

Abstract
Background and purpose:
Excessive inflammation and apoptosis are pathological features of endotoxaemic acute renal failure. Activation of glycogen synthase kinase-3 (GSK-3) is involved in inflammation and apoptosis. We investigated the effects of inhibiting GSK-3 on lipopolysaccharide (LPS)-induced acute renal failure, nuclear factor-κB (NF-κB), inflammation and apoptosis.
Experimental approach:
The effects of inhibiting GSK-3 with inhibitors, including lithium chloride (LiCl) and 6-bromo-indirubin-3′-oxime (BIO), on LPS-treated (15 mg·kg−1) C3H/HeN mice (LiCl, 40 mg·kg−1 and BIO, 2 mg·kg−1) and LPS-treated (1 µg·mL−1) renal epithelial cells (LiCl, 20 mM and BIO, 5 µM) were studied. Mouse survival was monitored and renal function was analysed by histological and serological examination. Cytokine and chemokine production, and cell apoptosis were measured by enzyme-linked immunosorbent assay and terminal deoxynucleotidyl transferase-mediated dUTP–biotin nick-end labelling staining, respectively. Activation of NF-κB and GSK-3 was determined by immunostaining and Western blotting, respectively.
Key results:
Mice treated with GSK-3 inhibitors showed decreased mortality, renal tubular dilatation, vacuolization and sloughing, blood urea nitrogen, creatinine and renal cell apoptosis in response to endotoxaemia. Inhibiting GSK-3 reduced LPS-induced tumour necrosis factor-α (TNF-α) and CCL5/RANTES (released upon activation of normal T-cells) in vivo in mice and in vitro in murine kidney cortical collecting duct epithelial M1 cells. Inhibiting GSK-3 did not block TNF-α-induced cytotoxicity in rat kidney proximal tubular epithelial NRK52E or in M1 cells.
Conclusions and implications:
These results suggest that GSK-3 inhibition protects against endotoxaemic acute renal failure mainly by down-regulating pro-inflammatory TNF-α and RANTES.
Keywords: lipopolysaccharide, acute renal failure, inflammation, apoptosis, GSK-3, TNF-α
Introduction
Acute renal failure is generally caused by infectious pathogens, hypoxia/ischaemia, toxins, obstructive insults and severe inflammation (Thadhani et al., 1996). Sepsis-induced acute renal failure (or septic acute renal failure) is commonly present in multiple organ failure (MOF) and multiple organ dysfunction syndrome (MODS) (Martin et al., 2003). Septic acute renal failure has a 70% mortality rate in hospital inpatients, whereas non-septic acute renal failure has a 45% mortality rate (Neveu et al., 1996; Schrier and Wang, 2004). Despite improvements in supportive care and renal replacement therapy, there is still no target-specific therapy for preventing septic acute renal failure. Clinically, septic acute renal failure is characterized by deregulated physiological responses, which include a lower renal blood flow and glomerular filtration rate as well as hypotension (Wan et al., 2003). In patients with septic acute renal failure, blood urea nitrogen (BUN) and creatinine levels are abnormally high (Thadhani et al., 1996). Histopathological changes to the kidneys, such as tubular cell necrosis and apoptosis, sloughing, vacuolization and tubular dilatation, are the hallmarks of septic acute renal failure (Cunningham et al., 2002; Wan et al., 2003).
The mechanisms involved in endotoxaemia-induced septic acute renal failure are diverse; they include uncontrolled systemic inflammatory activation, renal ischaemia, coagulopathy, cytokine/chemokine overproduction and deregulated cell apoptosis (Khan and Badr, 1999; Wan et al., 2003; Schrier and Wang, 2004). Pro-inflammatory tumour necrosis factor-α (TNF-α), which is overproduced in endotoxin lipopolysaccharide (LPS)-treated mice and renal mesangial cells, is crucial in the pathogenesis of septic acute renal failure (Baud et al., 1989; Wan et al., 2003). Down-regulating TNF-α using neutralizing antibodies prevents endotoxaemic acute renal failure (Knotek et al., 2001), and TNF receptor-knockout mice are protected from endotoxaemic acute renal failure (Cunningham et al., 2002). TNF-α causes endotoxaemic acute renal failure through a mechanism involving acute tubular apoptosis (Jo et al., 2002) and renal inflammation (Schrier and Wang, 2004; Anderson, 2007; Wu et al., 2007). Preventing caspase activation is a protective strategy against disease progression (Guo et al., 2004).
Glycogen synthase kinase-3 (GSK-3) regulates a variety of cellular responses, including cell growth, differentiation, apoptosis and inflammation (Frame and Cohen, 2001; Jope et al., 2007). GSK-3 mediates apoptotic signalling induced by staurosporine, heat shock, growth factor withdrawal, hypoxia, endoplasmic reticulum stress, mitochondrial complex I inhibitors and ceramide (Bijur et al., 2000; Hetman et al., 2000; King et al., 2001; Somervaille et al., 2001; Bhat et al., 2002; Loberg et al., 2002; Song et al., 2002; Hongisto et al., 2003; Lin et al., 2007). In addition, the activation of transcription factor NF-κB by GSK-3 is important for Toll-like receptor (TLR) and TNF-α receptor signalling. Therefore, GSK-3 is a cellular target for anti-inflammation (Takada et al., 2004; Dugo et al., 2005; 2006a,b; 2007a,b; Martin et al., 2005; Woodgett and Ohashi, 2005; Cuzzocrea et al., 2006; 2007; Ougolkov and Billadeau, 2006; Whittle et al., 2006). Using a sepsis model induced by LPS plus peptidoglycan, Dugo et al. (2005) first demonstrated the anti-inflammatory effects of GSK-3 inhibition by down-regulating NF-κB activation and cytokine production in lung inflammation. They also showed that treatment with GSK-3β inhibitors conferred protection against liver and renal injury. However, the protective mechanism due to inhibiting GSK-3 in endotoxaemic acute renal failure remains unclear. To study the pathological roles of TNF-α in endotoxaemic acute renal failure, we developed a mouse model of endotoxaemic acute renal failure and analysed the protective action of inhibiting GSK-3 on key pathological changes: cell death, renal dysfunction and cytokine/chemokine production. The role of GSK-3 in TNF-α-induced cytotoxicity was also investigated.
Methods
Animal treatment
The 8-week-old progeny of male C3H/HeNCrl mice were purchased from Charles River Japan, Inc. (Atsugi, Japan). They were fed standard laboratory chow and water ad libitum in the Laboratory Animal Center of National Cheng Kung University. The animals were raised and cared for according to the guidelines set up by the National Science Council, Taiwan. The experimental protocol adhered to the rules of the Animal Protection Act of Taiwan and was approved by the Laboratory Animal Care and Use Committee of National Cheng Kung University.
To establish the murine model of endotoxaemic acute renal failure, mice were injected i.p.with a total volume of 200 µL of 15 mg·kg−1 of Escherichia coli-derived LPS, dissolved in sterile phosphate-buffered saline (PBS), to obtain ∼50% mortality during the early phase of sepsis (within 24 h). To verify the role of GSK-3, the mice were treated with GSK-3 inhibitors at the indicated doses, diluted in a total volume of 200 µL PBS, for the time periods previously described (Dugo et al., 2005; 2006a; 2007b; Cuzzocrea et al., 2006; 2007; Whittle et al., 2006). PBS was used as the vehicle control. The survival rate of the mice was monitored for 48 h.
Pathological examination
To detect serum levels of TNF-α, RANTES, BUN and creatinine, we collected serum from peripheral blood via retro-orbital bleeding immediately before the LPS injection and post-injection at the indicated time periods under pentobarbital anaesthesia (40 mg·kg−1 i.p.). The mice were given a lethal overdose of pentobarbital (200 mg·kg−1 i.p.), and their kidney tissue was harvested at the indicated times post-injection. The levels of BUN and creatinine were measured using an automatic biochemical analyser. For histopathological observation, portions of kidney tissue were fixed in 10% neutral-buffered formalin solution and then dehydrated in graded alcohol. The fixed tissue was embedded in paraffin and sliced into 4 µm thick sections. The tissue sections were mounted on regular glass slides and stained with haematoxylin and eosin (H&E stain). The overall pathological changes, including renal vacuolization, dilatation, sloughing, renal cell necrosis and apoptosis, were diagnosed according to previous studies (Cunningham et al., 2002; Wan et al., 2003).
Morphometric analysis
To quantify the histopathological changes, morphometric analysis of photomicrographs of the kidney sections was performed using ImageJ software (version 1.41o) from W. Rasband (National Institutes of Health, Bethesda, MD, USA) (http://rsb.info.nih.gov/ij/). Vacuolization of kidney sections was determined by measuring the area of tissue destruction (negative H&E stain). The amount of vacuolization was calculated as % of the total area of the microscopic fields observed at ×20. Necrotic/Apoptotic cells were determined by quantifying nuclear and cytoplasmic destruction (negative H&E stain) around the nuclei. The numbers of necrotic/apoptotic cells were calculated as % of the total number of cells in the microscopic fields observed at ×20.
Cell culture
Healthy mouse kidney cortical collecting duct epithelial M1 cells and healthy rat kidney proximal tubular epithelial NRK52E cells were provided by Dr. Y-Y Chiou (Department of Pediatrics, National Cheng Kung University). The M1 and NRK52E cells were routinely grown on plastic in Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 medium and in DMEM only, respectively, with l-glutamine and 15 mM HEPES supplemented with 10% fetal bovine serum (FBS), 100 U penicillin and 100 µg·mL−1 streptomycin. M1 and NRK52E cells were used at a passage of 7 to 10 in this study.
Apoptosis assay
To detect apoptosis in kidney tissue, we fixed the paraffin-treated sections for 24 h in 3.7% formaldehyde and then removed the paraffin from them. We then used a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP–biotin nick-end labelling (TUNEL) staining DNA fragmentation assay kit according to the manufacturer's instructions. The sections were counterstained with 5 µg·mL−1 of 4,6-diamidino-2-phenylindole (DAPI) for nuclear counter-staining, and a blinded observer counted the number of positively stained nuclei per high-power field using a fluorescence microscope (IX71). Three different and randomly selected areas per specimen were analysed. Apoptosis was analysed using propidium iodide staining as described previously (Lin et al., 2007), and then measured using a flow cytometer with excitation set at 488 nm.
Enzyme-linked immunosorbent assay
The levels of TNF-α and RANTES in serum and cell culture supernatant were measured using TNF-α and RANTES enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's instructions. All measurements were done in triplicate. After the reaction, the plates were washed, and 100 µL of o-phenylenediamine substrate was added to each well and incubated for 30 min at room temperature, after which 50 µL per well of 4 N sulphuric acid was added. The plates were read at 490 nm on a microplate reader and the data analysed by use of Softmax Pro software.
Western blotting
Harvested cells were lysed with a buffer containing 1% Triton X-100, 50 mM of Tris (pH 7.5), 10 mM of EDTA, 0.02% NaN3 and a protease inhibitor cocktail. Following one cycle of freeze–thaw, cell lysates were centrifuged at 10 000×g at 4°C for 20 min. Lysates were boiled in sample buffer for 5 min. The proteins were then subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred to PVDF membrane using a semi-dry electroblotting system. After being blocked with 5% skim milk in PBS, the membranes were incubated with a 1/1000 dilution of primary antibodies, including phospho-GSK-3α (Ser21), phospho-GSK-3β (Ser9), GSK-3α, GSK-3β and β-actin, at 4°C overnight. The membranes were then washed with 0.05% PBS–Tween 20 and incubated with a 1/5000 dilution of horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. After being washed, the membranes were soaked in ECL solution for 1 min, and then exposed to film (BioMax, Rochester, NY, USA).
Immunocytochemistry staining
Cells were fixed in 3.7% formaldehyde in PBS for 10 min. After washing twice with PBS, the cells were mixed with anti-NF-κB p65 antibodies (Chemicon, Temecula, CA, USA) in antibody diluents (DAKO Corporation, Carpinteria, CA, USA), applied to the sections and incubated at 4°C overnight. The next day, the cells were washed with PBS and then incubated with Alexa Fluor 488-labelled secondary antibodies at room temperature for 1 h. For nuclei counter-staining, DAPI was added and stained at room temperature for 10 min. The cells were washed with PBS and then visualized under a fluorescent microscope (BX51). The positive cells in three fields viewed for each culture were measured.
Cytotoxicity assay
To evaluate cell damage, lactate dehydrogenase activity was assayed using a colorimetric assay kit according to the manufacturer's instructions. Aliquots of the culture media were transferred to 96-well microplates. A microplate reader (Spectra MAX 340PC, Sunnyvale, CA, USA) was used to measure the absorbance at 620 nm with a reference wavelength of 450 nm and data were analysed with Softmax Pro software. The levels of cytotoxicity were calculated as percentage increases compared with the control, and the control was normalized to 100% of the basal level.
Statistical analysis
Values are expressed as means ± SD. Groups were compared using Student's two-tailed unpaired t-test or one-way analysis of variance analysis followed by Dunnet's post hoc test, as appropriate. Statistical significance was set at P < 0.05.
Reagents and materials
GSK-3 inhibitors, including lithium chloride (LiCl), 6-bromo-indirubin-3′-oxime (BIO), or thiadiazolidine-8 (TDZD-8), SB216763 (3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione), SB415286 (3-[(3-chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrol-2,5-dione) and the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) were obtained from Sigma Chemical Company (St Louis, MO, USA) and dissolved in DMSO prior to dilution with PBS and use in mice or cells. PI, DAPI and o-phenylenediamine substrate were obtained from Sigma-Aldrich. Rabbit anti-mouse GSK-3α (Ser21), GSK-3β (Ser9), GSK-3α and GSK-3β were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). β-Actin antibodies and horseradish peroxidase-conjugated anti-rabbit IgG were obtained from Chemicon International, Inc. (Temecula, CA, USA). Recombinant mouse and human cytokine TNF-α were from PeproTech Inc. (Rocky Hill, NJ, USA). All drug treatments in cells were assessed for their cytotoxic effects using cytotoxicity assays before experiments. Non-cytotoxic doses were used in this study.
Escherichia coli-derived LPS was obtained from Calbiochem (San Diego, CA, USA). The automatic biochemical analyser (7080) from Hitachi Koki Co., Ltd., (Tokyo, Japan); DNA fragmentation assay kit (ApoAlert), Clontech (Mountain View, CA, USA); fluorescence microscopes, Olympus (Tokyo, Japan); flow cytometer (FACSCalibur), BD Biosciences (San Jose, CA); TNF-α and RANTES ELISA kits, R&D Systems (Minneapolis, MN, USA); microplate reader (Spectra MAX 340PC), Molecular Devices Corporation (Sunnyvale, CA, USA); Softmax Pro software, Molecular Devices Corporation; protease inhibitor cocktail, Roche Boehringer Mannheim Diagnostics (Mannheim, Germany); PVDF membrane, Millipore (Billerica, MA, USA); ECL solution, PerkinElmer Life Sciences Inc. (Boston, MA, USA); BioMax film, Eastman Kodak (Rochester, NY, USA); colorimetric assay (Cytotoxicity Detection kit), Roche Diagnostics (Lewes, UK); anti-NF-κB p65 antibodies, Chemicon International, Inc.; antibody diluents, DAKO Corporation.
Results
GSK-3 inhibitors reduce endotoxaemia-induced mortality in C3H/HeN mice
To set up a murine model of endotoxaemic acute renal failure, we injected 10 of the mice with 15 mg·kg−1 of E. coli-derived LPS, which caused three of them to die within 24 h. We found that LPS time-dependently up-regulated serum levels of BUN and creatinine within 12 h (data not shown). To determine the protective effects of inhibiting GSK-3 in vivo, we treated the mice with LPS (15 mg·kg−1) with or without GSK-3 inhibitors: lithium chloride (LiCl) (40 mg·kg−1), BIO (2 mg·kg−1) or TDZD-8 (1 mg·kg−1). Within 36 h, all 10 mice in the LPS-only group died. Notably, we found that all three of the GSK-3 inhibitors significantly (P < 0.05) reduced LPS-induced mortality (Figure 1A). We next examined the protective effects of inhibiting GSK-3 for various periods of time. Pretreatment (6 h before LPS treatment) and co-treatment with BIO (2 mg·kg−1), but not post-treatment (6 h after LPS treatment), significantly (P < 0.05) reduced LPS-induced mortality (Figure 1B). These results provide strong evidence that inhibiting GSK-3 reduces endotoxaemia-induced mortality.
Figure 1.
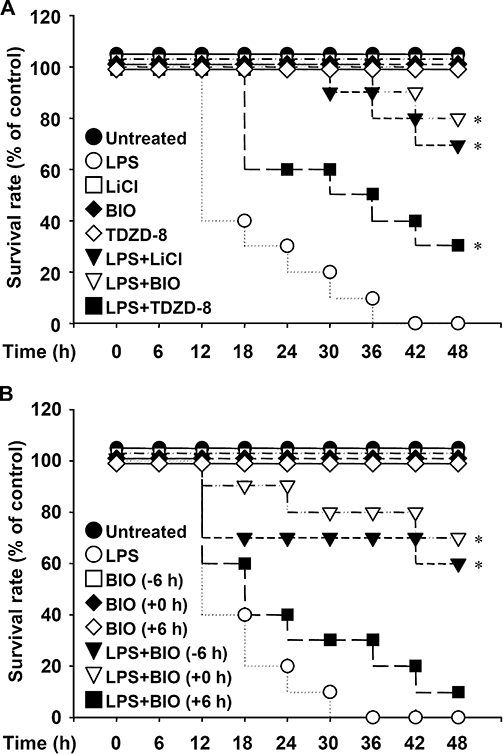
Glycogen synthase kinase-3 inhibitors reduce lipopolysaccharide (LPS)-induced mortality for the first 48 h post-LPS injection in C3H/HeN mice. Mice were injected with LPS (15 mg·kg−1 i.p.) with or without lithium chloride (LiCl) (40 mg·kg−1), 6-bromo-indirubin-3′-oxime (BIO) (2 mg·kg−1) or thiadiazolidine-8 (TDZD-8) (2 mg·kg−1) co-treatment (A), and with or without BIO (2 mg·kg−1) pretreatment (−6 h), co-treatment (+0 h) or post-treatment (+6 h) (B). The survival rate of each group (n= 10) was monitored for the indicated time periods. *P < 0.05 compared with the LPS group.
GSK-3 inhibitors reduce endotoxaemia-induced nephrotoxicity in C3H/HeN mice
Using a histopathological examination, we found that treating mice with LiCl (40 mg·kg−1) or BIO (2 mg·kg−1) down-regulated LPS (15 mg·kg−1)-induced renal vacuolization, dilatation and sloughing, as well as renal cell necrosis and apoptosis, for the first 12 h post-treatment (Figure 2A). Morphometric analysis was used to quantify the histopathological changes, including vacuolization (Figure 2B) and cell necrosis/apoptosis (Figure 2C), in LPS-treated groups with or without GSK-3 inhibition. All of the results demonstrated the renal protective effects of inhibiting GSK-3, and are summarized in Table 1. Treating the mice with combined LPS and LiCl (40 mg·kg−1) or BIO (2 mg·kg−1) significantly (P < 0.05) inhibited the LPS-induced up-regulation of serum BUN and creatinine production (Figure 2D). TUNEL staining showed that endotoxaemia time-dependently induced renal cell apoptosis in mice (data not shown). Other experiments showed that treating mice with BIO (2 mg·kg−1) significantly (P < 0.05) decreased LPS-induced renal cell apoptosis 3 h post-treatment (Figure 2E). These results indicate that inhibiting GSK-3 attenuates endotoxaemia-induced nephrotoxicity in vivo.
Figure 2.
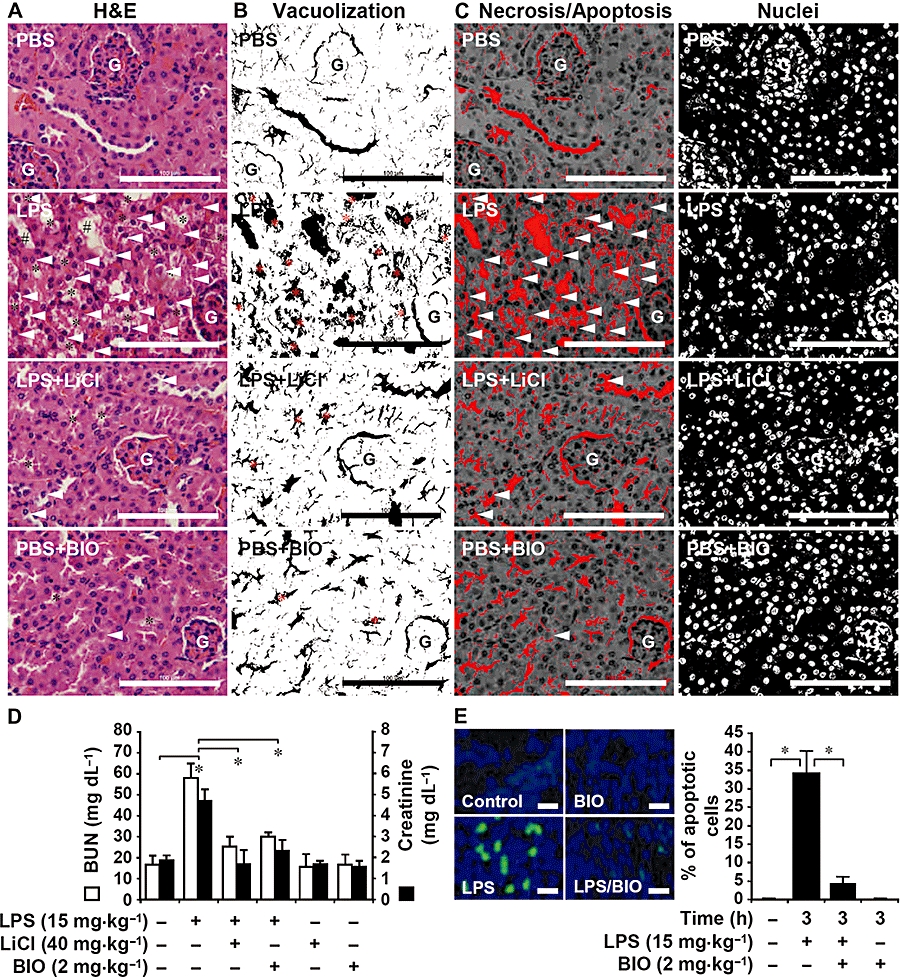
Glycogen synthase kinase-3 inhibitors reduce lipopolysaccharide (LPS)-induced nephrotoxicity in C3H/HeN mice. The mice were injected with LPS (15 mg·kg−1 i.p.) with or without lithium chloride (LiCl) (40 mg·kg−1) or 6-bromo-indirubin-3′-oxime (BIO) (2 mg·kg−1) co-treatment. (A) Twelve hours post-treatment, the mice were killed and their kidneys were removed and prepared in sections for haematoxylin–eosin staining. Compared with phosphate-buffered saline (PBS)-treated control mice, histopathological changes of vacuolization (*), brush border attenuation and sloughing (#), and necrotic/apoptotic cells (arrowheads) were significantly greater in the LPS-treated group than in the LPS + LiCl group. Data are representative of three mice. G, glomerulus. Scale bar is 100 µm. The area of vacuolization (*) (B) and the generation of necrotic/apoptotic cells (arrowheads) were determined (C) by morphometric analysis using ImageJ software version 1.41o. For numbers of nuclei, the total number of cells in the observed fields was counted. (D) Concentrations of blood urea nitrogen and creatinine were determined in sera. Data, obtained from three mice, are means ± SD *P < 0.05. (E) Three hours post-treatment, the mice were killed, and their kidneys were removed and prepared in cryosections to detect cell apoptosis using terminal deoxynucleotidyl transferase (TdT)-mediated dUTP–biotin nick-end labelling staining (green). 4,6-diamidino-2-phenylindole was used for nuclei staining (blue). Data are representative of three mice. Scale bar is 75 µm. The percentages of apoptotic cells were calculated, and data, obtained from three different areas, are means ± SD *P < 0.05.
Table 1.
Morphometric analysis and characterization of pathological changes in lipopolysaccharide (LPS)-treated C3H/HeN mice with or without glycogen synthase kinase-3 (GSK-3) inhibition
| Vacuolization (%) | Necrosis/Apoptosis (%) | Dilatation/Sloughing | |
|---|---|---|---|
| Untreated | 7.96 ± 3.19 | 2.09 ± 1.27 | 0/0/0 |
| LPS | 47.93 ± 7.99 | 36.69 ± 4.45 | 2/2/2 |
| LPS + LiCl | 18.05 ± 2.93* | 7.66 ± 1.56* | 0/0/1 |
| LPS + BIO | 13.27 ± 2.75* | 5.97 ± 1.86* | 0/0/0 |
The values of vacuolization area and necrosis/apoptosis represent the percentages (means ± SD) of three fields at ×20 and were obtained by ImageJ software version 1.41o analysis. For the analysis of vacuolization area, the % area relative to the total area of the observed field was calculated. For the quantification of necrotic/apoptotic cells, total cells were calculated. Grades of dilatation/sloughing of three fields are as follows: 0, no dilatation/sloughing; 1, weak; 2, severe. The effects of LPS (15 mg·kg−1) and GSK-3 inhibition (LiCl, 40 mg·kg−1 and BIO, 2 mg·kg−1) were assessed in kidney tissue 12 h after treatment.
P < 0.05.
LiCl, lithium chloride; BIO, 6-bromo-indirubin-3′-oxime.
Inhibiting GSK-3 suppresses LPS-induced cytokine production in vivo and in vitro
TNF-α-induced renal inflammation and apoptosis is involved in the progression of endotoxaemic acute renal failure (Schrier and Wang, 2004). To investigate the anti-inflammatory effects of GSK-3 inhibition, we used ELISA to determine the in vivo production of the cytokine TNF-α and the chemokine RANTES in LPS-treated (15 mg·kg−1) mice and in vitro in LPS-treated (1 µg·mL−1) mouse cortical collecting duct epithelial M1 cells. We found that co-treatment with either LiCl or BIO significantly (P < 0.05) reduced LPS-up-regulated TNF-α and RANTES in vivo (Figure 3A) and in vitro (Figure 3B). These results show that GSK-3 inhibitors suppress LPS-induced inflammation, and TNF-α and RANTES production.
Figure 3.
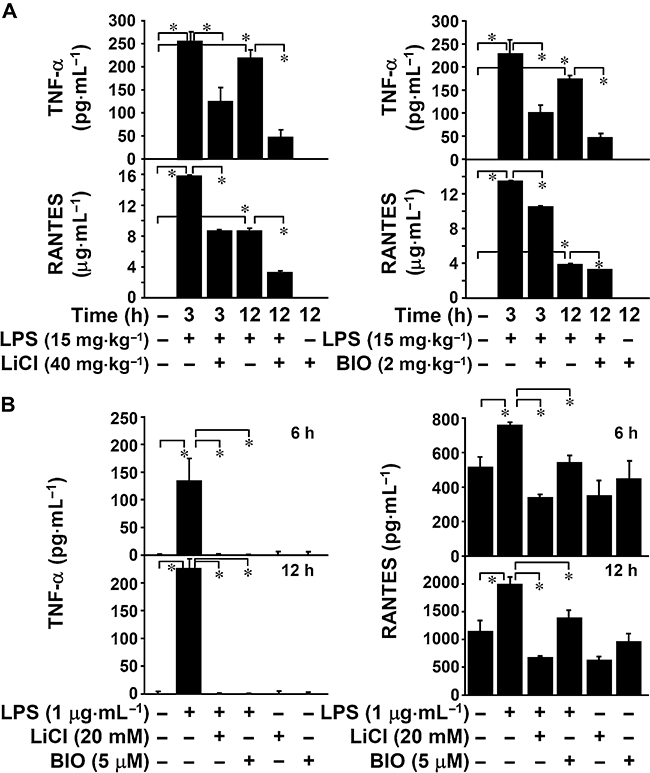
Inhibiting glycogen synthase kinase-3 reduced lipopolysaccharide (LPS)-induced tumour necrosis factor-α (TNF-α) and regulated on activation normal T cell expressed and secreted (RANTES) production. (A) C3H/HeN mice were injected with LPS (15 mg·kg−1 i.p.) for the indicated time periods with or without lithium chloride (LiCl) (40 mg·kg−1) or 6-bromo-indirubin-3′-oxime (BIO) (2 mg·kg−1) co-treatment. The mice were killed and their sera collected to determine levels of TNF-α and RANTES using enzyme-linked immunosorbent assay (ELISA). Data, obtained from three mice, are means ± SD *P < 0.05. (B) Mouse collecting duct epithelial M1 cells (5 × 104 cells/well in 96-well culture plates) were treated with LPS (1 µg·mL−1) for the indicated time periods with or without LiCl (20 mM) or BIO (5 µM) pretreatment for 0.5 h. Levels of TNF-α and RANTES in culture supernatants were determined by ELISA. Data, obtained from triplicate cultures, are means ± SD *P < 0.05.
Inhibiting NF-κB suppresses LPS-induced cytokine production in vivo and in vitro
To investigate the possible mechanism of the anti-inflammatory effect induced by inhibiting GSK-3, the effects of NF-κB, a cellular target of GSK-3 (Takada et al., 2004; Dugo et al., 2005; 2006a,b; 2007a,b; Martin et al., 2005; Woodgett and Ohashi, 2005; Cuzzocrea et al., 2006; 2007; Ougolkov and Billadeau, 2006; Whittle et al., 2006), on LPS-induced TNF-α and RANTES production were studied. We used ELISA to determine the in vivo production of the cytokine TNF-α and the chemokine RANTES in LPS-treated (15 mg·kg−1) mice, and in vitro in LPS-treated (1 µg·mL−1) mouse cortical collecting duct epithelial M1 cells. We found that co-treatment with the NF-κB inhibitor PDTC significantly (P < 0.05) reduced LPS-up-regulated levels of TNF-α and RANTES in vivo (Figure 4A) and in vitro (Figure 4B). These results show that LPS induces TNF-α and RANTES production in an NF-κB-regulated manner.
Figure 4.
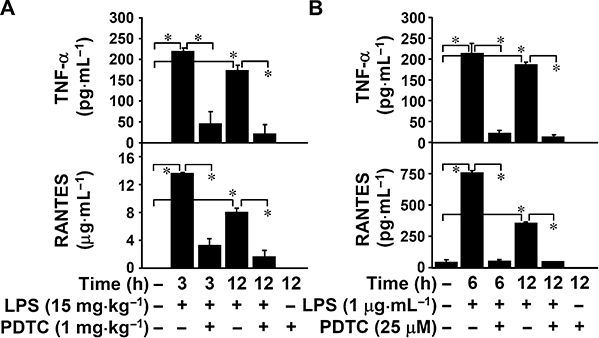
Inhibiting nuclear factor-κB reduced lipopolysaccharide (LPS)-induced tumour necrosis factor-α (TNF-α) and regulated on activation normal T cell expressed and secreted (RANTES) production. (A) C3H/HeN mice were injected with LPS (15 mg·kg−1 i.p.) for the indicated time periods with or without pyrrolidine dithiocarbamate (PDTC) (1 mg·kg−1) co-treatment. The mice were killed and their sera collected to determine levels of TNF-α and RANTES using enzyme-linked immunosorbent assay (ELISA). Data, obtained from three mice, are means ± SD *P < 0.05. (B) Mouse collecting duct epithelial M1 cells (5 × 104 cells/well in 96-well culture plates) were treated with LPS (1 µg·mL−1) for the indicated time periods with or without PDTC (25 µM) pretreatment for 0.5 h. Levels of TNF-α and RANTES in culture supernatants were determined by ELISA. Data, obtained from triplicate cultures, are means ± SD *P < 0.05.
Inhibiting GSK-3 blocks LPS-induced GSK-3 and NF-κB activation
Because we found that GSK-3 and NF-κB were essential for LPS-induced inflammation, we next investigated the activation of GSK-3 and its effects on LPS-induced inflammation. Western blot analysis showed that LPS induced the dephosphorylation of GSK-3α (Ser21) and GSK-3β (Ser9), which activated GSK-3 both in C3H/HeN mice kidney cells (Figure 5A) and in M1 cells (Figure 5B). Furthermore, LiCl and BIO potently blocked these effects. Although GSK-3 regulation of NF-κB is involved in TLR-mediated cytokine production (Martin et al., 2005), we found that LiCl (20 mM) and BIO (5 µM) significantly (P < 0.05) reduced LPS-induced NF-κB nuclear translocation in M1 cells (Figure 5C). Treatment with PDTC was used as a positive control. These results indicate that inhibiting GSK-3 suppresses the LPS-induced activation of GSK-3 and NF-κB.
Figure 5.
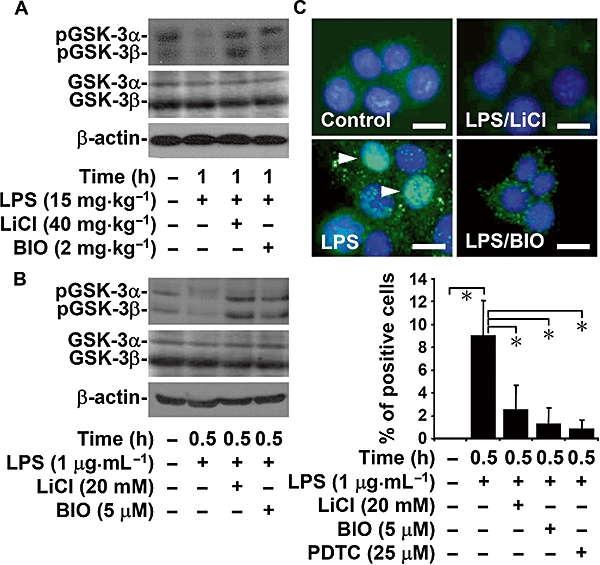
Inhibiting glycogen synthase kinase-3 (GSK-3) blocked lipopolysaccharide (LPS)-induced GSK-3 and nuclear factor-κB (NF-κB) activation. C3H/HeN mice were injected with LPS (15 mg·kg−1 i.p.) for 1 h with or without lithium chloride (LiCl) (40 mg·kg−1) or 6-bromo-indirubin-3′-oxime (BIO) (2 mg·kg−1) co-treatment. M1 cells (3 × 106 cells/10 cm culture dish) were treated with LPS (1 µg·mL−1) for 0.5 h with or without LiCl (20 mM) or BIO (5 µM) pretreatment for 0.5 h. Western blot analysis was used to determine the phosphorylation of GSK-3α (Ser21) and GSK-3β (Ser9) in vivo (A) and in vitro (B). β-actin was the internal control. Data are representative of three mice or three individual experiments. (C) Fluorescent microscopy was used to determine the expression of NF-κB (green) in M1 cells immunostained with anti-NF-κB p65 antibody (positive cells: arrowheads). 4,6-Diamidino-2-phenylindole was used for nuclei staining (blue). Pyrrolidine dithiocarbamate (PDTC) (25 µM) was used as a positive control. Scale bar is 25 µm. Data obtained from three different areas are means ± SD *P < 0.05.
Inhibiting GSK-3 does not reduce TNF-α-induced apoptosis in renal epithelial cells
Because we found that LPS treatment caused renal cell apoptosis in C3H/HeN mice kidneys and that inhibiting GSK-3 down-regulated apoptosis (Figure 2C), we investigated how inhibiting GSK-3 affected TNF-α-induced cell death and endotoxaemic acute renal failure (Schrier and Wang, 2004). A cytotoxicity analysis showed that TNF-α-induced rat kidney proximal tubular epithelial NRK52E cell death was time dependent (data not shown). However, we found that the GSK-3 inhibitors we used – LiCl (20 mM), BIO (5 µM), SB216763 (25 µM), SB415286 (25 µM) and TDZD-8 (25 µM) – did not reduce TNF-α-induced cytotoxicity in NRK52E or M1 cells (Figure 6A). Using PI staining and then flow cytometric analysis, we found that inhibiting GKS-3 did not decrease TNF-α-induced cell apoptosis (Figure 6B). These results indicate that GSK-3 is not involved in TNF-α-induced renal cell death.
Figure 6.
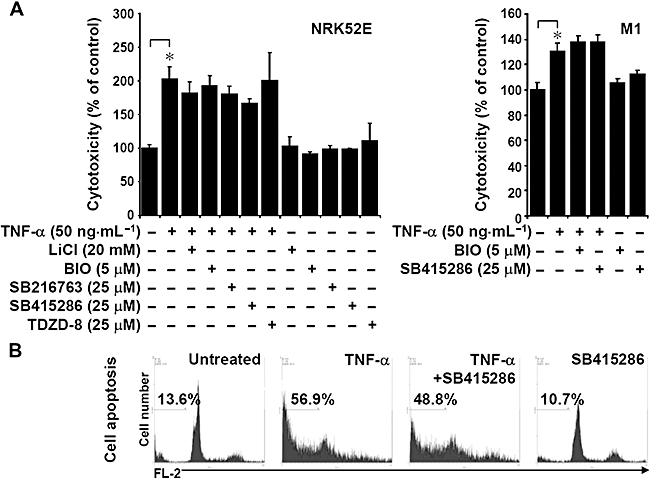
The effects of glycogen synthase kinase-3 inhibitors on tumour necrosis factor-α (TNF-α)-induced renal cell death. (A) Rat kidney proximal tubular epithelial NRK52E and M1 cells (3 × 105 cells/well in 12-well culture plates) were treated with TNF-α (50 ng·mL−1) for 72 h with or without lithium chloride (LiCl) (20 mM), 6-bromo-indirubin-3′-oxime (BIO) (5 µM), SB216763 (25 µM), SB415286 (25 µM) or thiadiazolidine-8 (TDZD-8) (25 µM) pretreatment for 0.5 h. Cell cytotoxicity was determined using an LDH assay. Data, obtained from triplicate cultures, are means ± SD *, P < 0.05. (B) Apoptosis in TNF-α-treated (50 ng·mL−1) NRK52E cells with or without SB415286 (25 µM) for 72 h was determined using PI staining and then flow cytometric analysis. Data shown are representative of three individual experiments. The percentages of apoptotic cells are shown.
Discussion and conclusions
Inhibiting GSK-3 is currently used to reduce MOF/MODS, including renal dysfunction, in endotoxaemia-treated mice (Dugo et al., 2005; 2007a,b). However, the mechanism through which GSK-3 inhibition provides renal protection remains unclear. We developed a murine model of experimental endotoxaemia-induced acute renal failure characterized by renal dysfunction, tissue injury, inflammation and cell death. We showed that inhibiting GSK-3 reduces endotoxaemia-induced mortality from sepsis and nephrotoxicity. GSK-3 inhibition also reduced LPS-induced overproduction of TNF-α and RANTES. GSK-3 inhibitors suppressed the LPS-induced activation of NF-κB. Notably, in vivo, GSK-3 inhibitors down-regulated LPS-induced renal cell apoptosis, but in vitro, they had no significant effect on TNF-α-mediated cell death. We hypothesize that inhibiting GSK-3 reduces endotoxaemia-induced acute renal failure by reducing the amount of TNF-α produced, resulting in a decrease in the amount of inflammation and apoptosis developing.
The pathogenesis of endotoxaemic acute renal failure is multimodal: it includes nephrotoxicity and excessive inflammation (Thadhani et al., 1996). TNF-α, a highly expressed pro-inflammatory and pro-apoptotic cytokine, generally causes various pathological effects in endotoxaemic acute renal failure (Baud et al., 1989; Knotek et al., 2001; Cunningham et al., 2002). Neutralizing TNF-α with the soluble TNF receptor p55 prevents endotoxaemia-induced acute renal failure (Knotek et al., 2001). TNF receptor knockout mice are resistant to endotoxaemia-induced acute renal failure (Cunningham et al., 2002). We found that LPS caused excessive TNF-α production in vivo and in vitro, but inhibiting GSK-3 blocked TNF-α production in both settings. These results are in contrast to those from a recent study where it was found that inhibiting GSK-3 increased LPS-induced TNF-α in cardiomyocytes (Shen et al., 2008), but are consistent with findings from previous studies (Martin et al., 2005; Woodgett and Ohashi, 2005; Cuzzocrea et al., 2006; Whittle et al., 2006), which showed that inhibiting GSK-3 had anti-inflammatory effects. Both our study and that of Martin et al. (2005) demonstrated that inhibiting GSK-3 blocked LPS-induced TNF-α production. We hypothesize that inhibiting GSK-3 reduces endotoxaemia-induced acute renal failure primarily by down-regulating TNF-α.
Recent studies (Cunningham et al., 2002; Ortiz et al., 2003; Wan et al., 2003; Guo et al., 2004; Gupta et al., 2007) have found that caspase inhibitors and activated protein C protect kidneys from endotoxaemia-induced acute renal failure. We showed that inhibiting GSK-3 reduced LPS-induced renal cell apoptosis in vivo in mice but not TNF-α-induced renal cell death in vitro. We hypothesize that inhibiting GSK-3 reduces renal cell apoptosis in vivo primarily by down-regulating TNF-α overproduction. LPS may also up-regulate Fas and FasL, which contributes to renal cell apoptosis (Ortiz-Arduan et al., 1996; Koide et al., 1999; Jo et al., 2002). The anti-apoptotic effects induced by inhibition of GSK-3 require further investigation.
Although we showed that LPS activated GSK-3 in vivo and in vitro, the mechanisms of TLR-mediated GSK-3 activation are still unclear. In general, protein phosphatase and Akt regulate GSK-3 activation by modulating protein phosphorylation in a positive or negative sense, respectively (Frame and Cohen, 2001; Jope et al., 2007; Lin et al., 2007). The mechanisms of LPS-induced GSK-3 activation need further investigation. We also showed that inhibiting GSK-3 reduced LPS-induced TNF-α and RANTES overproduction by as yet unknown mechanisms. GSK-3 regulates the signalling pathways that TNF and LPS use to activate NF-κB and is necessary for that activation (Takada et al., 2004; Martin et al., 2005; Woodgett and Ohashi, 2005). We found that inhibiting GSK-3 decreased NF-κB activation. Because NF-κB is essential for LPS-induced TNF-α and RANTES production (Trinchieri and Sher, 2007), we hypothesize that inhibiting GSK-3 down-regulates NF-κB activation, which is followed by TNF-α and RANTES down-regulation.
Clinically, GSK-3 inhibitors have been used for treating mood disorders and neurodegenerative diseases as they inhibit inflammation and apoptosis (Perry et al., 2007). Recent reports (Soos et al., 2006; Obligado et al., 2008) indicate that inhibitors of cyclin-dependent kinases and GSK-3 are useful for reducing mesangial proliferative glomerulonephritis, crescentic glomerulonephritis, proliferative lupus nephritis, collapsing glomerulopathy, polycystic kidney diseases, diabetic nephropathy and several forms of acute kidney injury. Inactivating GSK-3 with exogenous insulin and hepatocyte growth factor (HGF) is a promising therapeutic strategy for progressive renal diseases (Gong et al., 2005; 2006a,b; Giannopoulou et al., 2008). HGF inhibits the inflammation induced by overproduction of TNF-α and obstructive nephropathy, including that due to RANTES production and endothelial E-selectin expression. HGF may inactivate NF-κB after GSK-3 has been inhibited (Gong et al., 2008). Our results further suggest that inhibiting GSK-3 reduces endotoxaemic acute renal failure primarily by triggering anti-inflammatory and anti-apoptotic effects after TNF-α has been down-regulated. From the findings of Dugo et al. (2005; 2007a,b) and those of the present study, we hypothesize that inhibiting GSK-3, which induces a down-regulation in the activation of NF-κB and a reduction in the amount of TNF-α and RANTES produced, may be a strategy to reduce endotoxaemic acute renal failure.
Acknowledgments
The authors thank Dr. R. Anderson for his critical reading of the manuscript. This work was supported by grants NSC 96-2320-B-006-013 and 96-2320-B-006-018-MY3 from the National Science Council, Taiwan, and by the Landmark Project C020 of National Cheng Kung University, Taiwan.
Glossary
Abbreviations:
- BIO
6-bromo-indirubin-3′-oxime
- BUN
blood urea nitrogen
- GSK-3
glycogen synthase kinase-3
- LiCl
lithium chloride
- LPS
lipopolysaccharide
- MODS
multiple organ dysfunction syndrome
- MOF
multiple organ failure
- TDZD-8
thiadiazolidine-8
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP–biotin nick-end labelling
Conflict of interest
The authors have no financial conflict of interest.
References
- Anderson RJ. Kidney in sepsis. Crit Care Med. 2007;35:2223–2224. doi: 10.1097/01.CCM.0000281465.24130.E3. [DOI] [PubMed] [Google Scholar]
- Baud L, Oudinet JP, Bens M, Noe L, Peraldi MN, Rondeau E, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35:1111–1118. doi: 10.1038/ki.1989.98. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Leonov S, Luthman J, Scott CW, Lee CM. Interactions between GSK3beta and caspase signalling pathways during NGF deprivation induced cell death. J Alzheimers Dis. 2002;4:291–301. doi: 10.3233/jad-2002-4404. [DOI] [PubMed] [Google Scholar]
- Bijur GN, De Sarno P, Jope RS. Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817–5823. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Muia C, Crisafulli C, Dugo L, et al. Glycogen synthase kinase-3beta inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin Immunol. 2006;120:57–67. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Malleo G, Genovese T, Mazzon E, Esposito E, Muia C, et al. Effects of glycogen synthase kinase-3beta inhibition on the development of cerulein-induced acute pancreatitis in mice. Crit Care Med. 2007;35:2811–2821. doi: 10.1097/01.ccm.0000295303.62996.9f. [DOI] [PubMed] [Google Scholar]
- Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, et al. GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- Dugo L, Abdelrahman M, Murch O, Mazzon E, Cuzzocrea S, Thiemermann C. Glycogen synthase kinase-3beta inhibitors protect against the organ injury and dysfunction caused by hemorrhage and resuscitation. Shock. 2006a;25:485–491. doi: 10.1097/01.shk.0000209545.29671.31. [DOI] [PubMed] [Google Scholar]
- Dugo L, Collin M, Allen DA, Murch O, Foster SJ, Yaqoob MM, et al. Insulin reduces the multiple organ injury and dysfunction caused by coadministration of lipopolysaccharide and peptidoglycan independently of blood glucose: role of glycogen synthase kinase-3beta inhibition. Crit Care Med. 2006b;34:1489–1496. doi: 10.1097/01.CCM.0000215457.83953.E3. [DOI] [PubMed] [Google Scholar]
- Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala E, et al. Inhibiting glycogen synthase kinase 3beta in sepsis. Novartis Found Symp. 2007a;280:128–142. discussion 142–126, 160–124. [PubMed] [Google Scholar]
- Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock. 2007b;27:113–123. doi: 10.1097/01.shk.0000238059.23837.68. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulou M, Dai C, Tan X, Wen X, Michalopoulos GK, Liu Y. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-κB signaling. Am J Pathol. 2008;173:30–41. doi: 10.2353/ajpath.2008.070583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Rifai A, Dworkin LD. Activation of PI3K-Akt-GSK3beta pathway mediates hepatocyte growth factor inhibition of RANTES expression in renal tubular epithelial cells. Biochem Biophys Res Commun. 2005;330:27–33. doi: 10.1016/j.bbrc.2005.02.122. [DOI] [PubMed] [Google Scholar]
- Gong R, Rifai A, Dworkin LD. Anti-inflammatory effect of hepatocyte growth factor in chronic kidney disease: targeting the inflamed vascular endothelium. J Am Soc Nephrol. 2006a;17:2464–2473. doi: 10.1681/ASN.2006020185. [DOI] [PubMed] [Google Scholar]
- Gong R, Rifai A, Dworkin LD. Hepatocyte growth factor suppresses acute renal inflammation by inhibition of endothelial E-selectin. Kidney Int. 2006b;69:1166–1174. doi: 10.1038/sj.ki.5000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem. 2008;283:7401–7410. doi: 10.1074/jbc.M710396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Wang Y, Minto AW, Quigg RJ, Cunningham PN. Acute renal failure in endotoxemia is dependent on caspase activation. J Am Soc Nephrol. 2004;15:3093–3102. doi: 10.1097/01.ASN.0000145530.73247.F5. [DOI] [PubMed] [Google Scholar]
- Gupta A, Berg DT, Gerlitz B, Sharma GR, Syed S, Richardson MA, et al. Role of protein C in renal dysfunction after polymicrobial sepsis. J Am Soc Nephrol. 2007;18:860–867. doi: 10.1681/ASN.2006101167. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol Cell Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SK, Cha DR, Cho WY, Kim HK, Chang KH, Yun SY, et al. Inflammatory cytokines and lipopolysaccharide induce Fas-mediated apoptosis in renal tubular cells. Nephron. 2002;91:406–415. doi: 10.1159/000064280. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RZ, Badr KF. Endotoxin and renal function: perspectives to the understanding of septic acute renal failure and toxic shock. Nephrol Dial Transplant. 1999;14:814–818. doi: 10.1093/ndt/14.4.814. [DOI] [PubMed] [Google Scholar]
- King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- Knotek M, Rogachev B, Wang W, Ecder T, Melnikov V, Gengaro PE, et al. Endotoxemic renal failure in mice: role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int. 2001;59:2243–2249. doi: 10.1046/j.1523-1755.2001.00740.x. [DOI] [PubMed] [Google Scholar]
- Koide N, Narita K, Kato Y, Sugiyama T, Chakravortty D, Morikawa A, et al. Expression of Fas and Fas ligand on mouse renal tubular epithelial cells in the generalized Shwartzman reaction and its relationship to apoptosis. Infect Immun. 1999;67:4112–4118. doi: 10.1128/iai.67.8.4112-4118.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- Loberg RD, Vesely E, Brosius FC., 3rd Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J Biol Chem. 2002;277:41667–41673. doi: 10.1074/jbc.M206405200. [DOI] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu H, Kleinknecht D, Brivet F, Loirat P, Landais P. Prognostic factors in acute renal failure due to sepsis. Results of a prospective multicentre study. The French Study Group on Acute Renal Failure. Nephrol Dial Transplant. 1996;11:293–299. doi: 10.1093/oxfordjournals.ndt.a027256. [DOI] [PubMed] [Google Scholar]
- Obligado SH, Ibraghimov-Beskrovnaya O, Zuk A, Meijer L, Nelson PJ. CDK/GSK-3 inhibitors as therapeutic agents for parenchymal renal diseases. Kidney Int. 2008;73:684–690. doi: 10.1038/sj.ki.5002731. [DOI] [PubMed] [Google Scholar]
- Ortiz A, Justo P, Sanz A, Lorz C, Egido J. Targeting apoptosis in acute tubular injury. Biochem Pharmacol. 2003;66:1589–1594. doi: 10.1016/s0006-2952(03)00515-x. [DOI] [PubMed] [Google Scholar]
- Ortiz-Arduan A, Danoff TM, Kalluri R, Gonzalez-Cuadrado S, Karp SL, Elkon K, et al. Regulation of Fas and Fas ligand expression in cultured murine renal cells and in the kidney during endotoxemia. Am J Physiol. 1996;271:F1193–F1201. doi: 10.1152/ajprenal.1996.271.6.F1193. [DOI] [PubMed] [Google Scholar]
- Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- Shen E, Fan J, Peng T. Glycogen synthase kinase-3beta suppresses tumor necrosis factor-alpha expression in cardiomyocytes during lipopolysaccharide stimulation. J Cell Biochem. 2008;104:329–338. doi: 10.1002/jcb.21629. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Linch DC, Khwaja A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- Soos TJ, Meijer L, Nelson PJ. CDK/GSK-3 inhibitors as a new approach for the treatment of proliferative renal diseases. Drug News Perspect. 2006;19:325–328. doi: 10.1358/dnp.2006.19.6.985939. [DOI] [PubMed] [Google Scholar]
- Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem. 2004;279:39541–39554. doi: 10.1074/jbc.M403449200. [DOI] [PubMed] [Google Scholar]
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Wan L, Bellomo R, Di Giantomasso D, Ronco C. The pathogenesis of septic acute renal failure. Curr Opin Crit Care. 2003;9:496–502. doi: 10.1097/00075198-200312000-00006. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br J Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nat Immunol. 2005;6:751–752. doi: 10.1038/ni0805-751. [DOI] [PubMed] [Google Scholar]
- Wu X, Guo R, Wang Y, Cunningham PN. The role of ICAM-1 in endotoxin-induced acute renal failure. Am J Physiol Renal Physiol. 2007;293:F1262–F1271. doi: 10.1152/ajprenal.00445.2006. [DOI] [PubMed] [Google Scholar]