

Abstract
Sulforaphane (SFN) is an isothiocyanate found in cruciferous vegetables, such as broccoli and broccoli sprouts. This anticarcinogen was first identified as a potent inducer of Phase 2 detoxification enzymes, but evidence is mounting that SFN also acts through epigenetic mechanisms. SFN has been shown to inhibit histone deacetylase (HDAC) activity in human colon and prostate cancer lines, with an increase in global and local histone acetylation status, such as on the promoter regions of P21 and bax genes. SFN also inhibited the growth of prostate cancer xenografts and spontaneous intestinal polyps in mouse models, with evidence for altered histone acetylation and HDAC activities in vivo. In human subjects, a single ingestion of 68 g broccoli sprouts inhibited HDAC activity in circulating peripheral blood mononuclear cells 3-6 h after consumption, with concomitant induction of histone H3 and H4 acetylation. These findings provide evidence that one mechanism of cancer chemoprevention by SFN is via epigenetic changes associated with inhibition of HDAC activity. Other dietary agents such as butyrate, biotin, lipoic acid, garlic organosulfur compounds, and metabolites of vitamin E have structural features compatible with HDAC inhibition. The ability of dietary compounds to de-repress epigenetically silenced genes in cancer cells, and to activate these genes in normal cells, has important implications for cancer prevention and therapy. In a broader context, there is growing interest in dietary HDAC inhibitors and their impact on epigenetic mechanisms affecting other chronic conditions, such as cardiovascular disease, neurodegeneration and aging.
Keywords: Epigenetics, Epigenomics, Chromatin remodeling, Acetylated histones, Cancer prevention, Diet, Sulforaphane, Butyrate, Organosulfur compounds, Garlic, Cruciferous vegetables
1. HDAC inhibitors in cancer prevention and therapy
The classic view of cancer etiology is that genetic alterations damage the DNA structure and induce mutations (i.e. altered sequence information) resulting in non-functional proteins that lead to disease progression. More recently, there has been increasing attention given to the role of epigenetic alterations during disease development, including in the area of cancer biology. Epigenetic alterations affect gene expression without directly changing DNA sequences, thereby turning gene expression ‘on’ or ‘off’ via post-translational modifications. In particular, there is growing interest in the mechanisms that regulate chromatin remodeling, and their implications for cancer development. Silencing and unsilencing of genes can occur via changes in DNA methylation, as well as through epigenetic modifications at the level of the histones [1,2]. In addition to factors that govern the overall recruitment and release of histones (histone occupancy), there is a complex interplay of reversible histone modifications that govern gene expression, including histone acetylation, methylation, phosphorylation, ubiquitination and biotinylation [3]. One hallmark of human cancers is the loss of monoacetylation and trimethylation of histone H4 [4]. Selective agents are being sought that might target abnormal patterns of histone modification, as a means of destroying cancer cells. A particularly active avenue of research involves inhibitors of histone deacetylase (HDAC), such as trichostatin A (TSA) and its structural analogs, which are potent agents used clinically for cancer therapy. One HDAC inhibitor showing promise in the treatment of cutaneous T-cell lymphoma is vorinostat (suberoylanilide hydroxamic acid, SAHA [5]).
Recent interest in HDAC inhibitors has expanded into the realm of cancer chemoprevention, as distinct from cancer therapy, with evidence that dietary compounds such as butyrate, diallyl disulfide (DADS) and sulforaphane (SFN) act as weak ligands for HDAC and exhibit HDAC inhibitory activity [6-8]. The working hypothesis for both drugs and dietary agents (Fig. 1) is that DNA/chromatin interactions are kept in a constrained state in the presence of HDAC/co-repressor complexes, but HDAC inhibitors enable histone acetyltransferase/co-activator (HAT/CoA) complexes to transfer acetyl groups to lysine ‘tails’ in histones, thereby loosening the interactions with DNA and facilitating transcription factor access and gene activation. Among the epigenetically silenced genes that have received particular interest are P21 and bax due to their implications for cell cycle arrest and apoptosis, and because they are among a select cadre of genes frequently repressed in cancer cells and de-repressed following treatment with HDAC inhibitors [2-4].
Fig. 1.
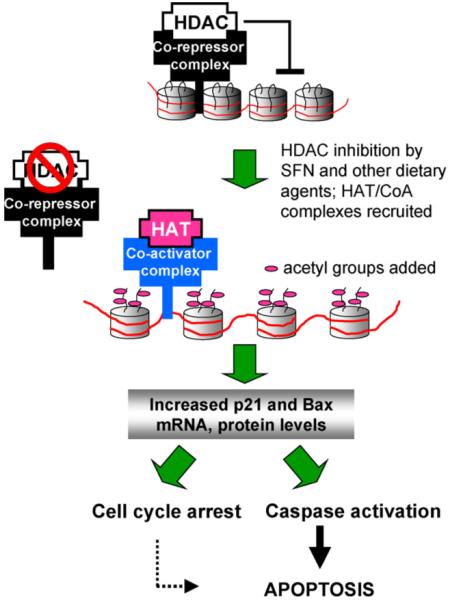
Working hypothesis for the role of dietary histone deacetylase (HDAC) inhibitors. HDAC/co-repressor complexes maintain a tightly restricted chromatin configuration, which limits access of transcription factors to DNA, and represses genes required for cell cycle checkpoint control and apoptosis. HDAC inhibition by SFN and other dietary agents enables histone acetyltransferase/co-activator (HAT/CoA) complexes to add acetyl groups to histone tails, loosening DNA/chromatin interactions, and allowing access of transcription factors to the promoters of genes such as P21 and bax. Re-expression of these genes facilitates cell cycle arrest and apoptosis in the context of cancer chemoprevention or therapy.
2. HDAC inhibition in cancer cells
HDAC inhibitors have been reported to disrupt the cell cycle in G2, allowing cells to prematurely enter the M phase, as well as interfering directly with the mitotic spindle checkpoint. Interestingly, HDAC inhibitors appear to trigger cell cycle arrest and apoptosis more effectively in cancer cells than in non-transformed cells, although the mechanisms are not well understood. Recent studies have implicated thioredoxin and intracellular thiol status, the accumulation of reactive oxygen species, and induction of TRAIL, DR4 and DR5 [9,10].
In the course of studying the mechanisms of cell cycle arrest triggered by dietary cancer chemopreventive agents in vitro, we observed inhibition of HDAC activity in nuclear extracts obtained from human HCT116 colon cancer cells treated with SFN [11]. By design, the experiments used concentrations of SFN in the range 3-15 μM (Fig. 2) to avoid the possible complications of oxidative stress and apoptosis, which occurs at higher doses of SFN in vitro [12-14]. Subsequent work revealed HDAC inhibition by SFN in HT-29 colon cancer cells and Nrf2-/- mouse embryonic fibroblasts, which lack significant endogenous Nrf2 protein expression, thereby supporting a mechanism distinct from the Keap1-Nrf2 pathway induced by SFN in other cell types [7,15,16]. In human colon and prostate cancer cells treated with SFN, inhibition of HDAC activity was accompanied by global increases in histone H3 and H4 acetylation [11,17], coupled with localized histone hyperacetylation on the promoter of the P21 gene (Fig. 2). There was a concomitant increase in p21WAF1 RNA and protein expression, including in PC-3 prostate cancer cells which lack p53 (Fig. 2, center panel). Taken together, these results with SFN provided evidence for HDAC inhibition, independent of Nrf2 and p53.
Fig. 2.

HDAC inhibition by SFN in human colon and prostate cells, and suppression of xenograft growth in mice. Human colon or prostate cell lines were seeded at ∼1 × 106 and 24 h later treated with SFN. Whole cell lysates or nuclear extracts were obtained after 48 h and examined for protein expression by immunoblotting, or HDAC activity using a commercial kit (BioMol). In chromatin immunoprecipitation (ChIP) assays, antibody to acetylated histone H4 was followed by primers to the promoter region of the P21 gene. In the xenograft studies, 5-week-old male athymic nude BALB/c (nu/nu) mice were randomized to 10 animals per group and fed control AIN93G diet or AIN93G diet containing 443 mg SFN/kg. Human prostate cancer PC-3 cells were mixed in a 1:1 ratio of complete media (RPMI 1640 + 10% FBS) and High Concentration Growth Factors Matrigel Matrix (Becton Dickinson). A suspension of 106 cells (50 μl) was injected subcutaneously into the right flank of each mouse. Tumor volume was calculated using the following formula for the volume of an ellipsoid: length × width2 × 0.5236 (π/6). After 21 days xenografts and host tissues were examined for HDAC activity using the BioMol kit. Error bars indicate mean±S.E.; *P < 0.05, **P < 0.01, ***P < 0.001. Full details of the studies in colon and prostate cells can be found in Refs. [11,17], respectively, and xenograft experiments were reported in Ref. [18]. PBMC, peripheral blood mononuclear cells.
3. HDAC inhibition in mice
To explore whether HDAC inhibition by SFN was also possible in vivo, we next implanted PC-3 cell xenografts subcutaneously into nude mice and examined their growth characteristics after feeding SFN in the diet for 21 days (Fig. 2, center panel). There was a significant retardation of tumor growth compared with animals given control diet [18], and most interestingly, in the xenografts recovered from mice at the end of the experiment there was significant inhibition of HDAC activity (Fig. 2, upper right). This suggested systemic distribution of SFN to the tumor implantation site. To test for systemic SFN effects in the host animal, blood samples and various mouse tissues also were examined (Fig. 2, lower right); there was significant inhibition of HDAC activity in the prostate and peripheral blood mononuclear cells (PBMCs).
In mice given a single oral dose of 10 μmol SFN, or 10 μmol of the metabolite SFN-N-acetylcysteine (SFN-NAC), HDAC activity was inhibited significantly in the colonic mucosa at 6 h (Fig. 3). In a longer-term study [19], Apcmin mice ingested ∼6 μmol SFN/day for 70 days, and this resulted in significant inhibition of spontaneous intestinal polyps, compared with controls fed AIN93 diet alone (Fig. 3 center). There was a concomitant increase in global histone H3 and H4 acetylation, and chromatin immunoprecipitation assays performed on mouse colon and intestinal tissues revealed an increase in acetylated histones associated with the promoter region of the P21 gene (Fig. 3, right), as well as bax [19]. Collectively, these findings supported a role for SFN as an HDAC inhibitor in vivo, with evidence for decreased HDAC activity in various tissues and increased global as well as local histone acetylation.
Fig. 3.

HDAC inhibition by SFN in mouse colon, and suppression of intestinal polyps. In a short-term pilot study, 3 mice/group were treated by single oral gavage with 10 μmol SFN, 10 μmol of SFN-N-acetylcysteine (SFN-NAC), or DMSO vehicle alone. Colonic mucosa was scraped 6 h later and examined in the BioMol HDAC activity assay. In a longer-term experiment, Apcmin mice ingested on average ∼6 μmol SFN/day for 70 days in AIN93 diet, whereas controls received AIN93 diet alone. Polyps were enumerated at the end of the study, and the intestinal mucosa was immunoblotted for acetylated histones and β-actin. In addition, intestinal mucosa was subjected to ChIP, as described in Fig. 2 legend. Error bars indicate mean±S.E.; **P < 0.01, ***P < 0.001. For further details, see Ref. [19].
4. HDAC inhibition in man
Given the level of HDAC inhibition in PBMCs obtained from mice fed SFN (Fig. 2, lower right), we conducted a pilot study of PBMCs in people following ingestion of a single dose of SFN-rich broccoli sprouts. Healthy volunteers in the age range 18-55 years, with no history of non-nutritional supplement use, refrained from cruciferous vegetable intake for 48 h. Each subject consumed 68 g (one cup) of broccoli sprouts, and blood was drawn at 0, 3, 6, 24 and 48 h following sprout consumption. In PBMCs of all subjects, HDAC activity was inhibited as early as 3 h after broccoli sprout intake, and returned to normal by 24 h (Fig. 4). This was the first study to show that a naturally consumed food in humans, namely broccoli sprouts, had such a marked effect on HDAC activity [18]. There was strong induction of histone H3 and H4 acetylation coincident with HDAC inhibition at 3 and 6 h, and whereas HDAC activities returned to normal by 24 h, histone hyperacetylation was evident for at least 48 h (Fig. 4). These findings provided the first evidence that dietary intake of broccoli sprouts, a SFN-rich food, influences HDAC activity in normal circulating blood cells of humans, with a level of HDAC inhibition and histone hyperacetylation equal to, or greater than, that achieved with clinically used HDAC inhibitors, such as vorinostat [5].
Fig. 4.
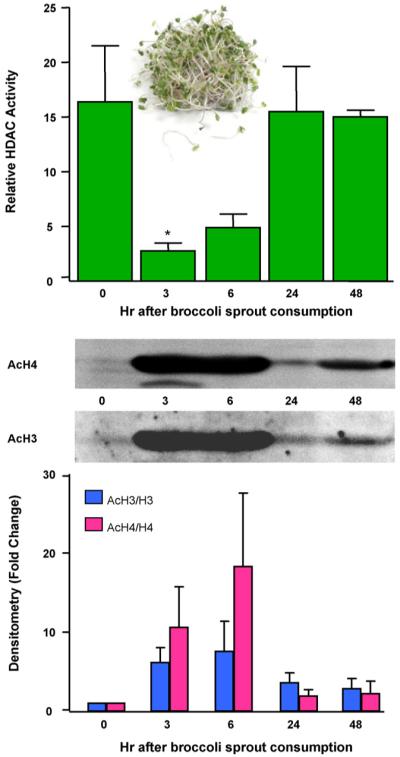
HDAC inhibition by SFN-rich broccoli sprouts in human volunteers. Three healthy human volunteers refrained from cruciferous vegetable intake for 48 h. Each subject consumed 68 g broccoli sprouts (∼105 mg SFN) with a bagel and cream cheese, and blood was collected at times indicated. PBMCs were immunoblotted for acetylated histones H3 and H4 (acH3, acH4), or the corresponding total histone (H3, H4), and HDAC activities were determined using the BioMol kit. Error bars indicate mean±S.E.; *P < 0.05, n=3. For further details, see Ref. [18].
5. Implications of HDAC inhibitors in the diet
Because PBMCs isolated from healthy human volunteers are considered ‘normal’ rather than transformed, a key question concerns the biological significance of histone modifications observed following intake of foods such as broccoli sprouts. What benefit might be derived from the rapid and transient reversal of histone ‘marks’ in normal cells, in terms of the genes silenced and unsilenced? We have proposed recently [8] that epigenetic changes induced by weak ligands might prime normal cells to respond effectively to exogenous insults (toxins, oxidative stress, etc.), activating genes such as P21 and bax to facilitate cell cycle arrest and/or apoptosis, thereby safeguarding against progression to neoplasia (Fig. 1). Rather than the current view of HDAC inhibitors as agents for cancer therapy, dietary HDAC inhibitors might be important for cancer chemoprevention, due to a lifetime of subtle modifications to the histone code.
If this view is indeed correct, how widespread might such HDAC inhibitors be in the human diet, and could they ameliorate other chronic conditions such as cardiovascular disease and neurodegeneration? This is an important question, because ‘epigenetics’ is now known to impact multiple areas, and the underlying mechanisms are central to basic stem cell biology, loss of pleuripotency during differentiation and cell fate determination, and developmental patterning [1,3,20-22].
Given such widespread implications, it is interesting to speculate further about SFN and other dietary HDAC inhibitors and their impact on development and chronic disease susceptibility. In addition to SFN, there are many other known or putative diet-derived HDAC inhibitors. Butyrate is the smallest known HDAC inhibitor (reviewed in Ref. [6]), and contains a simple three-carbon ‘spacer’ attached to a carboxylic acid group (Fig. 5). This compound is derived from the fermentation of dietary fiber and represents the primary metabolic fuel for the colonocytes, where it is present at millimolar concentrations in the large bowel. A second dietary agent reported to inhibit HDAC activity in vitro is the garlic compound DADS [23], which through metabolism can generate S-allylmercaptocysteine (Fig. 5) and related intermediates containing a spacer ending with a carboxylic acid functional group. As discussed elsewhere [8], deacetylation of SFN-NAC generates SFN-cysteine (SFN-Cys), a metabolite of SFN that fits well in the HDAC active site (Fig. 5, inset). Molecular modeling studies with other dietary compounds, such as biotin, α-lipoic acid, and metabolites of vitamin E and conjugated linoleic acids, also provided support for their role as putative HDAC inhibitors (Fig. 5). Sulforaphene, erucin, and phenylbutyl isothiocyanate, which contain a similar spacer length as SFN, each had comparable HDAC inhibitory activities [8], consistent with the Cys moiety occupying the active site and the isothiocyanate ‘cap’ group influencing accessibility to the binding pocket. Similar findings have been reported for structural analogs of TSA, in which the spacer and hydroxamic acid group were retained while substituting the cap group (reviewed in Ref. [8]). It is interesting to note that retinoic acid also has a cap group, spacer, and carboxylic acid functional group, but drug resistant cases of promyelocytic leukemia respond to retinoids only when coupled with potent HDAC inhibitors [24]. This might be due to poor fit of retinoids with HDACs that associate with the oncogenic RAR-PLZF fusion protein (reviewed in Ref. [8]).
Fig. 5.

Dietary HDAC inhibitors. HDAC inhibition has been reported in vitro and/or in vivo for butyrate, garlic organosulfur compounds, and metabolites of SFN, such as SFN-NAC and SFN-Cys, whereas other compounds shown are hypothetical HDAC inhibitors (see text). c9, t11-CLA, cis-9, trans-11-conjugated linoleic acid. Inset: SFN-Cys was manually docked into the active site of human HDAC8, according to the following constraints: (i) carboxylate binding in a bidentate fashion to the active site zinc, (ii) minimal steric conflict between substrate and enzyme, based on a fixed protein, (iii) favorable torsion angles, (iv) hydrogen-bond partners for buried polar atoms, and (v) following the favored position of a bound hydroxamic acid inhibitor [31].
HDAC inhibitors alone can de-repress epigenetically silenced genes in certain cancers, but there is growing interest in combining such compounds with agents that alter DNA methylation, thereby optimizing therapeutic efficacy through enhanced epigenetic gene activation. In theory, dietary HDAC inhibitors might cooperate with other food components known to inhibit DNA methyltransferases (DNMTs), such as soy isoflavones or tea catechins [25,26]. The tea polyphenol epigallocatechin-3-gallate (EGCG) was reported to inhibit DNMT in vitro [26], but a pilot study with SFN in combination with EGCG revealed no significant protection in Apcmin mice, even though each compound alone suppressed the growth of intestinal polyps [27]. Further studies are needed to explain this discrepancy, including the possible involvement of confounding pharmacokinetics and metabolism in vivo.
Finally, the mechanisms discussed herein are pertinent to class I and class II HDACs, but certain dietary agents might alter HDAC activities through other mechanisms, as reported for theophylline in alveolar macrophages from patients with chronic obstructive pulmonary disease [28], and for resveratrol in the activation of human SIRT1 [29]. The latter enzyme belongs to the NAD+-dependent SIR2 family, designated as class III HDACs, which do not typically respond to TSA. For more on this topic, the reader is directed to a discussion of sirtuin-activating compounds and their possible role in aging and neurodegenerative diseases [30].
In summary, interest in epigenetic mechanisms continues unabated and is impacting on treatment options in the clinic, such as with vorinostat (SAHA) in patients with cutaneous T-cell lymphoma. Potent HDAC inhibitors are seen as promising adjuncts to currently used chemotherapy, through epigenetic mechanisms that activate apoptosis and enhance the debulking of tumors and their subsequent regression. However, with the realization that HDAC inhibitors also exist in the diet, we must begin to expand our horizons and question what role epigenetic modifications might play in normal, non-transformed cells. We have hypothesized that the dietary agents such as SFN, DADS and butyrate prime normal cells epigenetically so that they respond most effectively to external insults. However, more work is needed to confirm or refute this idea, given the transient and reversible nature of the epigenetic changes detected (e.g. with broccoli sprouts in human volunteers). There also is a need to better define the precise mechanisms involved, such as the specific HDAC targets and the downstream pathways affected. These mechanisms could be cell-type specific, due to the unique epigenetic marks laid down in each tissue; thus, protection theoretically might be achieved with the same dietary agent against cancer development in the colon, or motor neuron loss in neurodegenerative disorders, or aberrant vascular changes leading to stroke. This is an optimistic view, but promising results obtained with HDAC inhibitors in Huntington’s disease, epilepsy, and bipolar disorder [32,33] suggest that ‘epigenetics’ will likely impact upon multiple disease areas, not simply cancer therapeutics.
Acknowledgments
We are indebted to members of the Dashwood and Ho laboratories for their contributions to the work presented, as well as to Drs. Joe Beckman, Mark Leid, Andy Karplus and Stephen Barnes for helpful discussions. Results presented here were from studies supported in part by NIH grants CA65525 (RHD), CA80176 (RHD), CA90890 (RHD), CA122906 (EH), CA107693 (EH), the Oregon Agricultural Experiment Station, as well as NIEHS center grant P30 ES00210.
References
- [1].Shames DS, Minna JD, Gazdar AF. DNA methylation in health, disease, and cancer. Curr Mol Med. 2007;7:85–102. doi: 10.2174/156652407779940413. [DOI] [PubMed] [Google Scholar]
- [2].Kortenhorst MS, Carducci MA, Shabbeer S. Acetylation and histone deacetylation inhibitors in cancer. Cell Oncol. 2006;28:91–222. doi: 10.1155/2006/760183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fraga MF, Ballestar, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys 16 and trimethylation at Lys 20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- [5].Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- [6].Myzak MC, Dashwood RH. Histone deacetylases as targets for dietary cancer preventive agents: lessons learned with butyrate, diallyl disulfide and sulforaphane. Curr Drug Targets. 2006;7:443–52. doi: 10.2174/138945006776359467. [DOI] [PubMed] [Google Scholar]
- [7].Myzak MC, Dashwood RH. Chemoprotection by sulforaphane: keep one eye beyond keap-1. Cancer Lett. 2006;233:208–18. doi: 10.1016/j.canlet.2005.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–9. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- [11].Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–74. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- [12].Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–21. doi: 10.1158/1535-7163.MCT-06-0494. [DOI] [PubMed] [Google Scholar]
- [13].Karmakar S, Weinberg MS, Banik NL, Patel SJ, Ray SK. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience. 2006;141:1265–80. doi: 10.1016/j.neuroscience.2006.04.075. [DOI] [PubMed] [Google Scholar]
- [14].Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280:19911–24. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- [15].Jeong WS, Keum YS, Chen C, Jain MR, Shen G, Kim JH, et al. Differential expression and stability of endogenous nuclear factor E2-related factor 2 (Nrf2) by natural chemopreventive compounds in HepG2 human hepatoma cells. J Biochem Mol Biol. 2005;38:167–76. doi: 10.5483/bmbrep.2005.38.2.167. [DOI] [PubMed] [Google Scholar]
- [16].Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171–84. doi: 10.1016/j.canlet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- [17].Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27:811–9. doi: 10.1093/carcin/bgi265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med. 2007;232:227–34. [PMC free article] [PubMed] [Google Scholar]
- [19].Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apcmin mice. FASEB J. 2006;20:506–8. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pleuripotency. Cell. 2007;128:747–62. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- [21].Kondo T. Epigenetic alchemy for cell fate conversion. Curr Opin Genet Dev. 2006;16:313–23. doi: 10.1016/j.gde.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [22].Simonsson S, Gurdon JB. Changing cell fate by nuclear reprogramming. Cell Cycle. 2005;4:513–5. doi: 10.4161/cc.4.4.1581. [DOI] [PubMed] [Google Scholar]
- [23].Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duee PH, et al. Diallyl disulfide (DADS) increases histone acetylation and p21waf1/cip1 expression in human colon cancer cell lines. Carcinogenesis. 2004;25:1227–36. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- [24].Cote S, Rosenauer A, Bianchini A, Seiter K, Vandewiele J, Nervi C, et al. Response to histone deacetylase inhibition of novel PML/RAR-α mutants detected in retinoic acid-resistant APL cells. Blood. 2002;100:2586–96. doi: 10.1182/blood-2002-02-0614. [DOI] [PubMed] [Google Scholar]
- [25].Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res. 2005;11:7033–41. doi: 10.1158/1078-0432.CCR-05-0406. [DOI] [PubMed] [Google Scholar]
- [26].Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cells. Cancer Res. 2003;63:7563–70. [PubMed] [Google Scholar]
- [27].Dashwood RH. Frontiers in polyphenols and cancer prevention. J Nutr. 2007;137(Suppl 1):267S–9S. doi: 10.1093/jn/137.1.267S. [DOI] [PubMed] [Google Scholar]
- [28].Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–95. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–95. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- [30].Sinclair D. Sirtuins for healthy neurons. Nat Genet. 2005;37:339–40. doi: 10.1038/ng0405-339. [DOI] [PubMed] [Google Scholar]
- [31].Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–96. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- [33].Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anti-convulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–41. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]