

Abstract
Background and purpose:
Tripterine is an inhibitor of heat shock protein 90 and an active component of Tripterygium wilfordii Hook F., which is used in traditional Chinese medicine to treat inflammatory diseases such as rheumatoid arthritis. We hypothesized that tripterine inhibits endogenous peroxynitrite formation and thereby prevents endothelial barrier dysfunction.
Experimental approach:
Effects of tripterine were investigated on endothelial barrier function, inducible nitric oxide synthase (iNOS) expression, nicotinamide adenine dinucleotide phasphate (NADPH) oxidase activity, 3-nitrotyrosine formation, protein phosphatase type 2A (PP2A) activity, activation of extracellular-regulated kinase (ERK), c-Jun terminal kinase (JNK) and Janus kinase (Jak2), and degradation of IκB in microvascular endothelial cells exposed to pro-inflammatory stimulus [lipopolysaccharide (LPS) + interferon γ (IFNγ)] and on vascular permeability in air pouches of mice injected with LPS + IFNγ.
Key results:
LPS + IFNγ caused an increase in monolayer permeability, induction of iNOS and NADPH oxidase type 1 (Nox1) proteins, formation of superoxide, nitric oxide and 3-nitrotyrosine, and increase in PP2A activity in endothelial cells. These effects of LPS + IFNγ were diminished by tripterine (50–200 nM). Further, LPS + IFNγ-induced expression of iNOS and Nox1 was attenuated by the mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor PD98059, the JNK inhibitor SP600125, the Jak2 inhibitor AG490 and the NFκB inhibitor MG132, but not by the p38 mitogen-activated protein kinase inhibitor SB203580. LPS + IFNγ stimulated phosphorylation of ERK, JNK and Jak2, and degradation of IκB, but only Jak2 phosphorylation was sensitive to tripterine (50–200 nM). Further, tripterine diminished the increased vascular permeability in inflamed air pouches.
Conclusion and implications:
Our results indicate that, by preventing Jak2-dependent induction of iNOS and Nox1, tripterine inhibits peroxynitrite precursor synthesis, attenuates the increased activity of PP2A and consequently protects endothelial barrier function.
Keywords: tripterine, permeability, hsp90, endothelial cells, iNOS, NADPH oxidase, peroxynitrite, inflammation, lipopolysaccharide, cytokines
Introduction
Endothelial barrier dysfunction is an important pathological process in inflammatory diseases, including rheumatoid arthritis and sepsis. In arthritis, increased permeability of capillaries leads to plasma extravasation, oedema formation and swelling of the joints (Middleton et al., 2004). Similarly in sepsis, the endothelial barrier dysfunction caused by a generalized inflammatory response to bacteria or bacterial endotoxin leads to life-threatening oedema in the lungs, kidneys, skeletal muscles and brain (Davies, 2002; Holbeck and Grande, 2003; Peters et al., 2003). Moreover, disruption of the endothelial barrier facilitates leucocyte emigration from the circulation into the subendothelial space, which further contributes to injury of target organs (Middleton et al., 2004).
Pro-inflammatory stimuli [e.g. endotoxin and the major cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β and interferon (IFN) γ] precede the onset of tissue injury in rheumatoid arthritis and sepsis (Titheradge, 1999; Peters et al., 2003; Middleton et al., 2004; McInnes and Schett, 2007). Endothelial cells respond to the pro-inflammatory stimuli by producing inflammatory effectors such as nitric oxide (NO), superoxide and peroxynitrite (Peters et al., 2003). NO generation by endothelial cells is necessary for the maintenance of the barrier function of endothelium (Cirino et al., 2003; Wong et al., 2004). However, the protective effect of NO on the barrier integrity is diminished under inflammatory conditions due to the simultaneous production of superoxide, which reacts with NO to form the powerful oxidant peroxynitrite (Salvemini et al., 2006). Peroxynitrite can oxidize and nitrate proteins, leading to the modulation of their function (Alvarez and Radi, 2003; Peluffo and Radi, 2007). It has been reported that nitration of cytoskeletal proteins by peroxynitrite is a cause of endothelial and epithelial barrier dysfunction (Knepler et al., 2001; Banan et al., 2003; Neumann et al., 2006). Further, our recent study indicates that increased production of peroxynitrite is associated with the activation of protein phosphatase type 2A (PP2A) that causes an increased permeability of the endothelial cell monolayer (Wu and Wilson, 2009). Inhibiting the production of superoxide or NO by administration of a superoxide dismutase (SOD) mimetic or NO synthase (NOS) inhibitor preserves the permeability barrier in cell monolayers challenged with endotoxin and pro-inflammatory cytokines (Forsythe et al., 2002; Duann et al., 2006).
Tripterygium wilfordii Hook F. (T. wilfordii), also known as ‘God of Thunder Vine’, is a traditional Chinese medicine that has been used for hundreds of years to treat rheumatoid arthritis and other rheumatic diseases (Setty and Sigal, 2005; Corson and Crews, 2007). Crude extracts of T. wilfordii roots have been shown to decrease the number of tender or swollen joints in arthritis patients and animal models (Asano et al., 1998; Tao et al., 2002). Adverse effects of these extracts have also been reported, most commonly, disturbances of the gastrointestinal tract and amenorrhoea (Setty and Sigal, 2005).
Tripterine (also known as celastrol), a pentacyclic triterpene, is an active component of T. wilfordii extracts that has anti-inflammatory activity (Setty and Sigal, 2005). Tripterine at nanomolar concentrations (20–200 nM) has no known cytotoxic effect (Zhang et al., 2006) but inhibits expression of inflammatory genes, including IL-1β, TNFα, prostaglandin E2 and inducible NOS (iNOS) (He et al., 1998; Allison et al., 2001). However, high concentrations (IC50 = 2.5 µM) of tripterine induce apoptosis and exhibit anti-tumour activity (Yang et al., 2006).
Tripterine is of particular interest because it is an inhibitor of heat shock protein 90 (hsp90) (Hieronymus et al., 2006) and the latter has been implicated in the pathophysiology of lipopolysaccharide (LPS)-induced vascular leakage (Chatterjee et al., 2007; 2008). Further, when used to treat arthritis, tripterine has been shown to suppress joint swelling and other manifestations of this condition without any obvious toxic effects (Allison et al., 2001; Li et al., 2008). Because peroxynitrite has been implicated as a causative agent of endothelial barrier dysfunction (Knepler et al., 2001; Zhang et al., 2005; Neumann et al., 2006; Wu and Wilson, 2009), it could be the target through which tripterine acts to prevent oedema (Allison et al., 2001; Li et al., 2008). In the present study, we determined whether tripterine prevents peroxynitrite formation and maintains barrier function in microvascular endothelium exposed to pro-inflammatory stimuli.
Methods
Cell cultures
The University at Buffalo Institutional Animal Care and Use Committee approved the procedures. Microvascular endothelial cells were isolated from hind limb skeletal muscle of male C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) using a cell-trapping technique we described previously (Wu et al., 2001). Endothelial cells were maintained in a growth medium consisting of Dulbecco's modified eagle medium (DMEM), 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 5 U·L−1 heparin, 25 µg·mL−1 endothelial cell growth supplement (ECGS), 100 U·L−1 penicillin and 100 µg·mL−1 streptomycin, in a CO2 incubator. For treatment of the cells for experiments, ECGS was omitted from the growth medium and the concentration of serum was reduced to 2%. Endothelial phenotype identification was carried out by immunocytochemical staining for von Willebrand factor, uptake of acetylated low-density lipoprotein labelled with 1,1-dioctadecyl-3,3,3-tetramethylindo-carbocyanine perchlorate and binding of fluorescently labelled Griffonia simplicifolia lectin I (Wilson et al., 1996). Experiments were performed using confluent monolayers (passages 3 through 8) obtained from at least three different mice for each experiment.
Endothelial monolayer permeability assay
Diffusion of Evans blue-coupled bovine serum albumin through endothelial monolayers was determined as previously described (Patterson et al., 1992). Cells were plated on gelatin-coated culture inserts (3 µm pore size) in 12-well companion plates and grown to confluency in the growth medium. For experiments, endothelial cells were incubated with various drugs (described in the figure legends) in DMEM containing 2% fetal bovine serum without phenol red. Subsequently, Evans blue-coupled bovine serum albumin was added to the upper chamber and bovine serum albumin was added to the lower chamber, so that no oncotic pressure gradient existed. After incubation of the monolayers with Evans blue-coupled bovine serum albumin for 1 h, medium was collected from the lower chamber and the absorbance of Evans blue was measured at 595 nm. The values of the absorbance were referenced to a standard curve in order to obtain trans-endothelial Evans blue-albumin leak as a flux of albumin in µg·h−1.
Cell viability assay
Endothelial cell viability was measured using the methlythiazolydiphenyltetrazolium bromide (MTT) assay. Cells were cultured in 12-well plates and treated with various drugs as indicated in the figure legends. One hour prior to end point, the culture medium was changed to phenol red-free DMEM containing 2% fetal bovine serum and 0.25 mg·mL−1 MTT. After 1 h incubation at 37°C, the medium was removed and 1 mL of dimethyl sulphoxide (DMSO) was added to each well, followed by trituration to dissolve the blue formazan. Two hundred microlitres from each well were transferred, in triplicate aliquots, to a 96-well plate and absorbance at 550 nm was read in a spectrophotometer.
Western blot analysis
Cells harvested from each well of the 12-well-plates were lysed in Laemmli sample buffer. The proteins were separated in 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel by electrophoresis and then transferred to polyvinylidene fluoride membrane. For protein detection, blocked membranes were incubated with primary antibodies in Tris-buffered saline containing 5% non-fat milk (or 5% bovine serum albumin for anti-phosphorylated protein antibodies) and 0.1% Tween 20. Subsequently, the membranes were washed and then further incubated with their respective horseradish peroxidase-conjugated secondary antibodies in Tris-buffered saline containing 5% non-fat milk and 0.1% Tween 20. Immunoreactive proteins were detected using the enhanced chemiluminescent (ECL) detection system and autoradiography film. Blots were further probed with anti-β-actin antibody to normalize for protein loading. The intensity of each protein band was normalized to the respective β-actin band and then expressed as percentage of the value for the LPS + IFNγ group, with the exception of p-eNOS and IκB. In the case of p-eNOS, the band intensity was normalized to the eNOS band. In the case of IκB, the band intensity was expressed as percentage of the value for the control group.
Measurement of superoxide production
Superoxide production was measured using a previously described cytochrome c reduction assay (Wu et al., 2007). Cells harvested from each 35 mm culture dish were disrupted by sonication in 200 µL lysis buffer composed of 5 mM potassium phosphate (pH 7.4), 250 mM sucrose, 0.1 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM phenylmethylsulphonylfluoride (PMSF). This mixture was added to 200 µL phenol red-free DMEM containing 40 µM cytochrome c and 200 µM NADPH, and incubated at 37oC for 30 min with or without manganese SOD (500 U·mL−1). The reduction of cytochrome c was measured spectrophotometrically at 550 nm. To calculate superoxide production, reduction of cytochrome c in the presence of SOD was subtracted from the value obtained without SOD.
Measurement of nitrate and nitrite
The concentration of NO metabolites in the culture medium was measured to estimate the total production of NO in cultured cells. An aliquot of the medium was filtered through a 10 kDa molecular weight cut-off filter to eliminate proteins. Nitrate was converted to nitrite by nitrate reductase. Total nitrite was then measured using a nitrate/nitrite colorimetric assay kit. Briefly, the reduced sample was mixed with an equal volume of Griess reagent, and the absorbance at 545 nm was measured immediately. The concentration of NO metabolites was determined by comparison to a standard curve based on serial dilution of a sodium nitrate standard.
Measurement of PP2A activity
PP2A activity was determined as okadaic acid-inhibitable phosphatase activity by the method we described previously (Wu and Wilson, 2009). Cells harvested from each well of 12-well plates were lysed by sonication in 100 µL assay buffer (50 mM Tris-Cl, pH 7.0, 0.1 mM EDTA and 1 mM PMSF). The cell extract was combined with 100 µL assay buffer containing 10 mM p-nitrophenyl phosphate (p-NPP) (final concentration = 5 mM) and 6 mM MnCl2 (final concentration = 3 mM), with or without 50 nM okadaic acid, and was incubated for 30 min at 30oC. The hydrolysis of p-NPP was measured at 405 nm, and PP2A activity was calculated as total phosphatase activity minus okadaic acid-insensitive activity.
Induction of air pouches
The University at Buffalo Institutional Animal Care and Use Committee approved the following procedures. To induce an air pouch, 8–9-week-old mice were injected subcutaneously on the back with 5 mL of air. After 3 days, the pouches were reinflated with 3 mL of air. On day 6, mice were injected with tripterine (3 mg·kg−1, i.p.) or vehicle (10% DMSO plus 90% cremophor; 10 mL·kg−1, i.p.), 1 h before injection of 1 mL of either LPS (25 ng·mL−1) + IFNγ (100 U·mL−1) or saline vehicle into the air pouch. The dose and route of administration of tripterine have been described previously (Cleren et al., 2005). Thirty minutes before the air pouch injections, mice were also injected with 0.1 mL of Evans blue dye (6.25 mg·mL−1 in saline) through the tail vein. After 6 h, the animals were killed, the air pouches were washed with 1 mL saline and the fluids were harvested. Blood was obtained by cardiac puncture. The absorbance of Evans blue in exudates and sera was measured at 595 nm. A standard curve was prepared by serial dilution of Evans blue dye. The value of the absorbance was referenced to the standard curve in order to calculate the concentration of Evans blue. To calculate the Evans blue leak, the concentration of Evans blue in the pouch exudates was expressed as the percentage of the concentration of Evans blue in the serum.
Statistics
Data are presented as mean ± standard error mean values. They were analysed with the Prism statistical program (GraphPad Software, San Diego, CA, USA). Comparisons between treatment groups were performed with one-way analysis of variance followed by the Tukey multiple comparison test. P < 0.05 was considered significant.
Chemicals and reagents
ECGS and the 12-well companion plates were obtained from BD Biosciences (San Jose, CA, USA); LPS (from Escherichia coli 055:B5) and mouse IFNγ (recombinant) were obtained from Sigma/Aldrich Chemical Co. (St. Louis, MO, USA). Apocynin, PD98059, 1400W, MG132, AG490, SB203580, SP600125 and tripterine were obtained from VWR (West Chester, PA, USA). Anti-IκB and anti-Nox1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phosphorylated extracellular-regulated kinase (pERK) and anti-phosphorylated c-Jun terminal kinase (pJNK) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA); anti-phosphorylated Janus kinase (pJak2), anti-PP2A catalytic subunit (PP2Ac) and anti-3-nitrotyrosine antibodies were obtained from Millipore Corporation (Temecula, CA, USA); anti-p-eNOS (Ser 1179), anti-eNOS and anti-iNOS antibodies were obtained from BD Biosciences; anti-β-actin antibody was obtained from EMD Chemicals (San Diego, CA, USA); nitrate/nitrite colorimetric assay kit was obtained from Cayman (Ann Arbor, MI, USA).
Results
Tripterine prevents LPS + IFNγ-induced endothelial barrier dysfunction
LPS (25 ng·mL−1) + IFNγ (100 U·mL−1) [i.e. optimal combination for iNOS induction in endothelial cells (Geiger et al., 1997)] induced a time-dependent increase in Evans blue-labelled albumin leak through the microvascular endothelial monolayer over a period of 48 h (Figure 1A). In the following experiments, exposure to LPS + IFNγ for 24 h was used to cause a moderate increase of endothelial monolayer permeability (Figure 1A).
Figure 1.
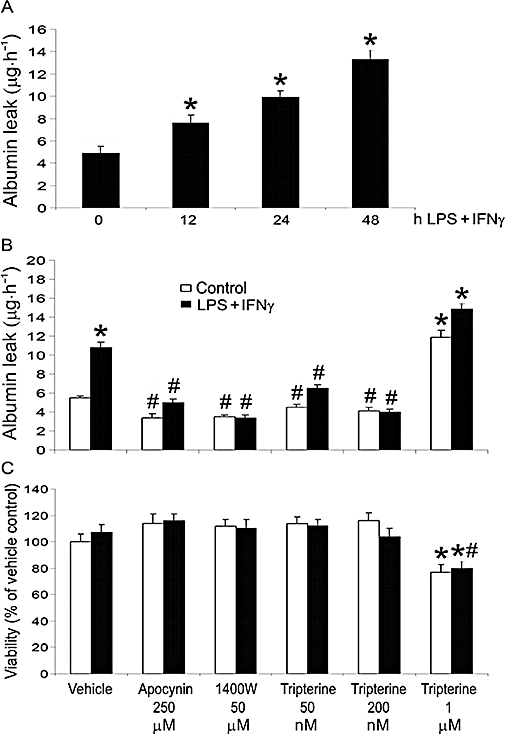
Tripterine inhibited endothelial monolayer leakage. (A) Time course of LPS+ IFNγ-induced endothelial monolayer leakage. Endothelial monolayers were incubated with vehicle (i.e. PBS) or LPS (25 ng·mL−1) + IFNγ (100 U·mL−1) for 0, 12, 24 and 48 h, and the permeability was then detected at each time point (n = 3). *P < 0.05 compared with 0 h group. For detection of permeability and cell viability, endothelial monolayers were pretreated for 2 h with vehicle (i.e. 0.05% DMSO), apocynin 250 µM, 1400W 50 µM, or tripterine 50, 200 nM and 1 µM, and then incubated with the same drugs and either LPS + IFNγ or vehicle for another 24 h. (B) A summary of permeability results (n = 3). (C) Determination of cell viability (n = 3). In both (B) and (C), *P < 0.05 compared with control; #P < 0.05 compared with LPS + IFNγ. DMSO, dimethyl sulphoxide; IFNγ, interferon γ; LPS, lipopolysaccharide; PBS, phosphate buffered saline.
The increased endothelial permeability induced by LPS + IFNγ was attenuated by the NADPH oxidase inhibitor apocynin (250 µM; based on Coyle and Kader, 2007), iNOS inhibitor 1400W (50 µM; based on Hortelano et al., 2000), and tripterine (50 nM and 200 nM; based on Allison et al., 2001) (Figure 1B). Neither apocynin, 1400W nor tripterine (50 nM and 200 nM) altered the endothelial permeability in the absence of a pro-inflammatory stimulus (Figure 1B). The observed effects of LPS + IFNγ, apocynin, 1400W and tripterine (50–200 nM) were not due to changes in cell viability, because these treatments did not influence cell survival (Figure 1C). However, tripterine at a concentration of 1 µM (based on Allison et al., 2001), either alone or together with LPS + IFNγ, significantly decreased cell viability, which may have caused the permeability increase observed for this high dose (Figure 1B,C).
LPS + IFNγ induces NO and superoxide production in microvascular endothelial cells (Wu et al., 2001; 2002; 2008). Tripterine may act by scavenging these free radicals and so protecting the integrity of the endothelial barrier. In this regard, we determined the effect of tripterine on the alterations in endothelial permeability induced by SIN-1, which generates simultaneously NO and superoxide (Knepler et al., 2001). SIN-1 (250–500 µM; based on Knepler et al., 2001) increased the monolayer permeability to albumin and this effect was not altered by tripterine (200 nM) (Figure 2A). SIN-1, either alone or together with tripterine, did not affect cell survival (Figure 2B).
Figure 2.
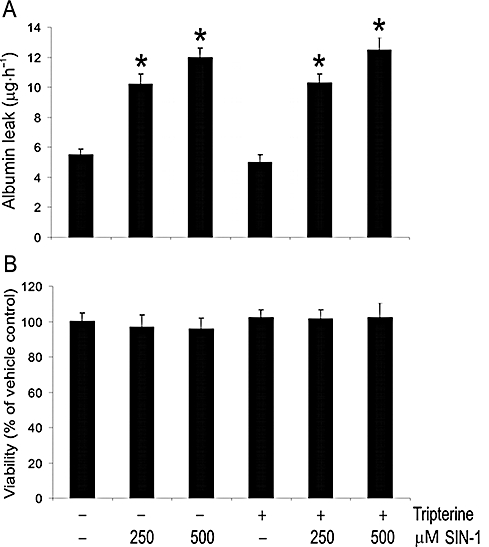
Tripterine had no effect on SIN-1-induced endothelial monolayer leakage. (A) Effect of tripterine on SIN-1-induced endothelial monolayer leak. Endothelial monolayers were pretreated for 2 h with vehicle or tripterine (200 nM), and then incubated with the same drugs and either SIN-1 (250 and 500 µM) or vehicle for another 8 h. Subsequently, the permeability was determined (n = 3). *P < 0.05 compared with vehicle control. (B) Effects of SIN-1 on cell viability (n = 3).
Tripterine inhibits endogenous peroxynitrite formation
Our previous study identified NADPH oxidase activity as the principal source of superoxide in LPS + IFNγ-stimulated microvascular endothelial cells (Wu et al., 2007; 2008). Nox1 is a catalytic subunit of NADPH oxidase that is expressed in endothelial cells (Sorescu et al., 2004; Wu et al., 2008). Our present results indicate that Nox1 is also expressed in microvascular endothelial cells and is up-regulated by LPS + IFNγ (Figure 3). Further, LPS + IFNγ-induced increases in Nox1 protein expression and superoxide production were diminished by treatment of the cells with 200 nM tripterine (Figure 3A,B,C). Tripterine alone did not alter Nox1 expression or superoxide production in the endothelial cells that had not been subjected to LPS + IFNγ stimulation (Figure 3A,B,C).
Figure 3.
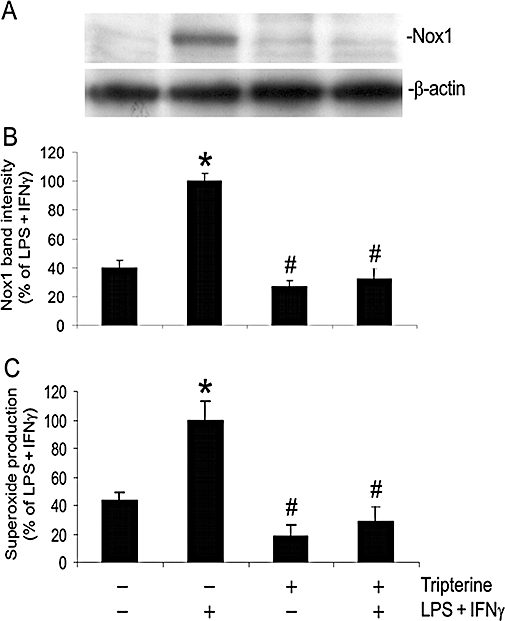
Tripterine inhibited the expression of Nox1 and superoxide production. Endothelial cells were pretreated for 2 h with vehicle or 200 nM tripterine, and then incubated with the same drugs and either LPS+IFNγ or vehicle for another 24 h. (A) Western blot examples for Nox1 and β-actin. (B) The summary of protein band intensities for Nox1 (n = 4). (C) The summary of superoxide production (n = 4). In both (B) and (C), *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. IFNγ, interferon γ; LPS, lipopolysaccharide; Nox1, NADPH oxidase type 1.
Endothelial cells synthesize NO by eNOS under basal conditions and produce large amounts of NO through induction of iNOS during inflammation. We analysed the actions of tripterine on the expression of p-eNOS (Ser 1179), total eNOS and iNOS, and the synthesis of NO in LPS + IFNγ-stimulated microvascular endothelial cells. Our results show that LPS + IFNγ and tripterine, either alone or in combination, failed to alter the levels of p-eNOS or total eNOS (Figure 4A,B). However, LPS + IFNγ markedly increased the expression of iNOS and the production of NO (Figure 4A,C,D). Further, these increases were abolished by 200 nM tripterine. Tripterine alone also decreased the basal production of NO in endothelial cells, in the absence of LPS + IFNγ stimulation (Figure 4D).
Figure 4.
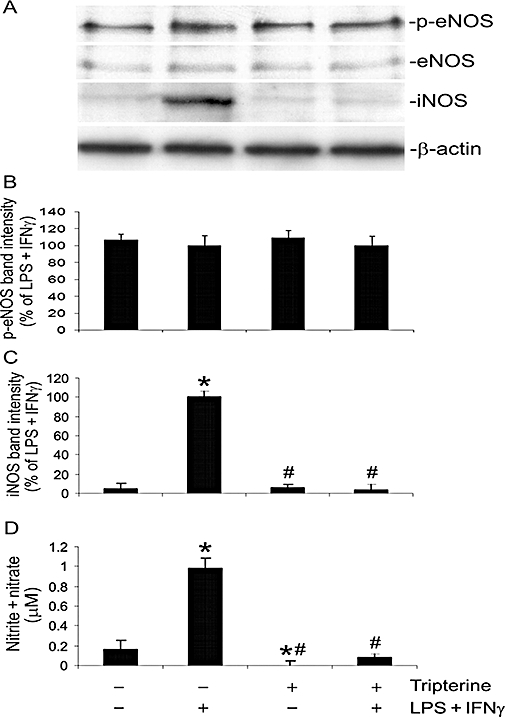
Tripterine inhibited the induction of iNOS and production of NO but had no effect on the expression and phosphorylation of eNOS. Endothelial cells were pretreated for 2 h with vehicle or 200 nM tripterine, and then incubated with the same drug and either LPS + IFNγ or vehicle for another 24 h for Western blot analysis. The culture media were also harvested for measurement of nitrite + nitrate. (A) Western blot examples for p-eNOS (Ser1179, phosphorylation site), eNOS, iNOS and β-actin. (B) The summary of p-eNOS protein band intensities (n = 4). (C) The summary of iNOS protein band intensities (n = 4). (D) The summary of nitrite + nitrate concentrations in the culture medium (n = 4). In both (C) and (D), *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. eNOS, endothelial nitric oxide synthase; IFNγ, interferon γ; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; NO, nitric oxide; p-eNOS, phosphorylated-eNOS.
iNOS-derived NO and NADPH oxidase-derived superoxide react to form peroxynitrite that can nitrate the tyrosine residues of target proteins, leading to the formation of 3-nitrotyrosine. We determined the effect of tripterine on the formation of 3-nitrotyrosine in LPS + IFNγ-stimulated endothelial cells. As shown in Figure 5, LPS + IFNγ induced a marked increase in total 3-nitrotyrosine; this appeared as several intense protein bands at positions of 92, 40, 35 and 25 kDa. This increase was diminished by apocynin, 1400W and tripterine (Figure 5A,B). Apocynin, 1400W and tripterine alone did not alter the 3-nitrotyrosine levels in endothelial cells in the absence of LPS + IFNγ stimulation (data not shown).
Figure 5.
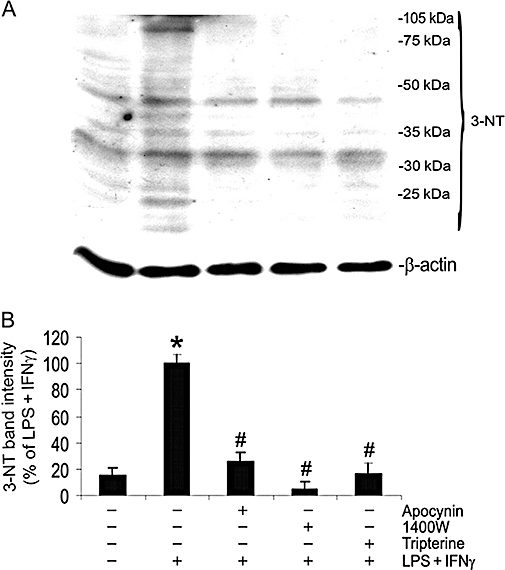
Tripterine inhibited the formation of 3-NT. Endothelial cells were pretreated for 2 h with vehicle, apocynin, 1400W or 200 nM tripterine, and then incubated with the same drugs and either LPS + IFNγ or vehicle for another 24 h for Western blot analysis. (A) Western blot examples for 3-NT and β-actin. (B) The summary of 3-NT band intensities (n = 3). *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. 3-NT, 3-nitrotyrosine; IFNγ, interferon γ; LPS, lipopolysaccharide.
Increased production of peroxynitrite is associated with the activation of PP2A; this causes an increase in the permeability of the endothelial cell monolayer (Wu and Wilson, 2009). Therefore, we determined the effect of tripterine on PP2A activity in LPS + IFNγ-stimulated endothelial cells. As shown in Figure 6A and B, treatment of endothelial cells with LPS + IFNγ, either alone or in the presence of 50–200 nM tripterine, did not alter the expression of the PP2Ac. However, LPS + IFNγ markedly increased the PP2A activity and, further, this increase was diminished by tripterine (Figure 6C).
Figure 6.
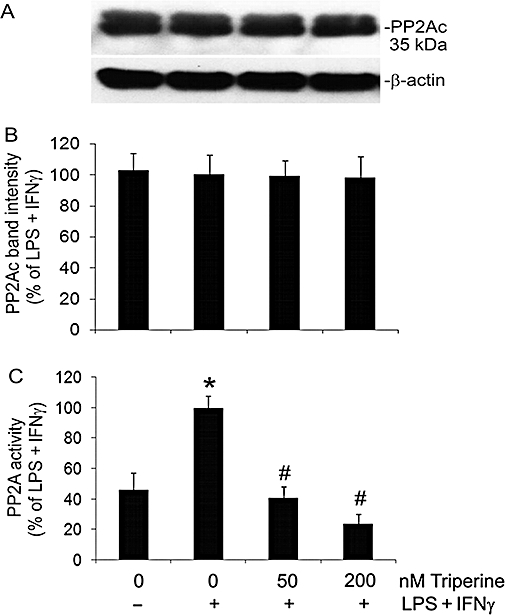
Tripterine inhibited PP2A activity without affecting the expression of PP2Ac protein. Endothelial cells were pretreated for 2 h with vehicle, 50 and 200 nM tripterine, and then incubated with the same drugs and either LPS + IFNγ or vehicle for another 24 h for PP2A activity assay and Western blot analysis. (A) Western blot examples for PP2Ac and β-actin. (B) The summary of PP2Ac protein band intensities (n = 3). (C) The summary of PP2A activity (n = 3). *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. IFNγ, interferon γ; LPS, lipopolysaccharide; PP2A, protein phosphatase type 2; PP2Ac, PP2A catalytic subunit.
Signalling pathways in the induction of Nox1 and iNOS
Exposure of the cells to LPS + IFNγ led to marked increases in the expression of iNOS and Nox1, which were attenuated by the MEK1/2 inhibitor PD98059 (30 µM; based on Chan and Riches, 2001), the JNK inhibitor SP600125 (20 µM; based on Lahti et al., 2003), the Jak2 inhibitor AG490 (100 µM; based on Kristof et al., 2000) and the NFκB inhibitor MG132 (10 µM; based on Barua et al., 2001) (Figure 7A,B,C). The p38 mitogen-activated protein kinase (p38 MAPK) inhibitor SB203580 (10 µM; based on Kristof et al., 2000) had no effect on the expression of either Nox1 or iNOS (Figure 7A,B,C). Treatment of endothelial cells with the drugs alone, in the absence of LPS + IFNγ, did not alter the baseline levels of Nox1 and iNOS (data not shown).
Figure 7.
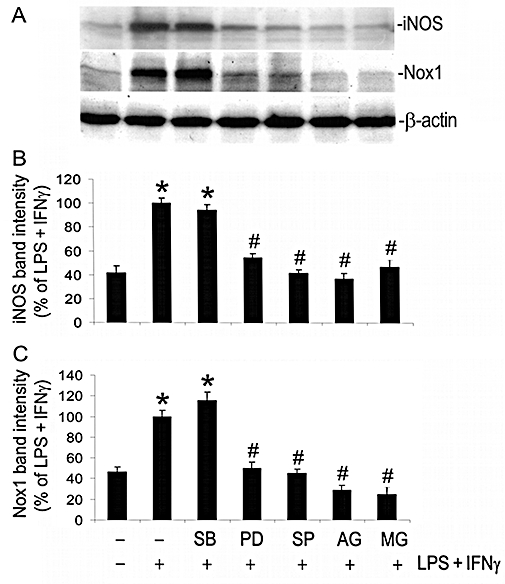
Signalling pathways involved in the induction of iNOS and Nox1 by LPS + IFNγ. Endothelial cells were pretreated for 2 h with vehicle, SB203580 (SB; 10 µM), PD98059 (PD; 30 µM), SP600125 (SP; 20 µM), AG490 (AG; 100 µM) or MG132 (MG; 10 µM), and then incubated with the same drugs and either LPS+IFNγ or vehicle for another 24 h for Western blot analysis. (A) Western blot examples for iNOS, Nox1 and β-actin. (B) and (C) The summaries of protein band intensities of iNOS and Nox1 (n = 3). *P < 0.05 compared with vehicle; #P < 0.05 compared with LPS + IFNγ. IFNγ, interferon γ; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; Nox1, NADPH oxidase type 1.
We next assessed the effects of tripterine on phosphorylation of ERK, JNK and Jak2, and degradation of IκB. LPS + IFNγ caused marked phosphorylation of Jak2, ERK and JNK, as well as complete degradation of IκB (Figure 8A,B,C,D,E). The maximal changes in ERK, JNK and Jak2 phosphorylation and IκB degradation occurred 30 min after the start of incubation with LPS + IFNγ (data not shown). Treatment of the cells with tripterine at nanomolar concentrations (50–200 nM) prevented the LPS + IFNγ-induced phosphorylation of Jak2, but had no effect on the phosphorylation of ERK and JNK or on the degradation of IκB (Figure 8A,B,C,D,E). However, tripterine at micromolar concentration (1 µM) inhibited LPS + IFNγ-induced phosphorylation of Jak2, ERK and JNK, and, to a large extent, the degradation of IκB (Figure 8A,B,C,D,E). In the absence of a pro-inflammatory stimulus, exposure of the cells to tripterine, either at nanomolar or micromolar concentrations, did not cause ERK, JNK and Jak2 phosphorylation or IκB degradation (Figure 8A,B,C,D,E)
Figure 8.
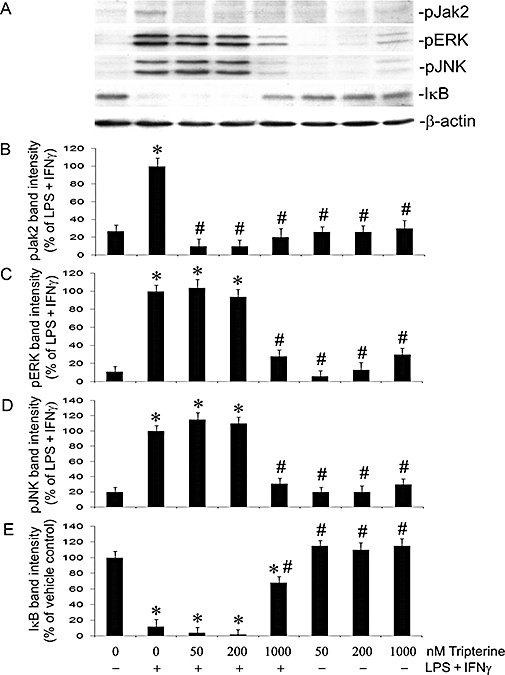
Effects of tripterine on the phosphorylation of Jak2, ERK and JNK, and the degradation of IκB. Endothelial cells were pretreated with vehicle or 50, 200 nM and 1 µM tripterine for 2 h, and then incubated with the same drugs and either LPS + IFNγ or vehicle for another 30 min for Western blot analysis. (A) Western blot examples for pJak2, pERK, pJNK, total IκB and β-actin. (B), (C), (D) and (E) The summaries of the band intensity of pJak2, pERK, pJNK and total IκB (n = 3) respectively. *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. IFNγ, interferon γ; LPS, lipopolysaccharide; pERK, phosphorylated extracellular-regulated kinase; pJak2, phosphorylated Janus kinase 2; pJNK, phosphorylated c-Jun terminal kinases.
Tripterine prevents vascular leakage
To elucidate the effect of tripterine in vivo, we determined LPS + IFNγ-induced vascular leakage to Evans blue in the air pouches of mice that were injected i.p. with tripterine. Our results show that injection of LPS + IFNγ significantly increased the air pouch vascular permeability because increased levels of Evans blue were detected in the exudates (Figure 9). Tripterine alone tended to decrease the baseline vascular permeability to Evans blue in the air pouches (Figure 9) and it abolished the vascular permeability response to LPS + IFNγ (Figure 9).
Figure 9.
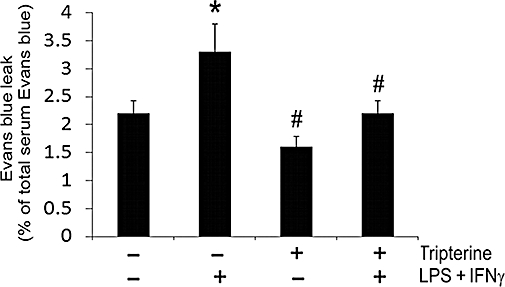
Tripterine prevented the increase in vascular permeability induced by LPS and IFNγ in mouse air pouches. Tripterine (3 mg·kg−1) or vehicle (10% DMSO plus 90% cremophor, 10 mL·kg−1) was administered i.p. to mice 1 h before injection of 1 mL of LPS (25 ng·mL−1) + IFNγ (100 U·mL−1) or 1 mL saline into the air pouch. Thirty minutes before the air pouch injections, 0.1 mL of Evans blue dye (6.25 mg·mL−1 in saline) was injected via the tail vein. After 6 h, the animals were killed, air pouches were washed with 1 mL saline and the fluids were collected. Blood was obtained by cardiac puncture. Evans blue leak in the pouch exudate was calculated as the percentage of total Evans blue in the serum. Data were obtained from five mice. *P < 0.05 compared with vehicle control; #P < 0.05 compared with LPS + IFNγ. DMSO, dimethyl sulphoxide; IFNγ, interferon γ; LPS, lipopolysaccharide.
Discussion
Endothelial barrier dysfunction is an important pathological process in inflammatory diseases. Increased permeability of the endothelium leads to plasma extravasation, oedema formation and swelling of the joints and tissues (Marx, 2003; Middleton et al., 2004). The effectiveness of T. wilfordii extracts in treatment of rheumatoid arthritis and other rheumatic diseases (Setty and Sigal, 2005; Corson and Crews, 2007) may be partially due to maintenance of the microvascular barrier by tripterine, through the mechanism described in the present study.
In unstimulated and LPS + IFNγ-stimulated endothelial cells, reactive oxygen species (ROS) are derived from NADPH oxidase, mitochondrial respiration and NOS (Wu et al., 2007; 2008). However, because inhibition of NADPH oxidase by apocynin, DPI and p47phox deficiency abolishes the LPS + IFNγ-induced increase in superoxide production, we presume that NADPH oxidase is the major source of ROS induced by LPS + IFNγ (Wu et al., 2007; 2008; and present study). On the other hand, iNOS is the principle enzyme that produces NO in LPS + IFNγ-stimulated endothelial cells (Wu et al., 2001; 2002; and present study). iNOS-derived NO and NADPH oxidase-derived superoxide react to form peroxynitrite. Peroxynitrite may alter endothelial permeability by two potential mechanisms. First, peroxynitrite is a powerful oxidant, which can oxidize cysteine residues in proteins, leading to barrier dysfunction (Alvarez and Radi, 2003; Peluffo and Radi, 2007). It has been shown that the increase in endothelial permeability induced by SIN-1 or authentic peroxynitrite is partially inhibited by cysteine, suggesting that protein cysteine residues are a target of peroxynitrite in this process (Knepler et al., 2001). Second, peroxynitrite can nitrate tyrosine residues in proteins, leading to a modulation of their function (Alvarez and Radi, 2003; Peluffo and Radi, 2007). For example, nitration of cytoskeletal proteins by peroxynitrite may contribute to endothelial barrier dysfunction (Knepler et al., 2001; Neumann et al., 2006). Nevertheless, the results from our recent study indicate that peroxynitrite also nitrates PP2Ac. This nitration is associated with increased PP2A activity (Wu and Wilson, 2009). PP2A activity may mediate dephosphorylation and redistribution of the tight junction proteins, leading to paracellular leakage (Nunbhakdi-Craig et al., 2002). The reason that LPS + IFNγ does not enhance PP2Ac protein expression but increases PP2A activity, as observed in Figure 6 in the present study, has been explained in our previous publication (Wu and Wilson, 2009).
We observed that LPS + IFNγ-increased PP2A activity was completely inhibited in cells treated with tripterine at concentrations of 50–200 nM. Our study further indicates that tripterine acts upstream from peroxynitrite formation, because it had no inhibitory effect on SIN-1-induced increase in endothelial permeability. Moreover, in our study, it was found that tripterine inhibited LPS + IFNγ-induced increase in Nox1 and iNOS and consequently attenuated the production of superoxide and NO as well as the formation of 3-nitrotyrosine. Although eNOS and p-eNOS (Ser 1179) are detectable, the levels of these proteins were not altered by LPS + IFNγ and tripterine. Nonetheless, the baseline levels of NO were attenuated by tripterine. It is known that eNOS activity can be modulated by the addition of hsp90 (Sessa, 2005). Therefore, the reduction of the basal levels of NO may be due to inhibition of hsp90 by tripterine. This inhibition may cause dissociation of hsp90 from eNOS, and as a consequence, decrease eNOS activity (Sessa, 2005). Alternatively, tripterine may directly inhibit basal PP2A activity; PP2A dephosphorylates eNOS at Thr495. Therefore, tripterine may prevent the activation of eNOS that is associated with dephosphorylation of Thr495 (Fleming et al., 2001; Thomas et al., 2002). Thus, inhibition of the production of peroxynitrite by tripterine may account for the decreased PP2A activity, because PP2A activity is peroxynitrite dependent in LPS + IFNγ-stimulated cells (Wu and Wilson, 2009).
LPS + IFNγ induce iNOS expression in macrophages through activation of ERK and JNK, but not through p38 MAPK signalling pathways (Chan and Riches, 2001). Consistent with this observation, our results with microvascular endothelial cells showed that the induction of iNOS by LPS + IFNγ involves ERK and JNK but not p38 MAPK. The activation of ERK and JNK leads to the activation of transcription factor AP1 that is necessary for the induction of iNOS (Kleinert et al., 2004). In addition to activator protein 1 (AP1), NFκB and Jak2-interferon regulatory transcription factor 1 (IRF1) have also been identified as necessary signalling molecules for the induction of iNOS by LPS + IFNγ (Kleinert et al., 2004; Lowenstein and Padalko, 2004). The findings of the present study that show inhibitors of Jak2 and NFκB attenuate iNOS expression are consistent with this mechanism.
The signalling pathways leading to the activation of the Nox1 gene are not completely understood. Our observation of simultaneous inhibition of iNOS and Nox1 expression by inhibitors of ERK, JNK, Jak2 and NFκB suggests that common signalling pathways regulate the activation of Nox1 and iNOS genes. Further, inhibition of Jak2 phosphorylation by tripterine suppressed the expression of Nox1 and iNOS, consistent with the findings of several studies that show Jak2 activation is essential for the expression of iNOS and NADPH oxidase subunits (Dell'Albani et al., 2001; Kakar et al., 2005; Uto et al., 2005; Wu et al., 2007).
In our study, it was found that Jak2 activation is a target of tripterine. Jak is the client protein of hsp90 (Shang and Tomasi, 2006), whereas tripterine is an inhibitor of hsp90 (Hieronymus et al., 2006). We postulate that inhibition of hsp90 by tripterine prevents the formation of the Jak-hsp90-CDC37 functional complex (Shang and Tomasi, 2006). This inhibitory effect of tripterine on the activation of Jak2 may attenuate the increased expression of Nox1 and iNOS, and PP2A activity induced by LPS + IFNγ, leading to the protection of endothelial barrier function. Indeed, inhibition of hsp90 by small interferring RNA (siRNA) or other pharmacological inhibitors blocks IFNγ-induced gene expression (Shang and Tomasi, 2006). Further, Jak inhibitors are capable of decreasing pulmonary vascular permeability in LPS-injected mice (Severgnini et al., 2005).
Tripterine, at 0.2–2.2 µM, has been found to inhibit NFκB activity by suppressing IκB degradation (Lee et al., 2006). This inhibition may increase the cytotoxic effect of TNFα (Lee et al., 2006). Consistently, we observed that tripterine at 1 µM prevented the degradation of IκB induced by LPS + IFNγ. However, in our study, this effect is not related to cytotoxicity because micromolar concentrations of tripterine cause cell death to the same extent in unstimulated and LPS + IFNγ-stimulated endothelial cells. Moreover, tripterine at micromolar concentrations has been shown to induce apoptosis by inhibiting proteasome activity (Yang et al., 2006). However, inhibition of the proteasome by MG132 prevents LPS-induced IκB degradation but enhances ERK and JNK activation (Cuschieri et al., 2004). In our study, it was shown that tripterine at 1 µM inhibits not only LPS + IFNγ-induced degradation of IκB but also the activation of ERK and JNK. Thus, tripterine at micromolar concentrations may cause cell death by a non-selective mechanism.
The prolonged increase in endothelial permeability seen in inflammatory diseases, such as arthritis and sepsis, leads to tissue and organ injury (Marx, 2003; Peters et al., 2003; Middleton et al., 2004). Peroxynitrite, produced in response to pro-inflammatory stimuli, oxidizes and nitrates proteins in microvascular endothelial cells, leading to barrier disruption and sustained leakage. Tripterine is an active component of T. wilfordii extracts that have been used in China for hundreds of years to treat rheumatoid arthritis and other rheumatic diseases (Setty and Sigal, 2005). In the present study, it was found that tripterine inhibits the synthesis of the peroxynitrite precursor, attenuates the increase in PP2A activity and prevents barrier dysfunction in microvascular endothelial cells exposed to LPS + IFNγ. Similarly, i.p. administration of tripterine diminished the increase in vascular permeability induced in air pouches injected with LPS + IFNγ. These results provide a scientific basis for the use of tripterine as a treatment for inflammatory diseases, such as arthritis and sepsis.
In conclusion, the results from this study demonstrate that tripterine prevents LPS + IFNγ-induced microvascular endothelial barrier dysfunction. This effect of tripterine is associated with inhibition of the expression of Nox1 and iNOS proteins, formation of 3-nitrotyrosine and increase in PP2A activity induced by LPS + IFNγ. Further, signalling pathways dependent on ERK, JNK, Jak2 and NFκB mediate the induction of Nox1 and iNOS, but only the activation of Jak2 was found to be sensitive to tripterine.
Acknowledgments
The project described was supported by Award Number R01AT003643 from the National Center For Complementary & Alternative Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Complementary & Alternative Medicine or the National Institutes of Health.
Glossary
Abbreviations:
- 1400W
N-(3-aminomethyl)benzylacetamidine
- AG490
α-cyano-(3,4-dihydroxy)-N-benzylcinnamide
- DPI
diphenylene iodinium
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular-regulated kinase
- hsp90
heat shock protein 90
- IFNγ
interferon γ
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- Jak
Janus kinase
- JNK
c-Jun terminal kinases
- LPS
lipopolysaccharide
- MG132
carbobenzoxy-L-leucyl-L-leucyl-L-leucinal
- MTT
methlythiazolydiphenyltetrazolium bromide
- NO
nitric oxide
- Nox1
NADPH oxidase type 1
- p38 MAPK
p38 mitogen-activated protein kinase
- PD98059
2′-amino-3′-methoxyflavone
- p-eNOS
phosphorylated-eNOS
- pERK
phosphorylated-ERK
- pJak2
phosphorylated-Jak2
- pJNK
phosphorylated-JNK
- PP2A
protein phosphatase type 2A
- PP2Ac
PP2A catalytic subunit
- ROS
reactive oxygen species
- SB203580
4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole
- SIN-1
3-morpholinylsydnoneimine
- SOD
superoxide dismutase
- SP600125
1,9-pyrazoloanthrone
- TNFα
tumour necrosis factor-α
- T. wilfordii
Tripterygium wilfordii Hook F
Conflict of interest
The authors state no conflict of interest.
References
- Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids. 2003;25:295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- Asano K, Matsuishi J, Yu Y, Kasahara T, Hisamitsu T. Suppressive effects of Tripterygium wilfordii Hook f., a traditional Chinese medicine, on collagen arthritis in mice. Immunopharmacology. 1998;39:117–126. doi: 10.1016/s0162-3109(98)00006-x. [DOI] [PubMed] [Google Scholar]
- Banan A, Farhadi A, Fields JZ, Zhang LJ, Shaikh M, Keshavarzian A. The delta-isoform of protein kinase C causes inducible nitric-oxide synthase and nitric oxide up-regulation: key mechanism for oxidant-induced carbonylation, nitration, and disassembly of the microtubule cytoskeleton and hyperpermeability of barrier of intestinal epithelia. J Pharmacol Exp Ther. 2003;305:482–494. doi: 10.1124/jpet.102.047308. [DOI] [PubMed] [Google Scholar]
- Barua M, Liu Y, Quinn MR. Taurine chloramine inhibits inducible nitric oxide synthase and TNF-alpha gene expression in activated alveolar macrophages: decreased NF-kappaB activation and IkappaB kinase activity. J Immunol. 2001;167:2275–2281. doi: 10.4049/jimmunol.167.4.2275. [DOI] [PubMed] [Google Scholar]
- Chan ED, Riches DW. IFNγ+LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280:C441–C450. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, et al. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med. 2007;176:667–675. doi: 10.1164/rccm.200702-291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol. 2008;294:L755–L763. doi: 10.1152/ajplung.00350.2007. [DOI] [PubMed] [Google Scholar]
- Cirino G, Fiorucci S, Sessa WC. Endothelial nitric oxide synthase: the Cinderella of inflammation? Trends Pharmacol Sci. 2003;24:91–95. doi: 10.1016/S0165-6147(02)00049-4. [DOI] [PubMed] [Google Scholar]
- Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem. 2005;94:995–1004. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–774. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle CH, Kader KN. Mechanisms of H2O2-induced oxidative stress in endothelial cells exposed to physiologic shear stress. ASAIO J. 2007;53:17–22. doi: 10.1097/01.mat.0000247157.84350.e8. [DOI] [PubMed] [Google Scholar]
- Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Implications of proteasome inhibition: an enhanced macrophage phenotype. Cell Immunol. 2004;227:140–147. doi: 10.1016/j.cellimm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Albani P, Santangelo R, Torrisi L, Nicoletti VG, de Vellis J, Giuffrida Stella AM. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J Neurosci Res. 2001;65:417–424. doi: 10.1002/jnr.1169. [DOI] [PubMed] [Google Scholar]
- Duann P, Datta PK, Pan C, Blumberg JB, Sharma M, Lianos EA. Superoxide dismutase mimetic preserves the glomerular capillary permeability barrier to protein. J Pharmacol Exp Ther. 2006;316:1249–1254. doi: 10.1124/jpet.105.092957. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Forsythe RM, Xu DZ, Lu Q, Deitch EA. Lipopolysaccharide-induced enterocyte-derived nitric oxide induces intestinal monolayer permeability in an autocrine fashion. Shock. 2002;17:180–184. doi: 10.1097/00024382-200203000-00004. [DOI] [PubMed] [Google Scholar]
- Geiger M, Stone A, Mason SN, Oldham KT, Guice KS. Differential nitric oxide production by microvascular and macrovascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 1997;273:L275–L281. doi: 10.1152/ajplung.1997.273.1.L275. [DOI] [PubMed] [Google Scholar]
- He W, Huang FC, Gavai A, Chan WK, Amato G, Yu KT, et al. Novel cytokine release inhibitors. Part III: truncated analogs of tripterine. Bioorg Med Chem Lett. 1998;8:3659–3664. doi: 10.1016/s0960-894x(98)00671-4. [DOI] [PubMed] [Google Scholar]
- Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Holbeck S, Grande PO. Endotoxin increases both protein and fluid microvascular permeability in cat skeletal muscle. Crit Care Med. 2003;31:560–565. doi: 10.1097/01.CCM.0000048620.88344.70. [DOI] [PubMed] [Google Scholar]
- Hortelano S, Castrillo A, Alvarez AM, Boscá L. Contribution of cyclopentenone prostaglandins to the resolution of inflammation through the potentiation of apoptosis in activated macrophages. J Immunol. 2000;165:6525–6531. doi: 10.4049/jimmunol.165.11.6525. [DOI] [PubMed] [Google Scholar]
- Kakar R, Kautz B, Eklund EA. JAK2 is necessary and sufficient for interferon-gamma-induced transcription of the gene encoding gp91PHOX. J Leukoc Biol. 2005;77:120–127. doi: 10.1189/jlb.0704429. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Knepler JL, Jr, Taher LN, Gupta MP, Patterson C, Pavalko F, Ober MD, et al. Peroxynitrite causes endothelial cell monolayer barrier dysfunction. Am J Physiol Cell Physiol. 2001;281:C1064–C1075. doi: 10.1152/ajpcell.2001.281.3.C1064. [DOI] [PubMed] [Google Scholar]
- Kristof AS, Marks-Konczalik J, Moss J. Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J Biol Chem. 2000;276:8445–8452. doi: 10.1074/jbc.M009563200. [DOI] [PubMed] [Google Scholar]
- Lahti A, Jalonen U, Kankaanranta H, Moilanen E. c-Jun NH2-terminal kinase inhibitor anthra(1,9-cd)pyrazol-6(2H)-one reduces inducible nitric-oxide synthase expression by destabilizing mRNA in activated macrophages. Mol Pharmacol. 2003;64:308–315. doi: 10.1124/mol.64.2.308. [DOI] [PubMed] [Google Scholar]
- Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang YY, Tan HW, Jia YF, Li D. Therapeutic effect of tripterine on adjuvant arthritis in rats. J Ethnopharmacol. 2008;118:479–484. doi: 10.1016/j.jep.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Lowenstein CJ, Padalko E. iNOS (NOS2) at a glance. J Cell Sci. 2004;117:2865–2867. doi: 10.1242/jcs.01166. [DOI] [PubMed] [Google Scholar]
- Marx G. Fluid therapy in sepsis with capillary leakage. Eur J Anaesthesiol. 2003;20:429–442. doi: 10.1017/s0265021503000681. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Middleton J, Americh L, Gayon R, Julien D, Aguilar L, Amalric F, et al. Endothelial cell phenotypes in the rheumatoid synovium: activated, angiogenic, apoptotic and leaky. Arthritis Res Ther. 2004;6:60–72. doi: 10.1186/ar1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Gertzberg N, Vaughan E, Weisbrot J, Woodburn R, Lambert W, et al. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am J Physiol Lung Cell Mol Physiol. 2006;290:L674–L684. doi: 10.1152/ajplung.00391.2005. [DOI] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson CE, Rhoades RA, Garcia JG. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Physiol. 1992;72:865–873. doi: 10.1152/jappl.1992.72.3.865. [DOI] [PubMed] [Google Scholar]
- Peluffo G, Radi R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res. 2007;75:291–302. doi: 10.1016/j.cardiores.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60:49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Doyle TM, Cuzzocrea S. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem Soc Trans. 2006;34:965–970. doi: 10.1042/BST0340965. [DOI] [PubMed] [Google Scholar]
- Sessa WC. Regulation of endothelial derived nitric oxide in health and disease. Mem Inst Oswaldo Cruz. 2005;100(Suppl. 1):15–18. doi: 10.1590/s0074-02762005000900004. [DOI] [PubMed] [Google Scholar]
- Setty AR, Sigal LH. Herbal medications commonly used in the practice of rheumatology: mechanisms of action, efficacy, and side effects. Semin Arthritis Rheum. 2005;34:773–784. doi: 10.1016/j.semarthrit.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, et al. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med. 2005;171:858–867. doi: 10.1164/rccm.200407-981OC. [DOI] [PubMed] [Google Scholar]
- Shang L, Tomasi TB. The heat shock protein 90-CDC37 chaperone complex is required for signaling by types I and II interferons. J Biol Chem. 2006;281:1876–1884. doi: 10.1074/jbc.M509901200. [DOI] [PubMed] [Google Scholar]
- Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- Tao X, Younger J, Fan FZ, Wang B, Lipsky PE. Benefit of an extract of Tripterygium wilfordii hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum. 2002;46:1735–1743. doi: 10.1002/art.10411. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- Titheradge MA. Nitric oxide in septic shock. Biochim Biophys Acta. 1999;1411:437–455. doi: 10.1016/s0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]
- Uto T, Fujii M, Hou DX. 6-(Methylsulfinyl)hexyl isothiocyanate suppresses inducible nitric oxide synthase expression through the inhibition of Janus kinase 2-mediated JNK pathway in lipopolysaccharide-activated murine macrophages. Biochem Pharmacol. 2005;70:1211–1221. doi: 10.1016/j.bcp.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Wilson JX, Dixon SJ, Yu J, Nees S, Tyml K. Ascorbate uptake by microvascular endothelial cells of rat skeletal muscle. Microcirculation. 1996;3:211–221. doi: 10.3109/10739689609148290. [DOI] [PubMed] [Google Scholar]
- Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. 2004;190:446–455. doi: 10.1016/j.expneurol.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Wu F, Cepinskas G, Wilson JX, Tyml K. Nitric oxide attenuates but superoxide enhances iNOS expression in endotoxin- and IFNγ-stimulated skeletal muscle endothelial cells. Microcirculation. 2001;8:415–425. doi: 10.1038/sj/mn/7800113. [DOI] [PubMed] [Google Scholar]
- Wu F, Schuster DP, Tyml K, Wilson JX. Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic Biol Med. 2007;42:124–131. doi: 10.1016/j.freeradbiomed.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Wu F, Tyml K, Wilson JX. Ascorbate inhibits iNOS expression in endotoxin- and IFNγ-stimulated rat skeletal muscle endothelial cells. FEBS Lett. 2002;520:122–126. doi: 10.1016/s0014-5793(02)02804-1. [DOI] [PubMed] [Google Scholar]
- Wu F, Tyml K, Wilson JX. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J Cell Physiol. 2008;217:207–214. doi: 10.1002/jcp.21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Wilson JX. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovasc Res. 2009;81:38–45. doi: 10.1093/cvr/cvn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese ‘Thunder of God Vine’, is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- Zhang DH, Marconi A, Xu LM, Yang CX, Sun GW, Feng XL, et al. Tripterine inhibits the expression of adhesion molecules in activated endothelial cells. J Leukoc Biol. 2006;80:309–319. doi: 10.1189/jlb.1005611. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhao S, Gu Y, Lewis DF, Alexander JS, Wang Y. Effects of peroxynitrite and superoxide radicals on endothelial monolayer permeability: potential role of peroxynitrite in preeclampsia. J Soc Gynecol Investig. 2005;12:586–592. doi: 10.1016/j.jsgi.2005.09.003. [DOI] [PubMed] [Google Scholar]