

Abstract
Background and purpose:
We have investigated the therapeutic effects of the selective cyclophilin inhibitor D-MeAla3-EtVal4-cyclosporin (Debio 025) in myopathic Col6a1−/− mice, a model of muscular dystrophies due to defects of collagen VI.
Experimental approach:
We studied calcineurin activity based on NFAT translocation; T cell activation based on expression of CD69 and CD25; propensity to open the permeability transition pore in mitochondria and skeletal muscle fibres based on the ability to retain Ca2+ and on membrane potential, respectively; muscle ultrastructure by electronmicroscopy; and apoptotic rates by terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling assays in Col6a1−/− mice before after treatment with Debio 025.
Key results:
Debio 025 did not inhibit calcineurin activity, yet it desensitizes the mitochondrial permeability transition pore in vivo. Treatment with Debio 025 prevented the mitochondrial dysfunction and normalized the apoptotic rates and ultrastructural lesions of myopathic Col6a1−/− mice.
Conclusions and implications:
Desensitization of the mitochondrial permeability transition pore can be achieved by selective inhibition of matrix cyclophilin D without inhibition of calcineurin, resulting in an effective therapy of Col6a1−/− myopathic mice. These findings provide an important proof of principle that collagen VI muscular dystrophies can be treated with Debio 025. They represent an essential step towards an effective therapy for Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy, because Debio 025 does not expose patients to the potentially harmful effects of immunosuppression.
Keywords: Mitochondria, permeability transition, cell death, cyclosporins, cyclophilins, collagen VI, muscular dystrophies
Introduction
Mitochondria have a crucial role in pathophysiology, and the mitochondrial permeability transition pore (PTP) stands out as an effector mechanism of cell death. Cyclosporin A (CsA) favours PTP closure after high affinity binding to cyclophilin (CyP) D, a matrix peptidyl-prolyl cis-trans isomerase, an effect that does not involve calcineurin (Rasola and Bernardi, 2007). In chronic diseases where the PTP plays a role, such as muscular dystrophies due to collagen VI deficiency (Irwin et al., 2003; Angelin et al., 2007; Merlini et al., 2008) and possibly other forms of muscular dystrophy (Millay et al., 2008; Reutenauer et al., 2008), CsA derivatives that bind CyPs and desensitize the PTP but do not inhibit calcineurin (Nicolli et al., 1996; Waldmeier et al., 2002; Hansson et al., 2004; Paeshuyse et al., 2006) should therefore maintain the beneficial effects of CsA without exposing patients to the long-term complications that may arise from immunosuppression. The CyP inhibitor D-MeAla3-EtVal4-cyclosporin (Debio 025) was developed from CsA by substituting Sar in position 3 and MeLeu in position 4 with D-MeAla and EtVal respectively (Hansson et al., 2004). Unlike CsA, Debio 025 does not display affinity for calcineurin. In vitro, Debio 025 is 7000 times less effective than CsA at inhibiting interleukin-2 production by Jurkat cells and at least 15 times less potent than CsA in mixed lymphocyte reaction tests (Ptak et al., 2008). In addition, Debio 025 inhibits the PTP in brain mitochondria (Hansson et al., 2004), a finding that has encouraged the study of its effects in PTP-dependent disease paradigms.
In this study, we have investigated the effects of Debio 025 in myopathic Col6a1−/− mice, a model of muscular dystrophy caused by defects of collagen VI. Our findings show that it lacks immunosuppressive activity but has therapeutic efficacy against the myopathy of this mouse disease model. Hence, they provide an important proof of principle that the in vivo effect of CsA is not due to inhibition of calcineurin (Irwin et al., 2003), and may pave the way to the long-term treatment of Ullrich Congenital Muscular Dystrophy (UCMD) and Bethlem Myopathy.
Methods
Expression of nuclear factor of activated T cells-green fluorescent protein (NFAT-GFP)
Jurkat T cells were transfected with the plasmid enhanced green fluorescent protein (pEGFP)/NFAT-1D (Boncristiano et al., 2003) using a modification of the diethylamino ethyl (DEAE)/dextran procedure described previously (Plyte et al., 2001). One million Jurkat T cells were then treated for 15 min at 37°C with 0.8 µM CsA, Debio 025 or vehicle, followed by 500 ng·mL−1 of the ionophore A23187. After 20 min of incubation at 37°C, the localization of NFAT-GFP was studied by laser scanning confocal microscopy (Plyte et al., 2001).
Preparation of mouse T lymphocytes by negative selection
Single cell suspensions were prepared from the spleen of the 129-strain of mice using Cell Stainer-BD Falcon. Monocytes were removed by adherence. Unstimulated splenic T lymphocytes were then negatively selected by immunomagnetic sorting using anti-CD22 antibody conjugated magnetic beads (Dynabeads Mouse pan-B). Purity of T cells (typically above 90%) was assessed by flow cytometry after labelling with fluorochrome conjugated anti-CD3 and anti-CD22 mAb. Two hundred thousand T cells in 0.1 mL of RPMI-7.5% bovine calf serum were treated for 15 min at 37°C with 0.8 µM CsA or Debio 025 or with an equal volume of vehicle in six-well plates. The plates were transferred on ice, treated with saturating amounts of hamster anti-CD3 mAb (2C11) or an equivalent volume of medium (unstimulated samples) and incubated for 30 min at 4°C. Cells were washed and resuspended in 0.2 mL of cold RPMI-7.5% bovine calf serum, and 0.8 µM CsA or Debio 025, or vehicle added back to the appropriate samples, which were transferred to 96-well plates previously coated with an antihamster secondary antibody (50 µg·mL−1) and incubated at 37°C for 30 h in a CO2 incubator. Cells were resuspended, spun, resuspended in 0.2 mL phosphate-buffered saline-1% bovine calf serum and subjected to flow cytometric analysis after being labelled with a combination of fluorochrome conjugated anti-CD3/anti-CD69 mAbs or anti-CD3/anti-CD25 mAbs.
Preparation of mitochondria and calcium retention capacity (CRC) test
Mitochondria were prepared from liver and muscle homogenates by differential centrifugation exactly as described previously (Costantini et al., 1995; Fontaine et al., 1998a). The CRC of mitochondrial preparations was assessed fluorimetrically in the presence of the Ca2+ indicator Calcium Green-5N (1 µM; excitation, 505 nm; emission, 535 nm) using a PerkinElmer LS50B spectrofluorimeter equipped with magnetic stirring and thermostatic control (Fontaine et al., 1998b). The incubation conditions are detailed in the legend to Figure 3.
Figure 3.

Effects of cyclosporin A and Debio 025 on the in vitro calcium retention capacity (CRC) of isolated mouse liver mitochondria. Five hours after the last injection, liver (A,B) and skeletal muscle (A',B') mitochondria were isolated from mice treated for 5 days with vehicle (A,A') or with 10 mg·kg−1·day−1 Debio 025 (B,B'). The incubation medium contained 0.2 M sucrose, 10 mM Tris-MOPS, 5 mM glutamate-Tris, 2.5 mM malate-Tris, 1 mM (A and B) or 10 mM (A' and B') Pi-Tris, 10 µM EGTA-Tris and 1 µM Calcium green-5N in the absence (traces 1) or presence (traces 2) of 0.8 µM Debio 025. Final volume was 2 mL, pH 7.4, 25°C. All the experiments were started with the addition of 0.5 mg·mL−1 mitochondria (not shown) followed 1 min later by the indicated pulses of Ca2+. Traces are representative of four replicates for each condition. (C) Shows the CRC/CRCmax of liver and skeletal muscle mitochondria for each individual animal (dot) treated with vehicle or Debio 025. Average values for Debio 025-treated are significantly different from those of vehicle-treated animals (P < 0.05, Student's t-test).
Isolation of skeletal myofibres and measurements of tetramethylrhodamine methyl ester (TMRM) accumulation
Fibres from flexor digitorum brevis (FDB) muscles were isolated exactly as described previously (Irwin et al., 2002). Fibres were then plated onto glass coverslips coated with laminin (3 µg·cm−2) and cultured in Dulbecco's modified Eagle medium containing 10% fetal bovine serum. During the experiment, FDB fibres were placed in 1 mL Tyrode's buffer and loaded them with 20 nM of TMRM as described previously (Irwin et al., 2003). The images of mitochondrial TMRM fluorescence were acquired with a Zeiss Axiovert 100 TV inverted microscope exactly as described previously (Merlini et al., 2008).
In vivo treatments
Col6a1−/− mice (Bonaldo et al., 1998) had free access to a standard diet and were kept under controlled conditions of temperature and humidity on a 12-h light/12-h dark cycle. For the experiments presented in Figures 3–6, mice received two daily doses of Debio 025 5 mg·kg−1, i.p. or an identical volume of vehicle (i.p.) for 5 days. For the experiments shown in Figure 7, the final stated doses of Debio 025 were administered through gavage in two daily doses of identical volumes. All in vivo experiments were approved by the competent Authority of the University of Padova and authorized by the Italian Ministry of Health.
Figure 6.

Incidence of apoptosis in diaphragms from Col6a1−/− mice treated with vehicle or with Debio 025. Cross sections of diaphragms from Col6a1−/− mice treated for 5 days with vehicle (A) or Debio 025 (B) were analysed. Apoptotic nuclei were easily detected in Col6a1−/− muscle fibres from mice treated with vehicle (arrowheads in A). Bar, 50 µM. In (C) and (D) the ordinate scale represents the number of apoptotic nuclei per mm2 observed in diaphragm sections from Col6a1−/− mice treated with vehicle or with Debio 025. (C) Each column refers to one individual; (D) average. *P < 0.05 versus vehicle, Student's t-test.
Figure 7.

Dose-dependence of the effects of Debio 025 on diaphragm apoptosis and skeletal muscle calcium retention capacity (CRC) in Col6a1−/− mice. (A) and (B) The ordinate scale represents the number of apoptotic nuclei per mm2 observed in diaphragm cross sections from Col6a1−/− mice treated with vehicle (V) or with Debio 025 (dose is in mg·kg−1·day−1). Between 20 and 30 sections per mouse were analysed. (A) Each column refers to one individual; (B) average. (C) Represents the average CRC/CRCmax of skeletal muscle mitochondria prepared from the same individuals treated with vehicle (V) or Debio 025. *P < 0.05 versus vehicle, Student's t-test.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay
We prepared 7 µm-thick sections of muscle diaphragm after 4% paraformaldehyde fixation and paraffin embedding. TUNEL was done using the ApopTag in situ apoptosis detection kit. Samples were stained with peroxidase-diaminobenzidine to detect TUNEL-positive nuclei and counterstained with Hoechst 33258 to identify all nuclei, as described previously (Irwin et al., 2003). The total number of nuclei and number of TUNEL-positive nuclei were determined in randomly selected fibres using a Zeiss Axioplan microscope equipped with a Leica DC 500 camera.
Electron microscopy
Diaphragm muscles from Col6a1−/− mice were isolated and gently stretched on a dental wax support in order to prevent contraction during fixation. Samples were fixed with 2.5% glutaraldehyde in phosphate buffer 0.1 M, pH 7.4 for 3 h at 4°C, washed overnight in phosphate buffer 0.15 M, post-fixed in 1% osmium tetroxide in veronal buffer, dehydrated and embedded in Epon 812 epoxidic resin. Transverse ultrathin sections were stained with uranyl acetate and lead citrate and observed in a Philips EM400 transmission electron microscope operating at 100 kV. For statistical analysis, we studied 300 muscle fibres from three different blocks of tissue for each sample.
Data analysis and statistical procedures
Data were analysed either with the Mann–Whitney test or with Student's unpaired t-test as indicated in the text and legends, and only values with P < 0.05 were considered significant. Data are presented as mean ± SEM or mean ± SD.
Materials
Cell Stainer-BD Falcon was obtained from BD Biosciences Europe (Belgium); Dynabeads Mouse pan-B – Dynal Biotech, Invitrogen (San Giuliano Milanese, Italy); fluorochrome conjugated anti-CD3 and anti-CD22 mAb, Becton-Dickinson (Milan, Italy); Calcium Green-5N and TMRM, Molecular Probes; ApopTag in situ apoptosis detection kit, Intergen; Hoechst 33258, Sigma.
Results
Effects of CsA and Debio 025 on NFAT translocation and T cell activation
We investigated whether Debio 025 is able to prevent activation of calcineurin through the reporter plasmid pEGFP/NFAT-1D encoding a fusion GFP-NFAT protein that undergoes calcineurin-dependent dephosphorylation and translocation to the nucleus (Plyte et al., 2001). Nuclear NFAT localization increased from 10% to about 50% after treatment with the divalent cation ionophore A23187 in a process that was blocked by CsA but not by Debio 025 (Figure 1A–C). Only CsA inhibited expression of the T lymphocyte activation markers CD69 and CD25 after stimulation with the T cell receptor agonist antibody 2C11 (Figure 2A and B respectively).
Figure 1.

Effects of A23187, cyclosporin A (CsA) and Debio 025 on nuclear factor of activated T cells-green fluorescent protein (NFAT-GFP) distribution in Jurkat T cells. (A) Jurkat T cells were scored for cytoplasmic versus nuclear localization of NFAT-GFP before and after treatment with 500 ng·mL−1 A23187 in the presence of vehicle, 0.8 µM CsA or 0.8 µM Debio 025. (B) and (C) Representative examples of cells with cytosolic and nuclear NFAT-GFP respectively. *P < 0.05 versus untreated, Student's t-test.
Figure 2.

Effects of cyclosporin A (CsA) and Debio 025 on activation of mouse T lymphocytes. Mouse spleen CD3+ lymphocytes were prepared by negative selection as described in Methods and scored for expression of the activation markers CD69 (A) and CD25 (B), in the presence of vehicle, 0.8 µM CsA or Debio 025. Where indicated, cells were treated with the stimulatory antibody 2C11. *P < 0.05 versus vehicle, Student's t-test.
Effects of Debio 025 on the CRC of isolated mitochondria after in vivo administration
We next assessed whether a mitochondrial effect of Debio 025 can be demonstrated after in vivo drug administration to mice. Liver and hind leg muscle mitochondria were isolated from individual Col6a1−/− mice treated for 5 days with vehicle or with Debio 025. Mitochondria were then incubated in the presence of substrates and supplemented with Calcium Green-5N, which monitors extramitochondrial Ca2+. A train of Ca2+ pulses was added, each pulse being taken up until a threshold was reached that caused opening of the PTP and precipitous release of the previously accumulated Ca2+ (Figure 3). The threshold Ca2+ needed for PTP opening was lower in liver than in muscle mitochondria (compare traces 1 in Figure 3A and A', respectively); and it was significantly increased by in vivo treatment with Debio 025 in both organs (traces 1 in Figure 3B and B'). The effect was particularly striking for muscle mitochondria (Figure 3B'), the CRC getting close to the maximum level that could be attained by adding Debio 025 directly to the cuvette (trace 2 in all panels). Statistical analysis revealed a highly significant desensitization of the PTP in both liver and muscle mitochondria (Figure 3C). It should be noted that the CRC measured in mitochondria treated with Debio 025 in vivo represents a lower limit, because some washout effect was expected to occur during the preparation of mitochondria.
Effects of in vivo administration of Debio 025 on oligomycin-dependent mitochondrial depolarization in FBD fibres
Next, we tested whether this pharmacological inhibition of the PTP can be demonstrated in FDB fibres isolated from Debio 025-treated Col6a1−/− mice. In order to explore the status of the PTP, we exploited our previous observation that most of these fibres display an anomalous depolarizing response to the F1FO ATPase inhibitor oligomycin, which can be corrected by CsA (Irwin et al., 2003). Addition of oligomycin to vehicle-treated animals caused decreased fluorescence of mitochondrial TMRM (a measure of mitochondrial depolarization) in a fraction of the fibres; if a threshold is arbitrarily set at 90% of the initial fluorescence value (Merlini et al., 2008), 18 out of 31 fibres (58%) and 10 out of 40 fibres (25%) depolarized in vehicle- and Debio 025-treated animals respectively (Figure 4, compare A and B).
Figure 4.
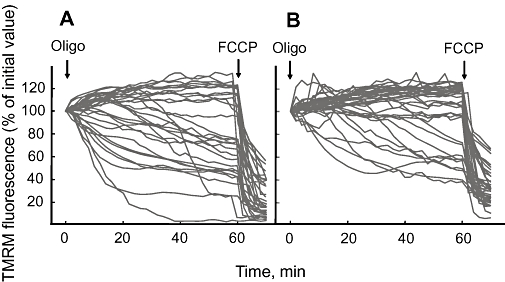
Effects of oligomycin and carbonylcyanide-p-trifluoromethoxyphenyl hydrazone (FCCP) on mitochondrial tetramethylrhodamine methyl ester (TMRM) fluorescence in flexor digitorum brevis (FDB) fibres isolated from Col6a1−/− mice treated with vehicle or with Debio 025. Mice were treated for 5 days with vehicle (A) or with 10 mg·kg−1·day−1 Debio 025 (B). FDB fibres were isolated and mitochondrial fluorescence images acquired as described in Methods. Where indicated, 5 µM oligomycin (Oligo) and 4 µM FCCP were added. Each trace shows the fluorescence of a single fibre, and data from all treated animals were pooled.
Effects of Debio 025 on mitochondrial ultrastructural defects and diaphragm apoptosis in vivo
Swelling, a typical feature of mitochondrial PTP opening, was easily detected in Col6a1−/− (Figure 5A) but not in wild-type diaphragm fibres (results not shown), and mitochondrial ultrastructure was normalized by treatment with Debio 025 (Figure 5B). Debio 025 caused a reduction in the number of abnormal mitochondria in all individuals (Figure 5C) with an average decrease of affected fibres from 13 to 2.5 % (Figure 5D). We also assessed whether treatment with Debio 025 was able to normalize the increased apoptotic rates observed in Col6a1−/− mice (Irwin et al., 2003). Vehicle-treated animals had an average of 55.4 ± 7.3 apoptotic nuclei per mm2, which is well within the range previously reported for these mice (Irwin et al., 2003). After treatment with Debio 025, the incidence dropped to 9.5 ± 2.6 nuclei per mm2 (Figure 6), which is the same value reported after treatment with CsA (Irwin et al., 2003). We finally studied the dose dependence of the effects of Debio 025 on diaphragm apoptosis after gavage. It can be seen that 0.66 mg·kg−1·day−1 was ineffective, while doses of 6.6 and 20 mg·kg−1·day−1 day effectively reduced the number of apoptotic nuclei, consistent with the CRC increase measured in muscle mitochondria (Figure 7). Doses of 60 mg·kg−1·day−1 were no longer effective at preventing apoptosis, but rather slightly increased the frequency of apoptotic nuclei (Figure 7 A,B); yet the effect of Debio 025 6.6 mg·kg−1·day−1 on the CRC was indistinguishable from that of 60 mg·kg−1·day−1 (Figure 7C). The overall increase of CRC was somewhat lower than that observed after i.p. administration of Debio 025 (compare with Figure 3C).
Figure 5.
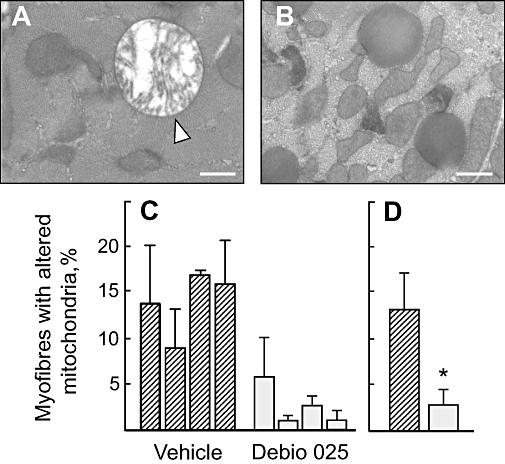
Ultrastructure of mitochondria in diaphragms from Col6a1−/− mice treated with vehicle or with Debio 025. Cross sections of diaphragms from Col6a1−/− mice treated for 5 days with vehicle (A) or Debio 025 (B) were analysed. Swollen mitochondria were easily detected in Col6a1−/− muscle fibres from mice treated with vehicle (arrowhead in A). Bar, 400 nm. In (C) and (D) the ordinate shows the percent of myofibres with altered mitochondria observed in diaphragm sections from Col6a1−/− mice treated with vehicle or with Debio 025. (C) Each column refers to one individual; (D) average. *P < 0.05 versus vehicle, Mann–Whitney test.
Discussion and conclusions
CyPA is the prototype of the CyP family of peptidyl prolyl cis-trans isomerases (Fischer et al., 1989), which in humans includes 17 unique proteins (Wang and Heitman, 2005). After binding to its inhibitory ligand CsA (Borel et al., 1977), the CsA/CyPA complex binds to and inhibits the cytosolic phosphatase calcineurin (Liu et al., 1991) resulting in immunosuppression (Clipstone and Crabtree, 1992; Walsh et al., 1992). Early work with active site mutants of human CyPA had already clearly separated the peptidyl prolyl cis-trans isomerase activity of the protein from CsA binding and calcineurin inhibition (Zydowsky et al., 1992), suggesting that CyPs have cellular functions that are independent of immunosuppression. This prediction has been fulfilled, and the emerging field of CyP biochemistry and pathophysiology is uncovering the great importance of these proteins for a variety of processes relevant to human disease (Wang and Heitman, 2005). These include inflammation and vascular dysfunction (Jin et al., 2004; Kim et al., 2004; 2005; Arora et al., 2005; Damsker et al., 2007), wound healing (Kong et al., 2007), innate immunity to HIV (Sokolskaja and Luban, 2006), hepatitis C infection (Flisiak et al., 2007), host-parasite interactions (Bell et al., 2006), tumour biology (Yao et al., 2005) and mitochondrial PTP-dependent dysfunction mediated by matrix CyPD (Connern and Halestrap, 1992; Nicolli et al., 1996; Woodfield et al., 1997; Waldmeier et al., 2003). These studies underline the importance of developing CyP ligands like Debio 025 that do not result in formation of calcineurin-inhibitory complexes and thus offer great promise for the therapy of CyP-dependent diseases such as HCV and HIV (Flisiak et al., 2008; Paeshuyse et al., 2006; Inoue et al., 2007; Ptak et al., 2008) and collagen VI muscular dystrophies (present manuscript).
We have shown (i) that Debio 025 does not inhibit calcineurin-dependent NFAT translocation to the nucleus in Jurkat T cells, nor does it prevent activation of resting mouse T lymphocytes; this is conclusive evidence that calcineurin is not involved in the protective effects displayed by CsA in the Col6a1−/− mouse (Irwin et al., 2003), an issue of importance because calcineurin affects mitochondrial fission through dephosphorylation of Drp-1 (Cereghetti et al., 2008); (ii) that this drug is an effective inhibitor of the PTP in vivo, as demonstrated by its desensitizing effects in mitochondria and FDB myofibres isolated from Debio 025-treated mice; and (iii) that treatment of Col6a1−/− mice with Debio 025 has a therapeutic efficacy matching that previously described with CsA (Irwin et al., 2003). Remarkably, and at variance from the decreased contractile performance of human and rabbit heart muscle preparations caused by CsA (Janssen et al., 2000), Debio 025 was cardioprotective in a mouse model of myocardial infarction (Gomez et al., 2007) suggesting that the toxic effects of CsA are rather due to inhibition of calcineurin.
It should also be noted that Debio 025 was effective after both i.p. and oral administration, and that a per os dose of 6.6 mg·kg−1·day−1 was extremely effective at inhibiting diaphragm apoptosis, with no further decrease at 20 mg·kg−1·day−1. The antiapoptotic effect was not observed at 60 mg·kg−1·day−1, a dose that slightly increased the incidence of apoptosis. This event appears to be independent of PTP desensitization because the CRC of skeletal muscle mitochondria was the same at 6.6, 20 and 60 mg·kg−1·day−1 of Debio 025. This point should be considered in the future use of Debio 025 in a clinical setting for muscular dystrophy, and deserves further study. It should be mentioned, however, that Debio 025 1000 mg daily is in general well tolerated and proved to be safe in HCV-infected patients, the most prominent side effect being transient hyperbilirubinemia caused by inhibition of biliary canalicular transporters (Flisiak et al., 2009).
Debio 025 inhibits all CyPs including CyPA and/or B, which appears to be responsible for the effects of the drug on HCV and HIV replication respectively (Chatterji et al., 2005; Bobardt et al., 2008; Kaul et al., 2008). The conclusion that Debio 025 prevents muscle cell apoptosis in the Col6a1−/− mouse through CyPD inhibition and PTP desensitization is considerably strengthened by our recent finding that genetic ablation of CyPD cures the myopathy of Col6a1−/− mice (Palma et al., 2009).
Taken together, our findings have major implications for UCMD patients, whose treatment with CsA has proven beneficial (Merlini et al., 2008). UCMD is a chronic muscle-wasting disease involving the diaphragm, and respiratory failure is a common complication which is worsened by pulmonary infections (Merlini and Bernardi, 2008). Long-term treatment with CsA must therefore be carefully weighed against the risks of immunosuppression, which may favour life-threatening infections. Debio 025 has already proved effective at restoring mitochondrial function and at decreasing apoptosis in myoblasts from patients affected by UCMD and Bethlem myopathy (Angelin et al., 2007). Since this drug is as effective as CsA in the Col6a1−/− mouse animal model, we believe that the present results represent an important and necessary step towards a therapy of human collagen VI muscular dystrophies with Debio 025.
Acknowledgments
Tania Tiepolo and Alessia Angelin contributed equally to this work. This work was supported in part by Grants from Telethon-Italy (grant GGP08107), AFM-France (grant 9398) and FIRC (Fellowship to Francesca Finetti).
Glossary
Abbreviations:
- CRC
calcium retention capacity
- CsA
cyclosporin A
- CyP
cyclophilin
- Debio 025
D-MeAla3-EtVal4-cyclosporin
- FDB
flexor digitorum brevis
- GFP
green fluorescent protein
- NFAT
nuclear factor of activated T cells
- PTP
permeability transition pore
- TMRM
tetramethylrhodamine methyl ester
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling
- UCMD
Ullrich Congenital Muscular Dystrophy
Conflict of interest
The Authors declare no conflict of interest.
References
- Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci USA. 2007;104:991–996. doi: 10.1073/pnas.0610270104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K, Gwinn WM, Bower MA, Watson A, Okwumabua I, MacDonald HR, et al. Extracellular cyclophilins contribute to the regulation of inflammatory responses. J Immunol. 2005;175:517–522. doi: 10.4049/jimmunol.175.1.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A, Monaghan P, Page AP. Peptidyl-prolyl cis-trans isomerases (immunophilins) and their roles in parasite biochemistry, host-parasite interaction and antiparasitic drug action. Int J Parasitol. 2006;36:261–276. doi: 10.1016/j.ijpara.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Bobardt M, Tang H, Sakamoto N, Coelmont L, Neyts J, Dumont J-M. The isomerase activity of cyclophilin A is critical for HCV replication. 15th International Symposium on Hepatitis C Virus & Related Viruses, San Antonio, Texas.
- Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- Boncristiano M, Rossi Paccani S, Barone S, Ulivieri C, Patrussi L, Ilver D, et al. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel JF, Feurer C, Magnee C, Stahelin H. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology. 1977;32:1017–1025. [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji U, Bobardt MD, Stanfield R, Ptak RG, Pallansch LA, Ward PA, et al. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J Biol Chem. 2005;280:40293–40300. doi: 10.1074/jbc.M506314200. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis- trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J. 1992;284:381–385. doi: 10.1042/bj2840381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini P, Petronilli V, Colonna R, Bernardi P. On the effects of paraquat on isolated mitochondria. Evidence that paraquat causes opening of the cyclosporin A-sensitive permeability transition pore synergistically with nitric oxide. Toxicology. 1995;99:77–88. doi: 10.1016/0300-483x(94)02997-9. [DOI] [PubMed] [Google Scholar]
- Damsker JM, Bukrinsky MI, Constant SL. Preferential chemotaxis of activated human CD4+ T cells by extracellular cyclophilin A. J Leukoc Biol. 2007;82:613–618. doi: 10.1189/jlb.0506317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- Flisiak R, Horban A, Gallay P, Bobardt M, Selvaraiah S, Wiercinska-Drapalo A, et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- Flisiak R, Dumont JM, Crabbe R. Cyclophilin inhibitors in hepatitis C viral infection. Expert Opin Investig Drugs. 2007;16:1345–1354. doi: 10.1517/13543784.16.9.1345. [DOI] [PubMed] [Google Scholar]
- Flisiak R, Feinman SV, Jablkowski M, Horban A, Kryczka W, Pawlowska M, et al. The cyclophilin inhibitor Debio 025 combined with peg-IFNa2a significantly reduces viral load in treatment naïve hepatitis C patients. Hepatology. 2009;49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex I. J Biol Chem. 1998a;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem. 1998b;273:25734–25740. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- Gomez L, Thibault H, Gharib A, Dumont J-M, Vuagniaux G, Scalfaro P, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1654–H1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- Hansson MJ, Mattiasson G, Mansson R, Karlsson J, Keep MF, Waldmeier P, et al. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J Bioenerg Biomembr. 2004;36:407–413. doi: 10.1023/B:JOBB.0000041776.31885.45. [DOI] [PubMed] [Google Scholar]
- Inoue K, Umehara T, Ruegg UT, Yasui F, Watanabe T, Yasuda H, et al. Evaluation of a cyclophilin inhibitor in hepatitis C virus-infected chimeric mice in vivo. Hepatology. 2007;45:921–928. doi: 10.1002/hep.21587. [DOI] [PubMed] [Google Scholar]
- Irwin W, Fontaine E, Agnolucci L, Penzo D, Betto R, Bortolotto S, et al. Bupivacaine myotoxicity is mediated by mitochondria. J Biol Chem. 2002;277:12221–12227. doi: 10.1074/jbc.M108938200. [DOI] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:267–271. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- Janssen PM, Zeitz O, Keweloh B, Siegel U, Maier LS, Barckhausen P, et al. Influence of cyclosporine A on contractile function, calcium handling, and energetics in isolated human and rabbit myocardium. Cardiovasc Res. 2000;47:99–107. doi: 10.1016/s0008-6363(00)00052-3. [DOI] [PubMed] [Google Scholar]
- Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1186–1191. doi: 10.1161/01.ATV.0000130664.51010.28. [DOI] [PubMed] [Google Scholar]
- Kaul A, Stauffer S, Schmitt J, Pertel T, Luban J, Bartenschlager R. Role of cyclophilins in hepatitis C virus replication. 15th International Symposium on Hepatitis C Virus & Related Viruses, San Antonio, Texas.
- Kim H, Kim WJ, Jeon ST, Koh EM, Cha HS, Ahn KS, et al. Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin Immunol. 2005;116:217–224. doi: 10.1016/j.clim.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lessner SM, Sakurai Y, Galis ZS. Cyclophilin A as a novel biphasic mediator of endothelial activation and dysfunction. Am J Pathol. 2004;164:1567–1574. doi: 10.1016/S0002-9440(10)63715-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Li S, Longaker MT, Lorenz HP. Cyclophilin C-associated protein is up-regulated during wound healing. J Cell Physiol. 2007;210:153–160. doi: 10.1002/jcp.20830. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JDJ, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Merlini L, Bernardi P. Therapy of collagen VI-related myopathies (Bethlem and Ullrich) Neurotherapeutics. 2008;5:613–618. doi: 10.1016/j.nurt.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini L, Angelin A, Tiepolo T, Braghetta P, Sabatelli P, Zamparelli A, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci USA. 2008;105:5225–5229. doi: 10.1073/pnas.0800962105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- Paeshuyse J, Kaul A, De Clercq E, Rosenwirth B, Dumont JM, Scalfaro P, et al. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- Palma E, Tiepolo T, Angelin A, Sabatelli P, Maraldi NM, Basso E, et al. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum Mol Genet. 2009;18:2024–2031. doi: 10.1093/hmg/ddp126. 126. [DOI] [PubMed] [Google Scholar]
- Plyte S, Boncristiano M, Fattori E, Galvagni F, Rossi Paccani S, Majolini MB, et al. Identification and characterization of a novel nuclear factor of activated T-cells-1 isoform expressed in mouse brain. J Biol Chem. 2001;276:14350–14358. doi: 10.1074/jbc.M007854200. [DOI] [PubMed] [Google Scholar]
- Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, et al. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob Agents Chemother. 2008;52:1302–1317. doi: 10.1128/AAC.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- Reutenauer J, Dorchies OM, Patthey-Vuadens O, Vuagniaux G, Ruegg UT. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br J Pharmacol. 2008;155:574–584. doi: 10.1038/bjp.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolskaja E, Luban J. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr Opin Microbiol. 2006;9:404–408. doi: 10.1016/j.mib.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- Walsh CT, Zydowsky LD, McKeon FD. Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J Biol Chem. 1992;267:13115–13118. [PubMed] [Google Scholar]
- Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226.1–226.6. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield KY, Price NT, Halestrap AP. cDNA cloning of rat mitochondrial cyclophilin. Biochim Biophys Acta. 1997;1351:27–30. doi: 10.1016/s0167-4781(97)00017-1. [DOI] [PubMed] [Google Scholar]
- Yao Q, Li M, Yang H, Chai H, Fisher W, Chen C. Roles of cyclophilins in cancers and other organ systems. World J Surg. 2005;29:276–280. doi: 10.1007/s00268-004-7812-7. [DOI] [PubMed] [Google Scholar]
- Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, et al. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]