

Abstract
BACKGROUND
No curative therapy is currently available for locally advanced or metastatic prostate cancer. Oncolytic viruses represent a novel class of therapeutic agents that demonstrates no cross-resistance with existing approaches and can therefore be combined with conventional treatment modalities. Measles virus strains deriving from the Edmonston (MV-Edm) vaccine strain have shown considerable oncolytic activity against a variety of solid tumers and hematologic malignancies. In this study, we investigated the antitumor potential of recombinant MV-Edm derivatives as novel oncolytic agents against prostate cancer.
METHODS
The susceptibility of prostate cancer cell lines (PC-3, DU-145, and LNCaP) to measles virus infection was demonstrated using an MV-Edm derivative expressing green fluorescent protein (GFP). MV-Edm replication in prostate cancer cell lines was assessed by one step viral growth curves. The oncolytic effect of an MV-Edm strain engineered to express the human carcinoembryonic antigen (CEA) was demonstrated in vitro by MTT assays and in vivo in subcutaneous PC-3 xenografts. CEA levels were quantitated in cell supernatants and mouse serum samples.
RESULTS
Recombinant MV-Edm strains can effectively infect, replicate in and kill prostate cancer cells. Intratumoral administration of MV-CEA at a total dose of 6 ×106 TCID50 resulted in statistically significant tumor growth delay (P = 0.004) and prolongation of survival (P = 0.001) in a subcutaneous PC-3 xenograft model. Viral growth kinetics paralleled CEA production.
CONCLUSIONS
MV-CEA has potent antitumor activity against prostate cancer cell lines and xenografts. Viral gene expression during treatment can be determined by monitoring of CEA levels in the serum; the latter could allow dose optimization and tailoring of individualized treatment protocols.
Keywords: CEA, measles virus, MV-CEA, prostate cancer, virotherapy
INTRODUCTION
Prostate cancer is a leading cause of male morbidity and mortality in the western world. It is currently the second most common cause of cancer-related deaths among American men with 186,320 new cases and 28,660 deaths expected to be recorded in 2008 [1]. Although the majority of patients present with clinically localized disease (T1 and T2), a sizeable proportion of these patients will ultimately relapse, with a 5-year failure rate ranging from 14% to 34% [2–4]. No curative therapy is currently available for locally advanced or metastatic prostate cancer. Therefore, novel therapeutic approaches are clearly needed. Prostate cancer is a promising target for virotherapy approaches because the primary tumor site is easily accessible for locoregional treatment administration. The virotherapy agent can easily be applied directly to the prostate tumor via ultrasound-guided needle injections and close monitoring of therapy can be achieved by non-invasive techniques including ultrasound and magnetic resonance imaging.
In contrast to non-replicating or non-viral vector systems, replicating viruses offer the advantage of increased tumor dissemination with potentially enhanced therapeutic benefit [5]. Measles virus (MV) is a negative strand RNA paramyxovirus. The typical cytopathic effect of MV is the formation of mononuclear cell aggregates (syncytia) caused by cell–cell fusion [6]. The virus enters cells via the interaction of the surface hemagglutinin (H) glycoprotein with the two known receptors: CD46 [7,8] which is ubiquitously present on nucleated human cells, but is frequently overexpressed in tumors [9], and the signaling lymphocyte-activation molecule (SLAM) which is predominantly found on activated B and T lymphocytes [10].
Infection by wild-type MV can result in potentially serious disease while on the other hand the measles vaccine strains have an excellent safety record with millions of vaccine doses having been safely administered in over 40 years of use [11]. In contrast to the wild-type MV which enters cells exclusively via the SLAM receptor, the attenuated Edmonston MV vaccine strain (MV-Edm) enters via CD46 and preferentially infects cells that overexpress the CD46 receptor, without significant cytopathic effect against cells expressing low receptor levels [12]. CD46 is a trans-membrane complement regulatory protein that protects human cells against autologous complement lysis by acting as a cofactor in the proteolytic inactivation of C3b and C4b complement products. The overexpression of CD46 in tumors [13,14], serves as a mechanism of tumor cell protection against complement mediated lysis. Thus, it has been shown that MV-Edm derivatives are tumor-selective, but cause minimal cytopathic effect in non-transformed cells, including normal ovarian surface epithelial cells, hepatocytes, dermal fibroblasts, astrocytes, mesothelial cells, peripheral blood lymphocytes and coronary artery smooth muscle cells [12,15–17].
A major challenge in the clinical application of cancer virotherapy is the lack of convenient methods to measure the kinetics of viral gene expression. Knowledge of these parameters would facilitate the optimization of the dosing and administration schedules between repeat treatment cycles as well as the tailoring of treatment protocols for individual patients. One of the advantages of MV as a virotherapy vector is that it can be genetically modified while retaining its considerable oncolytic activity [15,16,18–21]. Thus, the MV-Edm vector has been effectively engineered to express carcinoembryonic antigen (CEA), a soluble marker with no biological activity and normal serum concentration of less than 3 ng/ml. Replication and gene expression of the engineered MV strain (MV-CEA) following infection of the target cells results in CEA production [16]. Therefore, CEA measurement in the MV-CEA-treated patients’ sera could provide important feedback on the viral gene expression profile.
In the present study, we evaluated the therapeutic potential of recombinant MV-Edm strains against prostate cancer and demonstrated the potent antitumor activity of MV-CEA against prostate cancer cell lines and xenografts.
MATERIALS AND METHODS
Cell Culture
Vero (African green monkey) cell line and human prostate cancer cell lines PC-3, DU-145, and LNCaP were purchased from American Type Culture Collection (ATCC, Manassas, VA). All cell lines were grown at 37°C in media recommended by ATCC in a humidified atmosphere of 5% CO2. All media contained 100 U/ml penicillin–streptomycin and were supplemented with 10% heat-inactivated FBS.
Production of Recombinant Viruses
The construction of recombinant MV-Edm encoding the soluble extracellular domain of human CEA (MV-CEA) has been described elsewhere [22]. In summary, a c-DNA infectious clone derived from the Edmonston vaccine lineage Seed B [23] was engineered by inserting the human CEA gene upstream from the MV N gene (Fig. 1A). MV-Edm encoding the enhanced green fluorescent marker protein (MV-eGFP) was generated as previously described [24] by inserting the GFP gene upstream of N (Fig. 1B). For the generation of MV-Edm encoding the firefly luciferase gene, a codon-optimized derivative of the firefly luciferase reporter gene (pGL3, Promega) was inserted in the additional transcription unit upstream of the N gene using MluI and AatII. The resulting full-length anti-genomic cDNA of the MV-Edmonston strain, pMV-lucIIN, was then co-transfected with nucleoprotein (N) and polymerase (L) proteins expression plasmids into 293-3-46 cells [23] to recover the corresponding virus (MV-Luc).
Fig. 1.

Schematic representation of the recombinant MV-Edm viruses. A: The cDNA encoding for the human carcinoembryonic antigen (CEA) was inserted upstream of the N gene (22). B: The cDNA encoding for the enhanced green fluorescent protein (eGFP) was inserted upstream of the N gene. C: The cDNA encoding for the firefly luciferase gene (Luc) was inserted upstream of the N gene (N, nucleoprotein gene; P, phosphoprotein gene; M, matrix protein gene; F, fusion protein gene; L, large protein gene).
All viruses were propagated by infecting 1.5 ×106 Vero cells in T75 flasks at a multiplicity of infection (MOI) of 0.02 in 3 ml Opti-MEM (Invitrogen) at 37°C for 2 hr. The medium was then replaced with DMEM. The cells were incubated at 37°C for 2 days and then transferred to 32°C for 1 day, and subsequently harvested in 1 ml Opti-MEM. The viruses were then released by two cycles of freezing and thawing. The titers of viral stocks were determined by 50% end-point dilution assays (TCID50) on Vero cells in 96-well plates.
Preparations of inactivated viruses followed the same procedures as described above. The viruses were inactivated by exposure to UV light at 120,000 μJ/cm2 for 90 min. Viral inactivation was confirmed by titration on Vero cells.
Determination of CD46 Expression by Flow Cytometry
Cells were harvested, washed in 2% BSA-PBS, incubated with FITC-conjugated mouse antihuman CD46 antibody (PharMingen, San Diego, CA) for 1 hr on ice, washed with PBS and fixed in 4% paraformaldehyde. Samples were assayed on a Becton-Dickinson FACScan Plus cytometer. Analysis was performed using the CellQuest software (Becton-Dickinson, San Jose, CA).
Assessment of Recombinant MV-Edm Infectivity in Prostate Cancer Cell Lines
To assess the susceptibility to recombinant MV-Edm infection of the prostate cancer cell lines in comparison to Vero cells (the prototype permissive line), PC-3, DU-145, LNCaP, and Vero cells were plated at a density of 2 ×105 cells/well in 24-well plates. Twenty-four hours after seeding, the cells (100% confluent) were infected with 100 plaque-forming units (PFU) of MV-eGFP in 0.2 ml of Opti-MEM for 2 hr at 37°C. At the end of the incubation period, the virus was removed and the cells were maintained in their standard medium. Infected cells expressing GFP were identified 48 hr after infection using a fluorescent microscope (Nikon, Melville, NY). GFP-expressing cells were counted in six different wells per cell line and the mean ratio of infected prostate cancer cells versus the infected Vero cells was calculated. Results are presented as the means of at least three independent experiments ± SEM.
Assessment of the Cytopathic Effect (CPE) In Vitro
Cell viability was determined using the MTT colorimetric assay. PC-3, DU-145, and LNCaP cells were plated at a density of 104 cells/well in 96-well plates. Twenty-four h after seeding, the cells were infected with either MV-CEA or UV-inactivated MV-CEA at different MOIs (0.1, 1, and 10) in Opti-MEM for 2 hr at 37°C. At the end of the incubation period, the virus was removed and the cells were maintained in their standard medium. The MTT assay was performed at 48, 96, and 168 hr after infection. The cells were incubated with 10% MTT (ATCC) added directly to the medium for 4 hr at 37°C, followed by cell lysis with a detergent reagent (ATCC) overnight in the dark at room temperature. Absorbance was determined in a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA) at 570 nm in six different wells per group and results are calculated as the percent of OD in the infected wells versus the uninfected controls. Vero cells were used as positive controls in each experiment. Results are presented as the means of at least three independent experiments ± SEM.
Assessment of MV-CEA Replication in Prostate Cancer Cell Lines
PC-3, DU-145, and LNCaP cells were plated in 6-well plates at a density of 2 ×105 cells/well. Twenty-four hours after seeding, the cells were infected with either MV-CEA or UV-inactivated MV-CEA at a MOI of 1.0 in 1 ml of Opti-MEM for 2 hr at 37°C. At the end of the incubation period, the virus was removed and the cells were maintained in their standard medium. The cells were harvested at 24, 48, and 72 hr from two different wells per time point and the virus was released by two cycles of freezing/thawing. The viral titer at each well was determined by 50% end point dilution assay (TCID50) on Vero cells in a 96-well plate.
CEA Level Determination in the Supernatant of MV-CEA Infected Cells
PC-3, DU-145, and LNCaP cells were plated in 6-well plates at a density of 1 ×106 cells/well. Twenty-four hours after seeding, the cells were infected with either MV-CEA or UV-inactivated MV-CEA at a MOI of 1.0 in 1 ml of Opti-MEM for 2 hr at 37°C. At the end of the incubation period, the virus was removed and the cells were maintained in their standard medium. The medium was changed every 24 hr to track the daily production in cell supernatant CEA in each 24-hr period for 3 days. The CEA level at 24, 48, and 72 hr after infection was measured using the Bayer Centaur Immunoassay System (Bayer, Tarrytown, NY). Un-infected and UV-inactivated MV-CEA-treated cell supernatants at the same time points were used as negative controls.
Animal Experiments
All experimental protocols were approved by the Mayo Foundation Institutional Animal Care and Use Committee.
Subcutaneous PC-3 Tumor Model
Xenografts derived from the androgen-insensitive PC-3 cell line were established into the right flanks of 4- to 6-week-old BALB/c nude mice by subcutaneous injection of 3 ×106 PC-3 cells suspended in 0.1 ml PBS and 0.1 ml of BD Matrigel basement membrane matrix (BD Biosciences, Bedford, MA). The mice were examined daily for tumor growth. Tumor volume was measured with calipers twice weekly and calculated by the formula: volume = [(smallest diameter)2 × (largest diameter)]/2.
Visualization of In Vivo Infection of PC-3 Xenografts by Recombinant MV-Edm Strains
Mice were subcutaneously engrafted with PC-3 cells as described above. When all tumors reached 100 mm3, mice received either one intratumoral (IT) injection of 106 MV-Luc or one intratumoral injection of 106 MV-eGFP. Mice treated with MV-Luc had in vivo bioluminescence imaging performed 1, 2, 3, 10, and 13 days after injection. Before each imaging, animals received intraperitoneal injection of 3 mg D-luciferin (Gold Biotechnology, St. Louis, MO) with xylazine/ketamine anesthesia. Images were acquired 10 min after luciferin administration using the Xenogen Ivis 200 System (Caliper Life Sciences, Hopkinton, MA). The Ivis 100 cooled CCD camera system was used for emitted light acquisition. Luciferase activity was analyzed using Living Image Software (version 2.5; Xenogen), according to the manufacturer’s instructions. The level of firefly luciferase was expressed as photons per second per cm2. Mice treated with the MV-eGFP were euthanized 3 days later and tumors were harvested to confirm the presence of infection using a fluorescent microscope (Nikon).
Assessment of In Vivo MV-CEA Treatment Efficacy
Twenty BALB/c nude mice were subcutaneously engrafted with PC-3 cells as described above. The mice were randomly assigned into one of two groups: IT administration of MV-CEA (n = 10) or IT administration of UV-inactivated MV-CEA (n = 10). IT administration of MV-CEA (106 TCID50/100 μl) or an equivalent dose of UV-inactivated MV-CEA was initiated 11 days after tumor cell implantation when all tumor volumes were >100 mm3. Mice were treated twice weekly for a total of six doses (total dose of 6 × 106 TCID50). Venous blood draws (0.1 ml/10 g body weight) were performed on two or three mice from each group on days 1, 2, 3, 4, 7, 11, 14, 18, 21, and 24 after the first virus injection to determine CEA levels. The mice were euthanized if the tumor volume was >2,000 mm3, if the tumor ulcerated, if the mice became unable to drink and/or eat or if weight loss >20% of body weight was observed.
Statistical Analysis
Statistical analysis was performed with the GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA). The Mann–Whitney U-test was used for two-group tumor volume comparisons. Survival curves were plotted according to the Kaplan–Meier method. The log rank test was used to compare the survival times of the two in vivo treatment groups. A P-value ≤ 0.05 was considered statistically significant.
RESULTS
Recombinant MV-Edm Strains Infect Prostate Cancer Cells
FACS analysis confirmed that PC-3, DU-145, and LNCaP express abundant CD46 (Fig. 2). Relative levels of CD46 expression in other human cancer as well as non-cancer cells have been previously described [12,15,16,25]. In contrast to high CD46 expression levels in tumors, the latter being a mechanism that allows tumor cells to escape complement mediated lysis [12], low CD46 expression levels in normal cells such as normal dermal fibroblasts, astrocytes, mesothelial cells, ovarian surface epithelial cells, peripheral blood lymphocytes, monocytes and neutrophils, has been observed [12,15,16,25]. The difference in CD46 expression levels between tumor and normal cells represents a key mechanism explaining oncolytic specificity of measles derivatives [12]. Prominent syncytia formation and cytopathic effect was seen after infection of the three prostate cancer cell lines with MV-eGFP (Fig. 3) and MV-CEA, indicating activity of recombinant MV-Edm strains in both androgen-resistant (LNCaP) and androgen-resistant (PC-3, DU-145) prostate cancer lines.
Fig. 2.
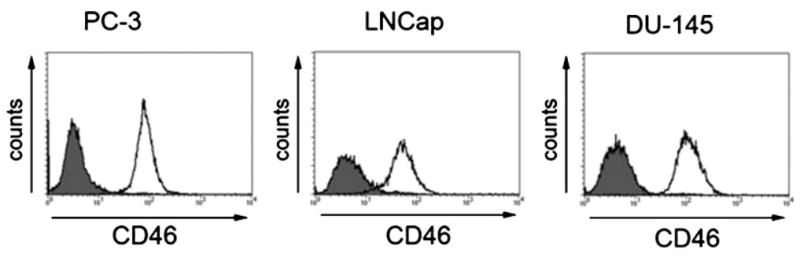
CD46 expression in prostate cancer cell lines. Cells were stained with anti-CD46 PE antibody (white histogram) or isotype control (gray histogram) and analyzed by flow cytometry.
Fig. 3.

MV-eGFP infection results in extensive syncytia formation in PC-3, DU-145, and LNCaP cells. Prostate cancer cells infected with engineered MV-Edm derivatives fuse with each other to form massive multinucleated syncytia that become non-viable (seventy-two hours after infection with MV-eGFP, 100× magnification).
MV-eGFP readily infected the prostate cancer cells which then fused with each other to form massive multinucleated syncytia (Fig. 3). A hundred infectious units of MV-eGFP (as assessed by Vero cell titration) were added to fully confluent wells and the number of infected counts in prostate cancer cells was compared to the number of infectious counts in fully confluent Vero cell controls. In this experiment, LNCaP cells demonstrated very high susceptibility to MV-eGFP infection (approximately 60% as compared to Vero cells; Fig. 4A) and productive infection that resulted in almost complete lysis of the LNCaP monolayers. Even at this low MOI (0.0005), very few LNCaP cells remained uninfected by day 7 after the MV-eGFP inoculation. The susceptibility to MV-eGFP infection of PC-3 and DU-145 cells was comparable to that of other solid tumors such as gliomas [15], and breast cancer [25].
Fig. 4.
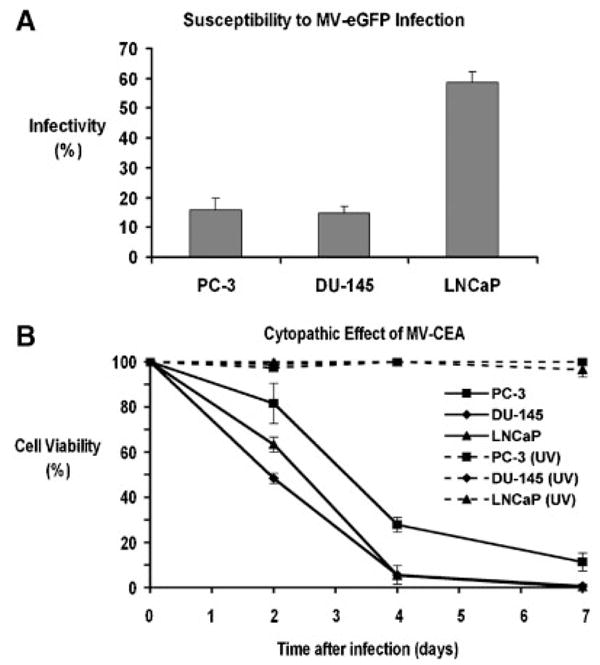
A: In vitro susceptibility of prostate cancer cell lines to MV-eGFP infection. Results are presented as the ratio of infected tumor cells compared to infected Vero cells. B: Cytopathic effect of MV-CEA on PC-3, DU-145, and LNCaP cells at MOI = 1.0. Cell viability was determined by the MTT colorimetric assay. Monolayer cell cultures were completely eradicated by day 7. All cell lines treated with UV-inactivated MV-CEA (UV) showed no cytopathic effect.
MV-CEA Causes Significant Cytopathic Effect in Prostate Cancer Cell Lines
MV-CEA showed significant in vitro oncolytic activity against prostate cancer. At an MOI as low as 1.0, in all three prostate cancer cell lines tested (PC-3, DU-145, and LNCaP), <20% of cells were viable by day 7 post-MV-CEA infection (Fig. 4B). LNCaP monolayers were completely lysed by day 7 at MOIs as low as 0.01. Differences in the level of CD46 expression did not parallel the antitumor effect, indicating that the observed CD46 levels were adequate for massive cell–cell fusion and death to occur. All cell lines treated with UV-inactivated MV-CEA showed no cytopathic effect.
MV-CEA Efficiently Replicates in Prostate Cancer Cell Lines
To determine whether MV-CEA replicates in prostate cancer cell lines, PC-3, DU-145, and LNCaP cells were infected and titers were obtained at 24, 48, and 72 hr. As shown in Figure 5, all prostate cancer cell lines supported robust replication of MV-CEA. The increase in MV-CEA titer correlated with increasing CEA titers in the cell supernatant, indicating that CEA levels represent a good correlate of viral replication. Virus titer was negative at all time points in all cell lines treated with UV-inactivated MV-CEA. CEA levels were undetectable in uninfected as well as treated with UV-inactivated MV-CEA prostate cancer cells.
Fig. 5.

Replication of MV-CEA in PC-3 cells (A) DU-145 cells (B), and LNCaP cells (C) is demonstrated by the titer increase obtained at 24,48, and 72 hr after infection at a MOI of 1.0. Titration was performed on Vero cells in a 96-well plate using the 50% end point dilution assay. The rise in viral titers paralleled the increasing CEA titers in the cell supernatant, thus indicating that CEA levels represent a good marker of viral gene expression. Virus titration and CEA production were negative at all time points in all cell lines treated with UV-inactivated MV-CEA.
Recombinant MV-Edm Strains Persistently Infect Prostate Cancer Xenografts and Express Viral Transgenes In Vivo
To monitor in vivo virus localization following IT administration of recombinant MV-Edm, we injected PC-3 xenografts with a measles vaccine strain engineered to express the firefly luciferase gene (MV-Luc). Using luciferin as a substrate we imaged the distribution of the virus in vivo. As illustrated in Figure 6A,B strong, persistent, localized measles infection was detected in the subcutaneous PC-3 xenografts for more than 2 weeks following infection. Peak uptake was observed at day 2 post-MV-Luc injection. Similarly, when PC-3 xenografts were infected with measles expressing green fluorescent protein (MV-eGFP), diffuse measles infection in the harvested tumors was observed using fluorescent microscopy (data not shown).
Fig. 6.

A: Mice bearing subcutaneous PC-3tumor xenografts on their right flank (day 3 after MV-Luc infection). Strong, localized expression of the firefly luciferase gene was observed. B: The level of firefly luciferase expression in all five mice peaked at day 2 post-MV-Luc injection and persisted for more than 2 weeks.
MV-CEA Has Potent Antitumor Activity When Administered Intravenously in Nude Mice Bearing Prostate Cancer Xenografts
The antitumor activity of MV-CEA was assessed in established subcutaneous PC-3 xenografts as shown in Figure 7. Mice treated by IT administration of MV-CEA at a total dose of 6 ×106 TCID50 had significant suppression of tumor growth as compared to mice treated with UV-inactivated MV-CEA (P = 0.004) (Fig. 7A).
Fig. 7.

Antitumor activity of MV-CEA administered IT in a subcutaneous PC-3 prostate cancer model. Eleven days after tumor implantation the mice were treated with either MV-CEA administered IT at a total dose of 6 ×106 TCID50 (n = 10) or UV-inactivated MV-CEA (n = 10). A: The mice had similar tumor volumes before treatment (P = 0.326). There was significant suppression of tumor growth in MV-CEA-treated animals (P = 0.004 at day 24). The plot was censored at 24 days because of deaths occurring in the UV-inactivated control group. B: Mice treated with MV-CEA had significantly longer survival compared to the UV-inactivated controls (P = 0.001).
Furthermore, MV-CEA-treated mice showed a statistically significant prolongation of survival compared to the UV-inactivated controls (P = 0.001) (Fig. 7B). The median survival of MV-CEA-treated mice was 77 days as compared with 45 days in the UV-inactivated controls. Sixty percent of mice were alive on day 90 in the MV-CEA group, as compared to 0% in the UV-inactivated MV-CEA control group. Complete regression of the subcutaneous tumors was observed in 20% of the MV-CEA treated mice while the remaining 80% showed slower tumor progression compared to the controls.
CEA was detected in the serum of the MV-CEA-treated animals. CEA peaked at 4 days post-treatment initiation (mean CEA value of 16.5 ng/ml) and was detectable up to 24 days following treatment initiation (7.5 ng/ml). The detection of elevated CEA levels indicated the persistence of MV-CEA infection up to day 24 post-treatment initiation. No CEA elevation was observed in the UV-inactivated control animals.
DISCUSSION
This study demonstrated the considerable antitumor potency of recombinant MV-Edm as a virotherapy agent against prostate cancer. MV-Edm is an attenuated live measles vaccine strain. While the wild-type MV is responsible for the potentially serious infectious disease, measles vaccine strains have an excellent safety record. In over 40 years of use, millions of vaccine doses have been administered without toxicity. Prostate cancer is an attractive target for measles-based virotherapy. Prostate tumors express the MV-Edm receptor CD46 [26–28] and our cell line data are consistent with these findings (Fig. 2). High CD46 expression protects the tumor cells from complement-mediated cell damage by blocking the complement components C3b and C4b [14,29].
As shown in Figures 3 and 4, the three well-studied prostate cancer cell lines [30], DU-145, PC-3, and LNCaP were substantially permissive to recombinant MV-Edm infection. Figures 4–5 demonstrate the significant cytopathic effect and robust replication of MV-CEA in a variety of prostate cancer cell lines, both androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU-145). MV-CEA infection at an MOI of 1.0 resulted in complete eradication of all prostate cancer monolayer cultures by day 7. Specifically, LNCaP cells were eliminated very quickly and efficiently in MOIs as low as 0.01.
For our animal efficacy experiments, we chose to use the cell line (PC-3) that represented the worst-case scenario for our study. PC-3 corresponds to advanced (androgen-resistant) prostate cancer and its infection with MV-CEA resulted to slower cytopathic effect in vitro compared to the other two lines tested (Fig. 4B). Nevertheless, as shown in Figure 6, recombinant MV-Edm strains successfully and persistently infected PC-3 xenografts, resulting in transgene expression for more than 2 weeks. Furthermore, MV-CEA treatment of PC-3 xenografts resulted in significant inhibition of tumor growth (P = 0.004) and prolongation of survival (P = 0.001). More specifically, the median survival time of MV-CEA-treated mice almost doubled compared to the controls and complete tumor regression was observed in one-fifth of treated animals. Rodents do not normally express the MV receptors CD46 and SLAM and therefore MV does not infect murine cells. Accordingly, while murine xenografts are informative models of in vivo oncolytic MV efficacy, they cannot be used for toxicity assessment of measles-based therapeutics. The recently developed Ifnarko CD46 Ge mouse strain is a transgenic model that expresses the CD46 receptor, and it is knockout for the Interferon alpha/beta receptor genes: these mice are permissive to measles virus infection and have been accepted by FDA for preclinical toxicity assessment of oncolytic MV-Edm strains [31–33]. No histopathological changes have been observed in vital organs (i.e., brain, heart, lung, liver, pancreas, kidneys, spleen, ovary, testes, peritoneum, and skeletal muscles) of Ifnarko CD46 Ge mice following intraperitoneal or CNS administration of MV-CEA [34,35], as well as intravenous administration of MV-NIS [36].
Viral growth in all three prostate cancer lines correlated with CEA titers, confirming that CEA levels parallel viral gene expression. CEA is an inert peptide and there are widely available clinical assays for CEA measurement. Even though CEA expression monitoring does not address questions pertaining to the localization and spread of the oncolytic virus, it is a highly safe, convenient and inexpensive method for measuring viral gene expression kinetics. Repeated CEA measurements following MV-CEA treatment can be performed with minimal risk to the patient, allowing for optimization of dosing as well as the tailoring of individualized treatment protocols. Our results were obtained with three well characterized CEA negative prostate cancer cell lines and serve as a proof of principle regarding the therapeutic potential of recombinant MV-Edm strains against prostate cancer. Prior to the routine use of PSA monitoring, moderately elevated CEA levels were found in approximately 30% of newly diagnosed prostate cancers [37]. Therefore, while MV-CEA would be well suited for the majority of prostate cancer patients who do not express CEA, alternative non-invasive monitoring strategies can be employed in patients with elevated CEA levels. A recombinant MV-Edm expressing the human sodium iodine symporter (NIS) gene has been developed [18]. NIS is a reporter gene that enables the non-invasive tracking overtime of viral localization, spread, gene expression and replication using PET, SPECT or gamma camera imaging.
The oncolytic potential of measles virotherapy was recently demonstrated in the first completed clinical trial using an unmodified MV vaccine strain as an oncolytic agent in patients with cutaneous lymphoma [38]. Our group and others have demonstrated the therapeutic potency of MV-Edm derivatives against a variety of preclinical animal models including ovarian cancer [16], glioblastoma multiforme [15], breast cancer [25], multiple myeloma [20], lymphoma [19], and hepatocellular carcinoma [17]. These promising results prompted the rapid translation of engineered MV-Edm strains in three clinical trials that are currently active in our institution. To date, no dose limiting toxicity has been observed. In the MV-CEA against ovarian cancer trial, which is the furthest advanced, evidence of biologic activity has been noted in refractory ovarian cancer patients. Furthermore, CEA elevation was observed in the peritoneal fluid and serum of some of the patients demonstrating the potential value of this monitoring strategy [39]. The results of our experiments can serve as a basis to support a translational effort using clinically approved measles virus derivatives such as MV-CEA as therapeutic agents in recurrent prostate cancer.
A notable hurdle of standard viral vectors is their poor delivery efficiency which emphasizes the importance of a potent bystander killing effect in order to destroy those tumor cells that the virus is unable to reach. The engineered MV-Edm derivatives we tested showed considerable local bystander effect that is mediated by massive cell–cell fusion resulting in syncytia formation. Anti-measles immunity could have a negative impact on the therapeutic efficacy of the virus. However, MV-Edm strains can propagate via the fusion of infected cells with the adjacent uninfected cells. It has been demonstrated that antibodies are much less efficient in neutralizing MV-Edm spread by direct cell-to-cell transfer compared to cell-free viruses [40]. It is therefore expected that once infection has been successfully established in the tumor, antibodies will not be able to halt the intratumoral spread. Although the ultimate goal is the development of an effective intravenous viral treatment for patients with hormone refractory metastatic prostate cancer, locoregional administration in patients with locally recurrent prostate cancer represents a logical first step. We are currently exploring novel strategies to circumvent anti-measles immunity following intravenous administration of the virus. Successful systemic delivery could be facilitated by MV-Edm-infected cell carriers, such as monocytoid cell lines, which would ideally traffic to tumors, protect measles from the immune system and efficiently deliver the virus to tumor cells by heterofusion [40–42].
In conclusion, we have shown that engineered MV-Edm is a novel and potent oncolytic agent against prostate cancer. We demonstrated the considerable therapeutic efficacy of MV-CEA against prostate cancer both in vitro and in vivo. This attenuated, live measles vaccine strain has an excellent safety profile and offers the opportunity for non-invasive tracking of viral gene expression via the easy and safe measurement of serum CEA. The finding of MV-CEA efficacy against prostate cancer irrespective of androgen-sensitivity supports the therapeutic promise of recombinant measles virotherapy in diverse populations of prostate cancer tumors. Our results set the foundation for additional studies in preparation for using engineered measles strains in a clinical trial for the treatment of prostate cancer.
Acknowledgments
The authors would like to acknowledge Dr. Donald J. Tindall for his support of this project. This study was supported by the Mayo Clinic Prostate SPORE grant P50 CA 091956.
Grant sponsor: Mayo Clinic Prostate SPORE; Grant number: P50 CA 091956.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Blute ML, Bergstralh EJ, Iocca A, Scherer B, Zincke H. Use of Gleason score, prostate specific antigen, seminal vesicle and margin status to predict biochemical failure after radical prostatectomy. J Urol. 2001;165(1):119–125. doi: 10.1097/00005392-200101000-00030. [DOI] [PubMed] [Google Scholar]
- 3.Shipley WU, Thames HD, Sandler HM, Hanks GE, Zietman AL, Perez CA, Kuban DA, Hancock SL, Smith CD. Radiation therapy for clinically localized prostate cancer: A multi-institutional pooled analysis. JAMA. 1999;281(17):1598–1604. doi: 10.1001/jama.281.17.1598. [DOI] [PubMed] [Google Scholar]
- 4.Wilt TJ, MacDonald R, Rutks I, Shamliyan TA, Taylor BC, Kane RL. Systematic review: Comparative effectiveness and harms of treatments for clinically localized prostate cancer. Ann Intern Med. 2008;148(6):435–448. doi: 10.7326/0003-4819-148-6-200803180-00209. [DOI] [PubMed] [Google Scholar]
- 5.Patel P, Ashdown D, James N. Is gene therapy the answer for prostate cancer? Prostate Cancer Prostatic Dis. 2004;7(Suppl 1):S14–S19. doi: 10.1038/sj.pcan.4500743. [DOI] [PubMed] [Google Scholar]
- 6.Wild TF, Malvoisin E, Buckland R. Measles virus: Both the haemagglutinin and fusion glycoproteins are required for fusion. J Gen Virol. 1991;72(Pt 2):439–442. doi: 10.1099/0022-1317-72-2-439. [DOI] [PubMed] [Google Scholar]
- 7.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67(10):6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75(2):295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 9.Cattaneo R. Four viruses, two bacteria, and one receptor: Membrane cofactor protein (CD46) as pathogens’ magnet. J Virol. 2004;78(9):4385–4388. doi: 10.1128/JVI.78.9.4385-4388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatsuo H, Ono N, Tanaka K, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406(6798):893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 11.Cutts FT, Markowitz LE. Successes and failures in measles control. J Infect Dis. 1994;170(Suppl 1):S32–S41. doi: 10.1093/infdis/170.supplement_1.s32. [DOI] [PubMed] [Google Scholar]
- 12.Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64(14):4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 13.Jurianz K, Ziegler S, Garcia-Schuler H, Kraus S, Bohana-Kashtan O, Fishelson Z, Kirschfink M. Complement resistance of tumor cells: Basal and induced mechanisms. Mol Immunol. 1999;36 (13–14):929–939. doi: 10.1016/s0161-5890(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 14.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40(2–4):109–123. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 15.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, Mishra PK, Macura SI, Russell SJ, Galanis EC. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003;63(10):2462–2469. [PubMed] [Google Scholar]
- 16.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62(16):4656–4662. [PubMed] [Google Scholar]
- 17.Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, Russell SJ, LaRusso NF. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology (Baltimore, MD) 2006;44(6):1465–1477. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 18.Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, Morris JC, Russell SJ. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 19.Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, Fielding AK. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97(12):3746–3754. doi: 10.1182/blood.v97.12.3746. [DOI] [PubMed] [Google Scholar]
- 20.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98(7):2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 21.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat Rev. 2008;6(7):529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng KW, Facteau S, Wegman T, O’Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med. 2002;8(5):527–531. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 23.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter MA. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14(23):5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duprex WP, McQuaid S, Roscic-Mrkic B, Cattaneo R, McCallister C, Rima BK. In vitro and in vivo infection of neural cells by a recombinant measles virus expressing enhanced green fluorescent protein. J Virol. 2000;74(17):7972–7979. doi: 10.1128/jvi.74.17.7972-7979.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald CJ, Erlichman C, Ingle JN, Rosales GA, Allen C, Greiner SM, Harvey ME, Zollman PJ, Russell SJ, Galanis E. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res Treat. 2006;99(2):177–184. doi: 10.1007/s10549-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 26.Buettner R, Huang M, Gritsko T, Karras J, Enkemann S, Mesa T, Nam S, Yu H, Jove R. Activated signal transducers and activators of transcription 3 signaling induces CD46 expression and protects human cancer cells from complement-dependent cytotoxicity. Mol Cancer Res. 2007;5(8):823–832. doi: 10.1158/1541-7786.MCR-06-0352. [DOI] [PubMed] [Google Scholar]
- 27.Liu AY. Differential expression of cell surface molecules in prostate cancer cells. Cancer Res. 2000;60(13):3429–3434. [PubMed] [Google Scholar]
- 28.Loberg RD, Wojno KJ, Day LL, Pienta KJ. Analysis of membrane-bound complement regulatory proteins in prostate cancer. Urology. 2005;66(6):1321–1326. doi: 10.1016/j.urology.2005.06.094. [DOI] [PubMed] [Google Scholar]
- 29.Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20(12):576–582. doi: 10.1016/s0167-5699(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 30.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: A compendium of old and new lines—Part 1. J Urol. 2005;173(2):342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- 31.Mrkic B, Odermatt B, Klein MA, Billeter MA, Pavlovic J, Cattaneo R. Lymphatic dissemination and comparative pathology of recombinant measles viruses in genetically modified mice. J Virol. 2000;74(3):1364–1372. doi: 10.1128/jvi.74.3.1364-1372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mrkic B, Pavlovic J, Rulicke T, Volpe P, Buchholz CJ, Hourcade D, Atkinson JP, Aguzzi A, Cattaneo R. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72(9):7420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roscic-Mrkic B, Schwendener RA, Odermatt B, Zuniga A, Pavlovic J, Billeter MA, Cattaneo R. Roles of macrophages in measles virus infection of genetically modified mice. J Virol. 2001;75(7):3343–3351. doi: 10.1128/JVI.75.7.3343-3351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng KW, Frenzke M, Myers R, Soeffker D, Harvey M, Greiner S, Galanis E, Cattaneo R, Federspiel MJ, Russell SJ. Biodistribution of oncolytic measles virus after intraperitoneal administration into Ifnar-CD46Ge transgenic mice. Hum Gene Ther. 2003;14(16):1565–1577. doi: 10.1089/104303403322495070. [DOI] [PubMed] [Google Scholar]
- 35.Allen C, Paraskevakou G, Liu C, Iankov ID, Msaouel P, Zollman P, Myers R, Peng KW, Russell SJ, Galanis E. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin Biol Ther. 2008;8(2):213–220. doi: 10.1517/14712598.8.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, Reid JM, Federspiel M, Ames MM, Dingli D, Schweikart K, Welch A, Dispenzieri A, Peng KW, Russell SJ. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82(6):700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neufeld L, Dubin A, Guinan P, Nabong R, Ablin RJ, Bush IM. Carcinoembryonic antigen in the diagnosis of prostate carcinoma. Oncology. 1974;29(5):376–381. doi: 10.1159/000224921. [DOI] [PubMed] [Google Scholar]
- 38.Heinzerling L, Kunzi V, Oberholzer PA, Kundig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood. 2005;106(7):2287–2294. doi: 10.1182/blood-2004-11-4558. [DOI] [PubMed] [Google Scholar]
- 39.Galanis E, Hartmann LC, Cliby W, Peethambaram PP, Long HJ, Kaur JS, Haluska JP, Sloan JA, Peng KW, Russell SJ. Phase I trial of intraperitoneal (IP) administration of a measles virus (MV) derivative expressing the human carcinoembryonic antigen (CEA) in recurrent ovarian cancer (OvCa) J Clin Oncol. 2008;26(15s):302s. [Google Scholar]
- 40.Iankov ID, Blechacz B, Liu C, Schmeckpeper JD, Tarara JE, Federspiel MJ, Caplice N, Russell SJ. Infected cell carriers: A new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15(1):114–122. doi: 10.1038/sj.mt.6300020. [DOI] [PubMed] [Google Scholar]
- 41.Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14(4):324–333. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 42.Munguia A, Ota T, Miest T, Russell SJ. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008;15(10):797–806. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]