These modelling approaches will provide information on the degree of receptor occupancy or biomarker change that can be achieved by a particular dose as a function of time; the investigator must then consider how much receptor occupancy or biomarker change is likely to be required to produce a therapeutic effect. Toxicity is more difficult to predict from PKPD models because its mechanisms are often unanticipated, and is therefore accounted for by deriving a NOAEL from toxicological studies in animals.
Estimating the volume of distribution
The volume of distribution depends on the plasma volume and the distribution of the drug between the plasma and tissues, represented by the partition coefficient. As discussed above, the distribution of a drug into tissues is determined by its own lipid solubility and protein binding, which arise from its own physicochemical properties. It is therefore possible to predict the distribution of a drug by using in silico modelling approaches that take into account the physicochemical properties of the drug (molecular weight, lipophilicity, ionization constant, polar surface area, number of hydrogen bond donors), the partition coefficient and protein binding of the drug, and the known protein and lipid compositions of the target tissues (Poulin and Theil, 2002a,b; Lowe et al., 2007). These models assume that the distribution of the drug is homogenous and mediated by diffusion alone and neglect the effects of active transport and changes in membrane permeability. If the results of the in silico modelling agree with the results of pharmacokinetic studies in two or more species, the in silico model can be used to predict the volume of distribution in humans without the need for scaling (Lowe et al., 2007). If the data disagree with the prediction, then the volume of distribution must be extrapolated using in vivo data. Multiple species scaling of the volume of distribution can be performed, although the results are typically less accurate than are obtained by scaling clearance (Lowe et al., 2007; Mahmood, 2007). Deriving both the volume of distribution and the clearance can improve accuracy by generating a predicted concentration–time profile in humans. Alternatively, a Dedrick plot can be used to adjust the concentration–time profile for differences in physiological time and derive a volume of distribution (Khor et al., 1997). Differences in membrane permeability and active transport can be investigated and accounted for by using PBPK models.
Example of the pharmacokinetically guided approach
The area under the curve obtained at the NOAEL for a particular drug is 23.5 µg·h−1·mL−1 in rats. The predicted clearance in humans is 20.0 L·h−1. The starting dose is given by the equation:
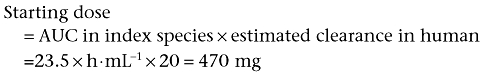 |
A suitable safety factor should also be applied. If a factor of 10 is used, the starting dose is 47 mg.
Other approaches
If the drug to be tested is in the same class as another drug that is already being used in humans, and if the pharmacokinetics and pharmacodynamics of both drugs are the same or very similar, the starting dose in humans can be estimated by the ‘similar drug’ approach (Blackwell and Martz, 1972). In this approach, it is assumed that the ratio of the starting dose to the NOAEL will be the same for both compounds. The optimal starting dose of the index drug is defined as a single dose that produces no pharmacodynamic response of any kind, including, by definition, toxic effects. More recently, it has been suggested that the dose-by-factor, pharmacokinetically guided and, if relevant, the similar-dose approaches should be all used to derive several candidate doses that can then be critically assessed for their relative merits and drawbacks (Reigner and Blesch, 2002).
An example of the similar-dose approach
Drug X, a compound never tested in humans, belongs to the same class as drug A, which is licensed for use in humans. Preclinical toxicological studies revealed that the most sensitive species was rats with a NOAEL of 2 mg·kg−1·day−1. The optimal starting dose of drug A is 30 mg·kg−1·day−1 and the NOAEL of drug A in rats was 14 mg·kg−1·day−1. The similar drug approach uses the equation:
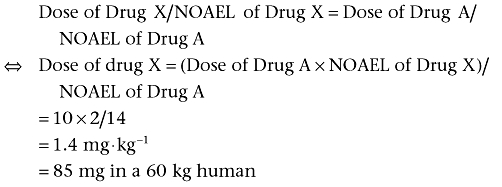 |
A safety factor should also be applied. If a factor of 10 is used, the starting dose will be 8.5 mg.
Deriving the first-in-human dose
As discussed above, the general approach recommended by the USFDA guidelines is that a maximum recommended safe dose (MRSD) be derived from a NOAEL in the most sensitive species by allometric scaling of the dose using an exponent of 0.67, followed by the application of a safety factor (USFDA, 2005). This is the dose-by-factor approach, but pharmacokinetically guided approaches are being increasingly used. When designing the first human trials, extra care must be taken in the estimation of the dose, with a firm emphasis on safety. The pharmacological, pharmacokinetic and toxicological data are carefully examined by experts. The targets of the drug, dose–response relationship, reversibility and exposure must be determined in preclinical studies, and the human exposure anticipated. It is important to consider how the MRSD relates to the pharmacologically active dose; if it is far below the MRSD, multiple dose escalations will be required to arrive at the active dose. If it is above the MRSD, part of the dose–response will be missed and there will be an increased risk of toxicity. In the latter case, it is important to start with a dose that is below the pharmacologically active dose. Pharmacokinetically guided and combined PKPD approaches may be more useful to predict the pharmacologically active dose and alert the investigator to situations in which it is too far above or below the MRSD. Because toxicological effects are difficult to predict, the minimum anticipated biological effect level (MABEL) is a safer level to use, as it is the lowest dose that produces a biological effect of any kind (ESG, 2006). The initial dose should be determined by either the MABEL or MRSD, whichever is lower, and dose escalation can then be determined from predictions of the pharmacologically active dose.
Dose extrapolation for biologicals
Biotechnologically derived drugs (proteins, peptides, antibodies, antibody fragments, antisense oligonucleotides, DNA for gene therapy) are receiving increasing interest as potential therapeutic agents. The pharmacokinetics and pharmacodynamics of these compounds differ from those of the smaller organic molecules with which we have more extensive experience. Peptides and proteins cannot pass biological membranes easily, and are therefore confined to the extracellular space. Their volumes of distribution are therefore small, and scale easily. Significant protein binding can occur with some biotech agents, and this should be corrected for when scaling. Peptide and protein drugs can be degraded by proteolytic enzymes that are present throughout the body, which can alter their biological activity (Tang et al., 2004). The elimination of the peptides and proteins from the body can occur in the kidney; following glomerular filtration, the proteins can be reabsorbed into endocytic vesicles in the proximal tubules and metabolized (the major route of elimination) or hydrolysed by brush border enzymes on the luminal membrane. A small percentage of protein may also be eliminated by peritubular extraction. The liver is also a site of elimination for proteins, where they are internalized and broken down. Finally, because many biological drugs are endogenous molecules, they can be eliminated by receptor-mediated endocytosis in the target tissues. This gives rise to a phenomenon of dose-dependent pharmacokinetics in which saturation of the receptor reserve decreases the clearance (see Tang et al., 2004 for review). Overall, lower molecular weight proteins (e.g. cytokines, calcitonin) are eliminated by the kidney or liver and have short half-lives; larger molecular weight proteins (e.g. IgG) are not filtered by the glumeruli and undergo metabolism following receptor-mediated endocytosis into the liver, vascular endothelium or, for endogenous proteins, their target tissues. IgG and albumin are endocytosed but are rescued from degradation and recycled (Tang et al., 2004; Lowe et al., 2007). Overall, the pharmacokinetics and indeed the pharmacodynamics of the biological agent can be predicted from the natural form of the protein. Because protein drugs undergo renal or flow-limited hepatic elimination, allometric scaling for these drugs is often more accurate than for smaller biomolecules. The USFDA guidelines recommend that scaling for biological compounds should be done on a mg·kg−1 basis rather than surface area; this is the one situation in which scaling doses based on weight is acceptable.
The crucial pitfall to avoid when scaling protein drugs is target binding and receptor occupancy. A tragic example of this is the case of TGN1412, a monoclonal antibody directed against T lymphocytes, which produced multi-organ failure in six healthy volunteers (ESG, 2006); this serious outcome led to the introduction of the MABEL. The MRSD calculated by the conventional allometric approach was 0.1 mg·kg−1. When receptor theory was used to investigate this dose, it was found that 0.1 mg·kg−1 would elicit greater than 90% receptor occupancy. In this situation, not only was the pharmacodynamic effect unacceptably high, producing a cytokine storm, but the increased receptor occupancy could have altered the pharmacokinetics of the antibody by decreasing the clearance, thereby further increasing the peak concentration of the antibody in the plasma and prolonging its effect. There are many lessons to be learned from this tragedy, but an important mechanistic lesson is that once receptor occupancy starts to increase, the pharmacodynamic and pharmacokinetic response to further dose escalations becomes non-linear; in this situation, allometric scaling, which was used for TGN1412, will not work. It is important to determine in preclinical studies whether target binding occurs and, if so, a MABEL must be derived by using models that account for target binding. The MABEL is useful for protein drugs because it defines a dose at which receptor occupancy is low.
Dosing in veterinary practice
Veterinarians are frequently faced with the need to estimate doses in larger animal species. The same principles discussed above for the derivation of human pharmacokinetic parameters also apply to scaling from small to large animals, although this is an area of research that has received less attention; data in large cats, camels and elephants are particularly scarce. Although the same equations used to scale doses from animals to humans can be used to scale from smaller to larger animals, the extrapolation obtained is often less accurate than for humans and further work is needed to refine the approaches for this situation (Mahmood, 2007). For practical reasons, simple allometry is widely used by veterinarians. Established practice is to use the exponent 0.75, in contrast to the exponent 0.67 that has historically been used in human clinical practice, and to scale the dose and the dosing interval separately as follows:
The reference dose in mg·kg−1 is converted into the total dose in mg (total dose = weight × dose in mg·kg−1·day−1) and the interval format (interval = 24 h/dosing frequency). Note that it is not the total daily dose that is calculated at this step. If a drug is given at a dose of 10 mg·kg−1 twice daily, the values 10 mg and 12 h are scaled.
-
The metabolic size, measured by the minimum energy cost (MEC), and the metabolic rate, measured by the minimum energy cost per unit weight (SMEC), is then calculated according to the equations:
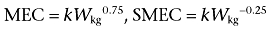
where Wkg is the weight of the animal species and k is the constant of proportionality, usually referred to as the factor. MEC and SMEC are calculated for the subject species and the reference species.
Veterinary formularies provide values for k for the major groups of animals, and a particular k value should only be used to scale between species belonging to the animal group for which the k value was calculated. The reference and subject species should therefore belong to the same group.
-
The MEC dose is calculated from the index species by using the equation:
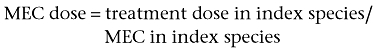
-
The treatment dose in the subject species is calculated by using the equation:
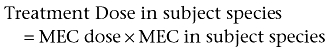
This is the total dose. The dose in mg·kg−1 can be easily derived.
-
The dosing interval is calculated from the SMEC. First, the SMEC interval is calculated in the index species by using the equation:
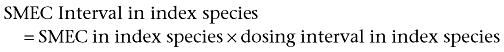
The Treatment Interval in the subject species is obtained by using the equation:
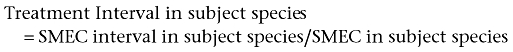
Dosing in experimental studies
In selecting an appropriate dose for experimental studies, investigators need to consider the same factors considered during drug development, namely the dose–response relationship and pharmacokinetics. If one is interested in using a clinically established drug as a pharmacological tool in animals, it may be necessary to scale the dose from a larger to a smaller species (e.g. humans to rats). The major focus of research into scaling and converting doses has been on scaling from smaller to larger species with an emphasis on safety. It is less clear how scaling of, for example, a human dose should be performed if one is interested in using a clinically used drug for research purposes in a smaller species. Although FDA conversion factors can easily be used to convert doses from humans to animals and to convert doses among animals, care must be taken to account for species differences in pharmacokinetics and pharmacodynamics. It is not clear what factor would be most appropriate to use for this, as it is now a therapeutic dose that is being scaled, not a NOAEL, and the effect of the adjustment factor would be to increase the estimated dose. However, there may be a case for using an adjustment factor in some instances. For example, the dose of metoprolol in a rat converted from the human dose using the FDA conversion factors is 10 mg·kg−1·day−1, and doses in this range have been used in rodents. However, we found that, for our studies, a dose of 75 mg·kg−1·day−1 was therapeutic in the rat, and doses as high as 250 mg·kg−1·day−1 have been reported (Maczewski and Mackiewicz, 2008; Sharma et al., 2008), so an additional adjustment factor is required to derive the dose in this case. Doses of established drugs used as pharmacological tools tend to be based on previously published studies and adjusted empirically as needed, so that scaling of the dose is not performed. However, there are clear conceptual advantages to using the effective dose of the drug in humans as a starting point. Further research is needed to develop a strategy for scaling clinical doses to smaller animals so that the time and resources spent optimizing the dose are minimized.
Dosing in children and the elderly
Physiological and biochemical differences can be just as marked within the same species as they are among species. The most striking example is the difference between children and adults. Some of these differences may be related to size during the neonatal period to adulthood, but many are not. The physiological and biochemical processes involved in drug pharmacokinetics mature significantly during paediatric development, resulting in increased or decreased drug clearance depending on the drug (Kearns et al., 2003; Rodriguez et al., 2008). The differences are, unsurprisingly, particularly extreme when considering the dosing of children younger than 18 months. In the neonatal period, the biochemical machinery is immature, and renal clearance is 50% of the adult value (adult clearance values are reached by 6 months). Furthermore, the total body water, a key determinant of the volume of distribution, is higher in neonates, starting at 80% in neonates and gradually declining to 60% by 1 year of age (the adult value is 55%). The drug metabolism systems of the body develop from birth to adulthood. Some CYP enzymes are expressed in the fetal liver and become down-regulated during development, some are only expressed within hours of birth, and some are expressed later in development. Glucuronidation is reduced in the neonate, reaching adult levels by approximately 3 years of age. Overall, drug metabolism is impaired in the first few months of life, but this gives way to a period of enhanced drug metabolism from 2 to 10 years in which drug metabolism is increased (see Johnson, 2003 for review). Pharmacodynamic differences also exist, but are extremely difficult to assess in the paediatric setting and information is therefore scarce.
The use of unlicensed medications in children is an increasing problem internationally, which has fuelled an increase in the incidence of adverse drug reactions. Regulatory authorities will currently not permit a dose regimen to be recommended in children without information on exposure and efficacy, and the USFDA has designed a decision tree to guide the design of the required studies based on prior knowledge of the disease state and the pharmacokinetics and pharmacodynamics of the drug (see Johnson, 2005 for review). Experience with paediatric dosing has allowed the development of a range of paediatric dosing formularies, and additional information services and online resources are becoming available in some countries. However, there are still occasional situations in which a first dose must be estimated for a drug for which there is no previous paediatric experience. PBPK modelling would be the superior method to use (Ginsberg et al., 2004; Bjorkman, 2005; Johnson et al., 2006). However, as a last resort, scaling approaches can also be used. A large number of children's drug dosage rules have been described, almost all of which involve scaling a dose in adults to a corresponding dose in children. Such approaches make the fundamentally false assumption that children are ‘little adults’ and are subject to the same difficulties as scaling on the basis of size among species. Having considered the complexities that arise in dose extrapolation, it is not surprising that a precise, universal and simple method for deriving paediatric doses was never developed. Three approaches are commonly used:
Scaling the adult dose on the basis of body surface area. This can be done by using allometric equations, calculated values of surface area or nomograms. A safety factor is often used in children younger than 18 months to account for the extreme differences in the maturity of physiological and biochemical systems.
-
The Salisbury Rule is designed to be a quick and practical method for approximating the body surface area calculation.
If child weight < 30kg, % adult dose = child weight in kg × 2.
If child weight > 30kg, % adult dose = child weight + 30.
Clark's Rule: Child dosage = (child's weight (kg) × adult dosage)/70. This is a simple weight proportional method. It provides as good a linear fit to the body surface area curve as can be obtained, but it tends to underestimate doses (Lack and Stuart-Taylor, 1997).
A recent study examined the performance of these scaling models for 30 different drugs and found that no single approach was appropriate for all age groups; Clark's rule was safer for neonates and infants, while allometric scaling was more appropriate for older children (Johnson, 2008). The study emphasized that these scaling approaches should only be used as a last resort. Increased recognition of the limitations of simple scaling is driving research into alternative methods of dose derivation in children such as PBPK and PKPD modelling. For a more detailed overview of these efforts, the reader is referred to a recent review (Bjorkman, 2006).
Issues of dose extrapolation also present themselves when dealing with elderly patients who are seldom represented in the clinical trials used to derive the recommended adult doses. However, scaling has nothing to offer in this setting. Despite the widely held belief that aging itself can profoundly alter a drug's pharmacokinetics, evidence for this is, in fact, lacking. For example, the activity of the main CYP enzymes, such as CYP3A4, do not change throughout adulthood (Hunt et al., 1992). Liver blood flow and liver size decline with age, fat content increases and serum albumin decreases slightly (Shah, 2004). However, it has been estimated that the change in pharmacokinetics produced by age alone is only of the order of 20–40%, which is modest compared with the effects of genotype, gender, ethnicity, polypharmacy and co-morbidities (see Shah, 2004 for review). These factors are unrelated to size of subjects and would not be amenable to scaling approaches. Drug pharmacodynamics in the elderly was relatively ignored as a study subject until recently, and information is only beginning to emerge. Drug pharmacodynamics do change with age; the elderly are more susceptible to the pharmacological effects of agents such as pro-arrhythmic, anti-cholinergic and dopaminergic drugs (Hammerlein et al., 1998). However, the major factors to specifically address when deriving drug doses in the elderly are co-morbidities and polypharmacy. PKPD modelling has much to offer in this setting, as does the use of genotyping methods to characterize the systems involved in drug pharmacokinetics and pharmacodynamics. The establishment of clear guidelines for the elderly as a group will be hampered by the fact that, unlike the case with children, age is not the main determinant of the changes in the system, and the range of possible scenarios in this group is vast.
Conclusion
As species increased in size, they adapted to limitations in their ability to gather and dissipate energy by decreasing their pace of life, giving rise to differences in physiological time relative to chronological time. The quarter power scaling that describes this phenomenon probably reflects the influence of fundamental physical and mathematical laws such as the fractal geometry of the transport networks over which evolution has no power. It is the difference in physiological time that allometry can account for. Viewed from an engineering standpoint, allometry accounts for changes in the number or size of functional units and the flow rate through the system. However, evolution has also endowed species with their own unique characteristics on the background of common anatomical, physiological and biochemical machinery. This uniqueness is reflected by inter-species differences in protein binding, drug metabolism and drug transport in the pharmacokinetic phase, and changes in receptor expression, affinity and distribution in the pharmacodynamic phase. From an engineering perspective, these represent differences in the flow scheme and redesigns of the system, which cannot be accounted for by allometry because they override the comparatively modest effects of size.
Overall, the following drugs are unlikely to be amenable to simple allometric scaling of their clearance or dose:
Drugs that are highly protein-bound, but this can be corrected for by considering fu.
Drugs that undergo extensive metabolism and active transport. Important species differences in drug-metabolizing systems must be considered.
Drugs that undergo significant biliary excretion (MW > 500, ampiphilic, conjugated)
Drugs whose targets are subject to inter-species differences in expression, affinity and distribution.
Drugs that undergo extensive renal secretion.
Biological drugs that exhibit significant target-binding effects.
By contrast, the following drugs are likely to be amenable to allometric scaling of their clearance or dose:
Drugs that are predominantly excreted renally.
Drugs that undergo minimal hepatic metabolism, or whose metabolism is primarily flow-limited.
Drugs whose targets are not subject to large inter-species differences in expression, affinity and distribution, or whose effects are not dependent on a receptor interaction (e.g. antibiotics).
Drugs that do not distribute extensively into tissues.
The industry approach to deriving a dose is to use the pharmacokinetically guided approach, using clearance and volume of distribution. The volume of distribution can be estimated initially using in silico approaches and compared with real data as they become available; allometric scaling, PKPD modelling or Dedrick plots can be used if the initial in silico predictions are inaccurate. For the estimation of clearance, physiological, in vitro and in vivo methods can be employed at an earlier stage of drug development, followed, if necessary, by a combination of scaling approaches and PBPK modelling. Extrapolation of concentration–time profiles may be superior in some cases. If allometric scaling is used, many scaling approaches are available (simple allometry, the Rule of Exponents, protein-binding correction, accounting for physicochemical properties, Dedrick plots). There is a logical conceptual basis for each of the methods, and the choice will depend on the situation. There is an advantage to using the Rule of Exponents with protein-binding correction in many cases. Dedrick plots are useful for scaling concentration–time profiles, and scaling of the volume of distribution can be improved by using Dedrick plots. The response–time relationship can be derived by integrating the pharmacokinetic and pharmacodynamic data to obtain an expected pharmacologically active dose. For biological compounds, scaling may not be necessary, and is often more accurate when used, but effects of receptor binding and occupancy must be sought and accounted for using the appropriate models.
We agree with the suggestion of Reigner and Blesch that candidate doses should be derived by using the dose-by-factor, pharmacokinetically guided and, if relevant, the similar-dose approach followed by consideration of their relative merits (Reigner and Blesch, 2002). Of these, we expect that the pharmacokinetically guided approach will be superior in most cases. The first dose in man should be estimated with an emphasis on safety and should be either a MABEL or a MRSD, whichever is lower. The pharmacologically active dose can then be used to inform dose escalation. It is important to consider how the MABEL and MRSD relate to the pharmacologically active dose. A problem with the MRSD is that it can be higher than the pharmacologically active dose in some situations, but we anticipate that this situation will not arise if a MABEL is used. Clinical trials sometimes yield equivocal or disappointing results for treatments, which were reasonably predicted to be beneficial. This is another situation in which careful consideration should be given to the administered dose and how close it really is to the pharmacologically active dose.
The same principles apply to the derivation of veterinary doses, although the practical approach is to scale the dose and dosing regimen separately using the exponent 0.75. More research is needed to provide species-specific dosing recommendations for the larger animal species. As regards scaling doses within the same species, scaling of doses in children should only be used as a last resort. In the short to medium term, PBPK and PKPD approaches should be used to derive first doses in children, followed by PKPD studies as outlined by the relevant guidelines.
In conclusion, it is important for the healthcare, scientific and wider communities to be aware that it is not appropriate to scale doses on the basis of body weight alone. There are only two very specific exceptions to this rule, which are made for practical rather than conceptual reasons; the first is scaling of biological compounds, because of their low tissue distribution, and the second is when performing allometric scaling of adult doses to neonates and infants, because of safety considerations. A large number of tools are available to estimate the first dose in human studies; scaling still plays an important role, but its position in the thought process of first-in-human dose estimation is being redefined as more advanced approaches are developed. For dose extrapolation from adults to children, scaling of the dose is considered the approach of last resort. Scaling is still frequently used in veterinary practice, but we recommend that the approaches that are being introduced to derive first-in-human and paediatric doses should also be used more extensively to derive doses for the larger animal species encountered in veterinary practice. In the end, dose estimation always requires careful consideration of the situation in hand; there is no universal approach that will work for every situation.
Acknowledgments
We would like to thank Dr Peter Seigl, Dr Thomas Chang and Dr Michael Walker for their helpful reviews of this manuscript. Studies quoted from this laboratory were supported by a grant from the Heart and Stroke Foundation of BC and Yukon.
Glossary
Abbreviations:
- CAR
constitutive androstane receptor
- CYP
cytochrome P450
- FDA
Food and Drug Administration of the United States
- fu
unbound fraction
- HED
human-equivalent dose
- LSD
lysergic acid diethylamide
- MABEL
minimum anticipated biological effect level
- MEC
minimum energy cost
- MLP
maximum lifespan potential
- NOAEL
no-observed adverse effect level
- PXR
pregnane X receptor
- SMEC
specific minimum energy cost (minimum energy cost per unit weight)
Conflict of interest
None declared.
References
- Adolph EF. Quantitative relations in the physiological constitutions of mammals. Science. 1949;109:579–585. doi: 10.1126/science.109.2841.579. [DOI] [PubMed] [Google Scholar]
- Agutter PS, Wheatley DN. Metabolic scaling: consensus or controversy? Theor Biol Med Model. 2004;1:13. doi: 10.1186/1742-4682-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken MM. Species differences in pharmacodynamics: some examples. Vet Res Commun. 1983;7:313–324. doi: 10.1007/BF02228640. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br J Clin Pharmacol. 2005;59:691–704. doi: 10.1111/j.1365-2125.2004.02225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman S. Prediction of cytochrome p450-mediated hepatic drug clearance in neonates, infants and children: how accurate are available scaling methods? Clin Pharmacokinet. 2006;45:1–11. doi: 10.2165/00003088-200645010-00001. [DOI] [PubMed] [Google Scholar]
- Blackwell B, Martz BL. For the first time in man. Clin Pharmacol Ther. 1972;13:812–826. doi: 10.1002/cpt1972135part2812. [DOI] [PubMed] [Google Scholar]
- Bleasby K, Castle JC, Roberts CJ, Cheng C, Bailey WJ, Sina JF, et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica. 2006;36:963–988. doi: 10.1080/00498250600861751. [DOI] [PubMed] [Google Scholar]
- Bokkers BG, Slob W. Deriving a data-based interspecies assessment factor using the NOAEL and the benchmark dose approach. Crit Rev Toxicol. 2007;37:355–373. doi: 10.1080/10408440701249224. [DOI] [PubMed] [Google Scholar]
- Bonate PL, Howard D. Prospective allometric scaling: does the emperor have clothes? J Clin Pharmacol. 2000;40:335–340. doi: 10.1177/00912700022009017. [DOI] [PubMed] [Google Scholar]
- Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm. 1982;10:201–227. doi: 10.1007/BF01062336. [DOI] [PubMed] [Google Scholar]
- Brody S. Bioenergetics and Growth. New York: Haffner; 1945. [Google Scholar]
- Brown JH, Gillooly AP, Allen VM, Savage M, West GB. Towards a metabolic theory of ecology. Ecology. 2004;85:1771–1789. [Google Scholar]
- Calder WA., 3rd Scaling of physiological processes in homeothermic animals. Annu Rev Physiol. 1981;43:301–322. doi: 10.1146/annurev.ph.43.030181.001505. [DOI] [PubMed] [Google Scholar]
- Calder WA., 3rd . Size, Function and Life History. Cambridge, MA: Harvard University Press; 1984. [Google Scholar]
- Callan WM, Sunderman FW., Jr Species variations in binding of 63 NI(II) by serum albumin. Res Commun Chem Pathol Pharmacol. 1973;5:459–472. [PubMed] [Google Scholar]
- Clark B, Smith DA. Pharmacokinetics and toxicity testing. Crit Rev Toxicol. 1984;12:343–385. doi: 10.3109/10408448409044214. [DOI] [PubMed] [Google Scholar]
- Clarke D, Burchell B. The uridine diphosphate glucuronyosyltransferase multigene family: function and regulation. In: Kauffman F, editor. Conjugation-Deconjugation Reactions in Drug Metabolism and Toxicity. Berlin: Springer-Verlag; 1994. pp. 3–43. [Google Scholar]
- Crozatier B, Su JB, Corsin A, Bouanani NE-H. Species differences in myocardial beta-adrenergic receptor regulation in response to hyperthyroidism. Circ Res. 1991;69:1234–1243. doi: 10.1161/01.res.69.5.1234. [DOI] [PubMed] [Google Scholar]
- Davis LE, Westfall BA. Species differences in biotransformation and excretion of salicylate. Am J Vet Res. 1972;33:1252–1262. [PubMed] [Google Scholar]
- Dedrick R, Bischoff KB, Zaharko DS. Interspecies correlation of plasma concentration history of methotrexate (NSC-740) Cancer Chemother Rep. 1970;54:95–101. [PubMed] [Google Scholar]
- Duncan RP, Forsyth DM, Hone J. Testing the metabolic theory of ecology: allometric scaling exponents in mammals. Ecology. 2007;88:324–333. doi: 10.1890/0012-9658(2007)88[324:ttmtoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Edginton AN, Theil FP, Schmitt W, Willmann S. Whole body physiologically-based pharmacokinetic models: their use in clinical drug development. Expert Opin Drug Metab Toxicol. 2008;4:1143–1152. doi: 10.1517/17425255.4.9.1143. [DOI] [PubMed] [Google Scholar]
- Edwards NA. Scaling of renal functions in mammals. Comp Biochem Physiol A. 1975;52:63–66. doi: 10.1016/s0300-9629(75)80128-9. [DOI] [PubMed] [Google Scholar]
- ESG. Expert Scientific Group on Phase One Clinical Trials Final Report. London: HMSO; 2006. [Google Scholar]
- Feng MR, Lou X, Brown RR, Hutchaleelaha A. Allometric pharmacokinetic scaling: towards the prediction of human oral pharmacokinetics. Pharm Res. 2000;17:410–418. doi: 10.1023/a:1007520818956. [DOI] [PubMed] [Google Scholar]
- Ginsberg G, Hattis D, Russ A, Sonawane B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children's risks from environmental agents. J Toxicol Environ Health A. 2004;67:297–329. doi: 10.1080/15287390490273550. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- Hammerlein A, Derendorf H, Lowenthal DT. Pharmacokinetic and pharmacodynamic changes in the elderly. Clinical implications. Clin Pharmacokinet. 1998;35:49–64. doi: 10.2165/00003088-199835010-00004. [DOI] [PubMed] [Google Scholar]
- Hill AV. The dimensions of animals and their muscular dynamics. Sci Prog. 1950;38:209–230. [Google Scholar]
- Hollenberg NK. Implications of species difference for clinical investigation: studies on the renin-angiotensin system. Hypertension. 2000;35:150–154. doi: 10.1161/01.hyp.35.1.150. [DOI] [PubMed] [Google Scholar]
- Hunt CM, Westerkam WR, Stave GM. Effect of age and gender on the activity of human hepatic CYP3A. Biochem Pharmacol. 1992;44:275–283. doi: 10.1016/0006-2952(92)90010-g. [DOI] [PubMed] [Google Scholar]
- Huxley JS. Problems of Relative Growth. London: Methuen; 1932. [Google Scholar]
- Ishizuka H, Konno K, Shiina T, Naganuma H, Nishimura K, Ito K, et al. Species differences in the transport activity for organic anions across the bile canalicular membrane. J Pharmacol Exp Ther. 1999;290:1324–1330. [PubMed] [Google Scholar]
- Johnson TN. The development of drug metabolising enzymes and their influence on the susceptibility to adverse drug reactions in children. Toxicology. 2003;192:37–48. doi: 10.1016/s0300-483x(03)00249-x. [DOI] [PubMed] [Google Scholar]
- Johnson TN. Modelling approaches to dose estimation in children. Br J Clin Pharmacol. 2005;59:663–669. doi: 10.1111/j.1365-2125.2005.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TN. The problems in scaling adult drug doses to children. Arch Dis Child. 2008;93:207–211. doi: 10.1136/adc.2006.114835. [DOI] [PubMed] [Google Scholar]
- Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45:511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–1167. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- Khor SP, Amyx H, Davis ST, Nelson D, Baccanari DP, Spector T. Dihydropyrimidine dehydrogenase inactivation and 5-fluorouracil pharmacokinetics: allometric scaling of animal data, pharmacokinetics and toxicodynamics of 5-fluorouracil in humans. Cancer Chemother Pharmacol. 1997;39:233–238. doi: 10.1007/s002800050566. [DOI] [PubMed] [Google Scholar]
- Kirkwood JK. Influence of body size in animals on health and disease. Vet Rec. 1983;113:287–290. doi: 10.1136/vr.113.13.287. [DOI] [PubMed] [Google Scholar]
- Kleiber M. Body size and metabolism. Hilgardia. 1932;6:315–353. [Google Scholar]
- Lack JA, Stuart-Taylor ME. Calculation of drug dosage and body surface area of children. Br J Anaesth. 1997;78:601–605. doi: 10.1093/bja/78.5.601. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lave T, Dupin S, Schmitt C, Chou RC, Jaeck D, Coassolo P. Integration of in vitro data into allometric scaling to predict hepatic metabolic clearance in man: application to 10 extensively metabolized drugs. J Pharm Sci. 1997;86:584–590. doi: 10.1021/js960440h. [DOI] [PubMed] [Google Scholar]
- LeCluyse E, Rowlands J. Species Differences in Receptor-Mediated Gene Regulation. In: Ekins S, editor. Computational Toxicology. Hoboken, NJ: John Wiley & Sons Inc; 2007. pp. 71–98. [Google Scholar]
- Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Lin JH. Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos. 1998;26:1202–1212. [PubMed] [Google Scholar]
- Lindstedt L, Schaeffer PJ. Use of allometry in predicting anatomical and physiological parameters of mammals. Lab Anim. 2002;36:1–19. doi: 10.1258/0023677021911731. [DOI] [PubMed] [Google Scholar]
- Lowe PJ, Hijazi Y, Luttringer O, Yin H, Sarangapani R, Howard D. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica. 2007;37:1331–1354. doi: 10.1080/00498250701648008. [DOI] [PubMed] [Google Scholar]
- Maczewski M, Mackiewicz U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc Res. 2008;79:42–51. doi: 10.1093/cvr/cvn057. [DOI] [PubMed] [Google Scholar]
- Mahmood I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv Drug Deliv Rev. 2007;59:1177–1192. doi: 10.1016/j.addr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Mahmood I, Balian JD. Interspecies scaling: predicting pharmacokinetic parameters of antiepileptic drugs in humans from animals with special emphasis on clearance. J Pharm Sci. 1996;85:411–414. doi: 10.1021/js950400y. [DOI] [PubMed] [Google Scholar]
- Mandelbrot B. The Fractal Geometry of Nature. New York: W.H. Freeman and Company; 1982. [Google Scholar]
- Muller M, Jansen PL. Molecular aspects of hepatobiliary transport. Am J Physiol. 1997;272:G1285–G1303. doi: 10.1152/ajpgi.1997.272.6.G1285. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Kusuhara H, Kondo T, Iwaki M, Shiroyanagi Y, Nakayama H, et al. Species difference in the inhibitory effect of nonsteroidal anti-inflammatory drugs on the uptake of methotrexate by human kidney slices. J Pharmacol Exp Ther. 2007;322:1162–1170. doi: 10.1124/jpet.107.121491. [DOI] [PubMed] [Google Scholar]
- Peters SA. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet. 2008a;47:261–275. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- Peters SA. Identification of intestinal loss of a drug through physiologically based pharmacokinetic simulation of plasma concentration-time profiles. Clin Pharmacokinet. 2008b;47:245–259. doi: 10.2165/00003088-200847040-00003. [DOI] [PubMed] [Google Scholar]
- Peters SA, Hultin L. Early identification of drug-induced impairment of gastric emptying through physiologically based pharmacokinetic (PBPK) simulation of plasma concentration-time profiles in rat. J Pharmacokinet Pharmacodyn. 2008;35:1–30. doi: 10.1007/s10928-007-9073-1. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci. 2002a;91:129–156. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci. 2002b;91:1358–1370. doi: 10.1002/jps.10128. [DOI] [PubMed] [Google Scholar]
- Price EM, Lingrel JB. Structure-function relationships in the Na,K-ATPase alpha subunit: site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry. 1988;27:8400–8408. doi: 10.1021/bi00422a016. [DOI] [PubMed] [Google Scholar]
- Price PS, Conolly RB, Chaisson CF, Gross EA, Young JS, Mathis ET, et al. Modeling interindividual variation in physiological factors used in PBPK models of humans. Crit Rev Toxicol. 2003;33:469–503. [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Reigner BG, Blesch KS. Estimating the starting dose for entry into humans: principles and practice. Eur J Clin Pharmacol. 2002;57:835–845. doi: 10.1007/s00228-001-0405-6. [DOI] [PubMed] [Google Scholar]
- Rennen MAJ, Hakkert BC, Stevenson H, Bos PMJ. Data-based derived values for the inter-species extrapolation, a quantitative analysis of historical toxicity data. Comments Toxicol. 2001;7:423–436. [Google Scholar]
- Resetar A, Spector T. Glucuronidation of 3′-azido-3′-deoxythymidine: human and rat enzyme specificity. Biochem Pharmacol. 1989;38:1389–1393. doi: 10.1016/0006-2952(89)90177-9. [DOI] [PubMed] [Google Scholar]
- Riviere JE, Martin-Jimenez T, Sundlof SF, Craigmill AL. Interspecies allometric analysis of the comparative pharmacokinetics of 44 drugs across veterinary and laboratory animal species. J Vet Pharmacol Ther. 1997;20:453–463. doi: 10.1046/j.1365-2885.1997.00095.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez W, Selen A, Avant D, Chaurasia C, Crescenzi T, Gieser G, et al. Improving pediatric dosing through pediatric initiatives: what we have learned. Pediatrics. 2008;121:530–539. doi: 10.1542/peds.2007-1529. [DOI] [PubMed] [Google Scholar]
- Schmidt-Nielsen K. Scaling: Why is Animal Size so Important? Cambridge: Cambridge University Press; 1984. [Google Scholar]
- Shah RR. Drug development and use in the elderly: search for the right dose and dosing regimen (Parts I and II) Br J Clin Pharmacol. 2004;58:452–469. doi: 10.1111/j.1365-2125.2004.02228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Dhillon P, Wambolt R, Parsons H, Brownsey R, Allard MF, et al. Metoprolol improves cardiac function and modulates cardiac metabolism in the streptozotocin (STZ) diabetic rat. Am J Physiol Heart Circ Physiol. 2008;294:H1609–H1620. doi: 10.1152/ajpheart.00949.2007. [DOI] [PubMed] [Google Scholar]
- Shi Q, Black TA, Gross KW, Sigmund CD. Species-specific differences in positive and negative regulatory elements in the renin gene enhancer. Circ Res. 1999;85:479–488. doi: 10.1161/01.res.85.6.479. [DOI] [PubMed] [Google Scholar]
- Sinha VK, De Buck SS, Fenu LA, Smit JW, Nijsen M, Gilissen RA, et al. Predicting oral clearance in humans: how close can we get with allometry? Clin Pharmacokinet. 2008;47:35–45. doi: 10.2165/00003088-200847010-00004. [DOI] [PubMed] [Google Scholar]
- Smith DA. Pharmacokinetics and pharmacodynamics in toxicology. Xenobiotica. 1997;27:513–525. doi: 10.1080/004982597240479. [DOI] [PubMed] [Google Scholar]
- Stanley LA, Horsburgh BC, Ross J, Scheer N, Wolf CR. PXR and CAR: nuclear receptors which play a pivotal role in drug disposition and chemical toxicity. Drug Metab Rev. 2006;38:515–597. doi: 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- Tang H, Mayersohn M. A mathematical description of the functionality of correction factors used in allometry for predicting human drug clearance. Drug Metab Dispos. 2005a;33:1294–1296. doi: 10.1124/dmd.105.004135. [DOI] [PubMed] [Google Scholar]
- Tang H, Mayersohn M. A novel model for prediction of human drug clearance by allometric scaling. Drug Metab Dispos. 2005b;33:1297–1303. doi: 10.1124/dmd.105.004143. [DOI] [PubMed] [Google Scholar]
- Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J Pharm Sci. 2004;93:2184–2204. doi: 10.1002/jps.20125. [DOI] [PubMed] [Google Scholar]
- Tobin T, Brody TM. Rates of dissociation of enzyme-ouabain complexes and K 0.5 values in (Na ++ K +) adenosine triphosphatase from different species. Biochem Pharmacol. 1972;21:1553–1560. doi: 10.1016/0006-2952(72)90305-x. [DOI] [PubMed] [Google Scholar]
- Toutain PL. Pharmacokinetic/pharmacodynamic integration in drug development and dosage-regimen optimization for veterinary medicine. AAPS PharmSci. 2002;4:E38. doi: 10.1208/ps040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USFDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Adult Healthy Volunteer. Rockville, MD: US Food and Drug Administration; 2005. [Google Scholar]
- Van Miert AS. Extrapolation of pharmacological and toxicological data based on metabolic weight. Arch Exp Veterinarmed. 1989;43:481–488. [PubMed] [Google Scholar]
- Wajima T, Fukumura K, Yano Y, Oguma T. Prediction of human clearance from animal data and molecular structural parameters using multivariate regression analysis. J Pharm Sci. 2002;91:2489–2499. doi: 10.1002/jps.10242. [DOI] [PubMed] [Google Scholar]
- West GB, Brown JH. The origin of allometric scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J Exp Biol. 2005;208:1575–1592. doi: 10.1242/jeb.01589. [DOI] [PubMed] [Google Scholar]
- West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276:122–126. doi: 10.1126/science.276.5309.122. [DOI] [PubMed] [Google Scholar]
- West LJ, Pierce CM, Thomas WD. Lysergic acid diethylamide: its effects on a male asiatic elephant. Science. 1962;138:1100–1103. doi: 10.1126/science.138.3545.1100. [DOI] [PubMed] [Google Scholar]
- Willmann S, Hohn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34:401–431. doi: 10.1007/s10928-007-9053-5. [DOI] [PubMed] [Google Scholar]
- Woodman D. Laboratory Animal Endocrinology: Hormonal Action, Control Mechanisms and Interactions with Drugs. Hoboken, NJ: John Wiley & Sons; 1997. [Google Scholar]
- Yang F, Tong X, McCarver DG, Hines RN, Beard DA. Population-based analysis of methadone distribution and metabolism using an age-dependent physiologically based pharmacokinetic model. J Pharmacokinet Pharmacodyn. 2006;33:485–518. doi: 10.1007/s10928-006-9018-0. [DOI] [PubMed] [Google Scholar]