

Abstract
Astrocytes are coupled via gap junctions (GJs) comprising connexin 43 (Cx43) (Gja1) and Cx30 (Gjb6), which facilitate intercellular exchange of ions. Astrocyte connexins also form heterotypic GJs with oligodendrocytic somata and lamellae. Loss of oligodendrocyte gap junctions results in oligodendrocyte and myelin pathology. However, whether loss of astrocyte GJs affects oligodendrocytes and myelin is not known. To address this question, mice with astrocyte-targeted deletion of Cx43 and global loss of Cx30 [double knock-out (dKO)] were studied using Western blotting, immunohistochemistry, electron microscopy, and functional assays. Commencing around postnatal day 23 and persisting into old age, we found widespread pathology of white matter tracts comprising vacuolated oligodendrocytes and intramyelinic edema. In contrast, gray matter pathology was restricted to the CA1 region of the hippocampus, and consisted of edematous astrocytes. No differences were observed in synaptic density or total NeuN+ cells in the hippocampus, or olig2+ cells in the corpus callosum. However, in dKO mice, fewer CC1-positive mature oligodendrocytes were detected, and Western blotting indicated reduced myelin basic protein. Pathology was not noted in mice expressing a single allele of either Cx43 or Cx30. When compared with single connexin knock-outs, dKO mice were impaired in sensorimotor (rotarod, balance beam assays) and spatial memory tasks (object recognition assays). We conclude that loss of astrocytic GJs can result in white matter pathology that has functional consequences.
Introduction
Gap junctions (GJs) allow the intercellular exchange of ions and small molecules and facilitate signaling important in the maintenance of brain homeostasis. Cells of the astrocytic lineage express the GJ proteins connexin 30 (Cx30) and Cx43, whereas oligodendrocytes express Cx47, Cx32, and Cx29 (for review, see Dermietzel et al., 2000; Nagy et al., 2004; Theis et al., 2005). The formation of heterotypic GJs between astrocytes and oligodendrocytic somata and lamellae (Kamasawa et al., 2005; Orthmann-Murphy et al., 2007b) has led to the suggestion that GJ connectivity forms the molecular basis for a panglial syncytium important in supplying glucose to neurons (Rouach et al., 2008), and in redistributing potassium (K+) released into the periaxonal compartment after neuronal activity (Kettenmann and Ransom, 1988; Newman, 1995; Wallraff et al., 2006). Indeed, ablation of oligodendrocyte GJs (Sutor et al., 2000; Menichella et al., 2003; Odermatt et al., 2003) and/or the potassium channel Kir4.1 (Neusch et al., 2001; Menichella et al., 2006; Djukic et al., 2007) causes vacuolation of myelin.
The syncytium becomes disrupted by neuroinflammatory conditions that alter connexin expression. For example, Cx43 is downregulated in astrocytes in response to interleukin-1 (John et al., 1999; Duffy et al., 2000) or the double-stranded RNA homolog polyI:C (Zhao et al., 2006), as well as in an animal model of multiple sclerosis (MS) (Brand-Schieber et al., 2005; Roscoe et al., 2007). However, how loss of astrocyte GJs might affect myelin and oligodendrocytes is not known.
The astrocyte-targeted Cx43 knock-out mouse (Theis et al., 2003) has enhanced stroke volume after middle cerebral artery occlusion (Nakase et al., 2004), accelerated spreading depression, and motor impairments (Theis et al., 2003; Frisch et al., 2003). The remaining astrocyte:astrocyte coupling was abolished after crosses with a mouse deficient in the other major astrocyte connexin, Cx30 (Wallraff et al., 2006). These double knock-out (dKO) mice have elevated hippocampal [K+]out in response to neuronal activity, slower decay of [K+]out (even when normalized for larger initial [K+]out), a decreased threshold for epileptiform events (Wallraff et al., 2006), and display more severe pathology in ischemia/reperfusion paradigms (Lin et al., 2008). Together, these data indicate that astrocyte connexins play an important role in maintaining ion homeostasis and minimizing damage in the CNS.
In this study, we tested the effects of loss of astrocyte GJs on myelin and oligodendrocytes in Cx43fl/fl Cx30−/− mice (Theis et al., 2003; Teubner et al., 2003) expressing Cre under the murine GFAP promoter (Garcia et al., 2004). Our findings revealed marked glial edema in nonmyelinated tissues that was restricted to the CA1 region of the hippocampus, and widespread edema and vacuolation of oligodendrocytes and astrocytes in myelinated tissues of the brain and spinal cord. Expression of one allele of either Cx43 or Cx30 was sufficient to protect against these aberrations.
Materials and Methods
Animals.
mGFAP-Cre Cx43fl/fl Cx30−/− [double knock-out (dKO)] mice were obtained by breeding mGFAP-Cre mice (backcrossed to C57BL/6 background for 9 generations) with Cx43fl/fl (Gja1) Cx30−/− (Gjb6) mice (backcrossed for 9 generations) resulting in deletion of Cx43 in cells of the astrocytic lineage and global deletion of Cx30. Mice expressing Cre under a promoter cassette containing the full murine GFAP gene sequence were used to enhance specificity of transgenic targeting to GFAP expressing cells (line 73.12, a gift from Dr. Michael Sofroniew; Garcia et al., 2004; Herrmann et al., 2008). Cx43fl/fl Cx30−/− mice were a gift from Dr. Klaus Willecke (Theis et al., 2003; Teubner et al., 2003).
Appropriate deletion of Cx43 and Cx30 in the adult CNS was determined using Western blotting, immunostaining, and quantitative PCR (supplemental Fig. S1, available at www.jneurosci.org as supplemental material, and data not shown). Supplemental Table 1, available at www.jneurosci.org as supplemental material, lists the examined genotypes. Homozygous floxed animals in the absence of Cre have ∼75% reduced Cx43 expression compared with Cx43+/+ mice (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Therefore, we used two control populations: mGFAP-Cre and Cx43fl/fl Cx30−/−. Expression of the GJ protein Cx26, detected in astrocytes by some groups (Nagy et al., 2001), was unchanged (supplemental Fig. S1, available at www.jneurosci.org as supplemental material, and data not shown).
Mice were allowed ad libitum food and water in an SPF facility. Protocols designed to minimize discomfort were approved by the IACUC of Albert Einstein College of Medicine. Tail DNA was isolated using Qiagen DNeasy columns. Genotyping for Cx30 was performed using the following primers: 5′ GGT ACC TTC TAC TAA TTA GCT TGG; 5′ AGG TGG TAC CCA TTG TAG AGG AAG; 5′ AGC GAG TAA CAA CCC GTC GGA TTC; transgenic band 460 bp, WT band 544 bp. Primers for Cx43 were: 5′ GGC ATA CAG ACC CTT GGA CTC C; 5′ TCA CCC CAA GCT GAC TCA ACC G; transgenic band 650 bp, WT band 500 bp. Primers for Cre were: 5′ ACC AGC CAG CTA TCA ACT C; 5′ TAT ACG CGT GCT AGC GAA GAT CTC CAT CTT CCA GCA G; 350 bp.
Tissue collection.
Anesthetized animals were transcardially perfused with PBS. Spinal cords were removed by insufflation and cut into cervical, thoracic and lumbar segments. For immunohistochemistry, tissue was fixed overnight in 4% paraformaldehyde. For Western blotting, cerebrum, hippocampus, cerebellum (brainstem removed), and cervical spinal cord were sonicated on ice in lysis buffer (50 mm HEPES, 250 mm NaCl, 0.1% Triton X-100, Protease Inhibitor Cocktail (Roche), 5 mm EDTA, 5 mm NaF, 1 mm Na3VO4, 5 mm Na4P2O7, 1 mm PMSF) and stored at −20°C.
Western blotting.
Protein samples were separated on 10% or 4–15% SDS-polyacrylamide gels (Mini-PROTEAN; Bio-Rad) and transferred onto polyvinylidene difluoride membranes (Millipore). After blocking in 5% milk/TBS/0.05% Tween 20, membranes were incubated with antibody overnight at 4°C, followed by HRP-coupled secondary (Pierce) and enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Pierce) on light-sensitive film (Biomax L; Sigma). Blots were quantified using ImageJ (NIH). Protein was normalized to β-tubulin or to total protein on the membrane using 0.05% Coomassie Brilliant Blue.
Immunohistochemistry.
Five micrometer paraffin sections were rehydrated and antigen retrieval was performed in sodium citrate buffer heated to 100°C for 25 min. Representative sections were stained with hematoxylin and eosin (H&E). For bright-field immunohistochemistry using paraffin or 50 μm vibratome sections, endogenous peroxidase was quenched using 3% H2O2 for 30 min. Nonspecific binding was blocked with 2.5% horse serum. Sections were incubated with primary antibody in PBS for 1 h at 22°C, incubated with ImmPRESS anti-mouse or anti-rabbit Ig (Vector Laboratories), developed with diaminobenzidine (Vector Laboratories), counterstained with hematoxylin or toluidine blue, dehydrated, cleared, and mounted in Permount. Sections were examined using a Leica microscope with an attached Olympus DP12 camera. Pixel density was measured with ImageJ.
For fluorescence immunohistochemistry, sections were blocked in 10% donkey serum for 1 h before incubation with primary antibody in 5% serum/PBS overnight at 4°C followed by Alexa-conjugated secondary antibodies (Invitrogen) for 1.5 h at 22°C and 4′,6′-diamidino-2-phenylindole (DAPI) to visualize nuclei (Invitrogen). Sections were mounted using Prolong Gold AntiFade Reagent (Invitrogen) and examined using an Olympus IX 81 microscope with motorized stage, 20× or 60× optics, and a Cooke Sensicam QE air-cooled CCD camera in the Einstein Analytic Imaging Facility.
Cell counting.
Image fields selected for examination were randomized with respect to X, Y, and Z planes and cells were counted using the “optical dissector” stereological technique (Mouton, 2002). Images were collected with IPLab 4.0.8 and assembled in Adobe Photoshop 7.0 using global manipulations to optimize signal-noise ratios. For some experiments, z-series were deconvolved using ImageJ.
Antibodies.
Primary antibodies were as follows: rabbit anti-Cx43 (1:1000, Sigma); rabbit anti-Cx43 (1:100, Cell Signaling Technology); mouse anti-Cx43 (1:250, Millipore); GFAP (1:2000, Cell Signaling Technology catalog #3670), GFAP (1:100, DAKO), Iba1 (1:500, Wako Chemicals USA), Olig2 (1:1000, Millipore), APC-CC1 (1:20, Calbiochem), MBP (1:2000, Covance, clone SMI99), cleaved caspase 3 (1:200, Cell Signaling Technology catalog #9661), cathepsin D (1:200, Millipore), CNPase (1:1000, Covance, clone SMI91), Cx26 (1:1000, Abcam), Cx30 (1:100, Invitrogen), NeuN (1:80, Millipore), PCNA (1:1000, Sigma), β-tubulin (1:2000, Sigma). Control sections were processed using species- and isotype-matched irrelevant primary antibodies, IgG fractions, or no primary antibody.
Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed on paraffin slides using In-Situ Cell Death Detection Kit with alkaline phosphatase (Roche) according to manufacturer instructions and developed using NBT/BCIP (Roche) in 0.1% Tween 20/dH2O with neutral red counterstain. In all experiments, morphology was examined for evidence of apoptosis (pyknotic chromatin, membrane blebbing) and necrosis.
Electron microscopy.
Anesthetized animals were perfused with phosphate-buffered 2.5% glutaraldehyde. Thin slices were taken from cerebral hemispheres, cerebellum/brainstem, and spinal cord at C7, Th3, L2, L5, L6, and S1, postfixed in cold 1% osmium tetroxide for 1 h to preserve lipids, dehydrated, and embedded in epoxy resin (Epon 812). One micrometer sections were stained with toluidine blue for light microscopy. Ultrathin sections were mounted on copper grids for examination in a Hitachi H-600 electron microscope and H-6010A scanning system.
G-ratios.
The ratio of myelin sheath thickness to axon diameter (g-ratio) was determined for at least 100 fibers per field for 5 fields per genotype in electron micrographs using ImageJ. The g-ratio was plotted against the axon diameter to directly compare the extent of myelination around a fixed axon size. Because not all axons had a perfectly round shape, we averaged the largest and smallest diameters of each myelinated fiber.
Motor coordination.
Latency to fall from a rotating rod was scored using a Rotamex 5 rotarod (Columbus Instruments). Motor coordination was determined by comparing the latency to fall on the first trial (Lalonde et al., 2003) and motor learning by increased latency to fall over repeated trials (Kuhn et al., 1995). In the balance beam task, a blinded observer recorded the number of times the mouse slipped and the time elapsed while crossing a beam.
Spatial memory.
In the initial 3 min of testing, the time (sec) the mouse attended to two identical objects was recorded. After a 10 min retention period in the home cage, one object was relocated, the mouse was returned to the arena, and the time spent attending to each object was again recorded for 3 min. To ensure scores were not influenced by center-avoidance (Dere et al., 2003) objects were not located in the center of the open field. We presented data as a ratio of time spent exploring the relocated object to the total object exploration time to avoid bias from the increased locomotor activity reported in Cx43-deficient mice (Frisch et al., 2003).
An object preference score of 50% reflects equal probability that the animal will investigate either object at any given time and indicates the animal does not discriminate between the unmoved and the relocated object. A higher preference score indicates better memory performance (Mumby et al., 2002). Six-month-old male mice were tested in random order, n = 5–6 per genotype. Groups were Cre, CreCx43fl/fl Cx30+/−, and dKO. Animals that failed to explore both objects during initial testing were excluded from analysis. The visual memory task was similar, except that during the retention period, one of the objects was replaced with a new, novel object in the same location. These tests are based on those described by Ennaceur and Aggleton (1994).
Statistical analysis.
Statistical analysis was performed with Prism 4 software (GraphPad), using ANOVA followed by Newman–Keuls post hoc test for multiple comparisons, or two-tailed Student's t test. p < 0.05 was considered significant.
Results
Loss of astrocyte gap junctions is associated with widespread cellular vacuolation
Coded slides of hematoxylin and eosin (H&E) stained sagittal sections of brains of 6-month-old animals were evaluated by two independent investigators. This analysis revealed marked vacuolization of white matter and edema in gray matter regions in dKO mice (Fig. 1). Using ImageJ software, we found a significant increase in white space present in the dKO corpus callosum/subcortical white matter, internal capsule, cerebellar white matter, and within the CA1 region of the hippocampus. Other gray matter regions including adjacent areas of the hippocampus were relatively spared. Despite extensive vacuolation, the laminar organization of dKO cerebellum was preserved. This is consistent with previous reports of astrocyte GJ depletion on the C57BL/6 background (Wiencken-Barger et al., 2007). Importantly, mice with other genotypes did not display edematous pathology, indicating that hypomorphic expression of Cx43 (as noted in the Cx43fl/fl mice) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) or heterotypic expression of Cx30 was sufficient to preserve wild-type (WT)-like cellular structure.
Figure 1.

Gray and white matter pathology in H&E-stained sagittal brain sections of 6-month-old dKO mice. A–C, Low-power views of mGFAP-Cre (A), Cx43del Cx30+/− (B), and dKO (C) mice show vacuolation in the corpus callosum and CA1 region of hippocampus in dKO mice only. D, E, High-power views of CA1 in mGFAP-Cre and dKO mice. F, Percentage area white space in CA1 is significantly greater (p < 0.01) in dKO compared with either Cx43del Cx30+/− or mGFAP-Cre mice. G–I, Low-power views of cerebellum in the same animals as above show vacuolation in cerebellar white matter in dKO mice. J, K, High-power views of cerebellar white matter from mGFAP-Cre and dKO mice. L, Percentage area of white space in deep cerebellar white matter is significantly greater (p < 0.05) in dKO compared with either Cx43del Cx30+/− or mGFAP-Cre mice. Scale bars are 100 and 10 μm, respectively, for low- and high-power figures.
Onset of pathology during development
To determine the relationship between age, Cx43 levels, and extent of tissue vacuolation, mouse brains and spinal cords were studied on postnatal day 2 (P2), P4, P15, P23, P30, and at 2, 4, 6 and 12 months (Fig. 2 and data not shown). Protein analysis showed that Cx43 expression was markedly decreased by P15 in the spinal cord, cerebellum, and spinal cord in dKO mice, compared with Cx43fl/fl Cx30−/− controls. The earliest age at which edematous pathology was noted was P23. The extent of pathology remained comparable in brains from P23 to 13 months. Increased immunoreactivity for GFAP occurred in dKO gray and white matter, but not in controls (GFAP-Cre or Cx43del Cx30+/−) (Fig. 3).
Figure 2.
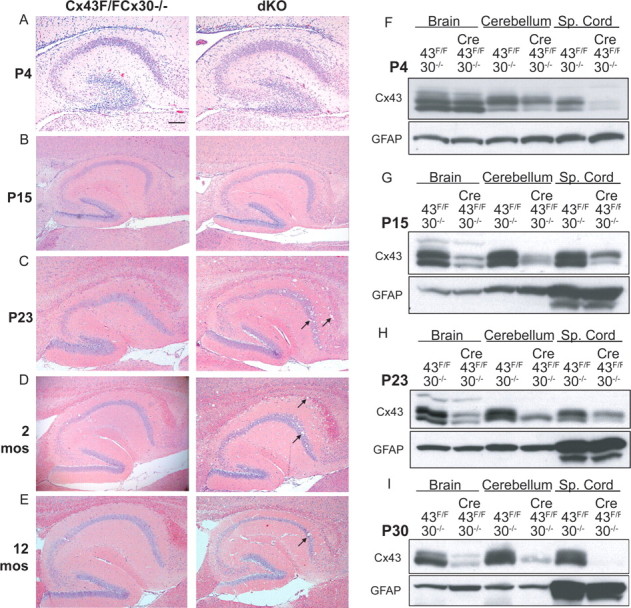
Onset of vacuolation and deletion of Cx43 occurs approximately at postnatal day 23 and persists into adulthood. A–E, H&E-stained sagittal brain sections through the hippocampus and corpus callosum of dKO and non-Cre (Cx43fl/fl Cx30−/−) littermates at P4 (A), P15 (B), P23 (C), 2 months (D) and 12 months (E). F–I, Western blots of dKO and non-Cre brain, cerebellum, and spinal cord at P4 (F), P15 (G), P23 (H), and P30 (I) demonstrate that Cx43 expression becomes reduced in dKO pups between P15 and P23. Western blots were stripped and stained for GFAP. mos, Months; Sp., spinal. Scale bar: (A), 400 μm; (B–E), 200 μm.
Figure 3.

Pathology in dKO animals is associated with astrogliosis in gray and white matter. A–C, Low-power views of GFAP immunostaining in hippocampus and corpus callosum in mGFAP-Cre (A), Cx43del Cx30+/− (B), and dKO (C) mice. D, E, Higher-power views of boxed region in C illustrating GFAP immunoreactivity in association with vacuolated areas of CA1. F, Percentage area GFAP immunoreactivity in the CA1 region of the hippocampus is significantly greater (p < 0.01) in dKO versus control (mGFAP-Cre and Cx43del Cx30+/−) mice. G, H, Higher-power views of vacuolated areas of the corpus callosum (CC) in dKO mice. I, Percentage area GFAP immunoreactivity in the corpus callosum was significantly greater (p < 0.05) in dKO versus control (mGFAP-Cre and Cx43del Cx30+/−) mice. J–L, Low-power views of GFAP immunoreactivity in cerebellum of mGFAP-Cre (J), Cx43del Cx30+/− (K), and dKO (L) mice. M, N, Higher-power views of vacuolated areas of the cerebellar white matter in dKO mice. O, Percentage area GFAP immunoreactivity in the deep cerebellar white matter was significantly greater (p < 0.01) in dKO versus control (mGFAP-Cre and Cx43del Cx30+/−) mice. Scale bar: (A–C, J–L), 200 μm; (D, G, M), 50 μm; (E, H, N), 10 μm.
Ultrastructural analysis of white matter pathology
To determine the structural correlates of this pathology in myelinated tracts, areas of the deep cerebellar white matter, corpus callosum and spinal cord were studied by light and electron microscopy (LM and EM). Three types of pathology were observed: edematous astrocytes; oligodendrocyte nuclei surrounded by large cytoplasmic vacuoles; and intramyelinic edema (Figs. 4, 5 Astrocytes were identified by their characteristic elliptical nuclear morphology with marginal heterochromatin, and bundles of filaments in the cytoplasm. Oligodendrocytes were identified by nuclei with clumped heterochromatin, relatively electron-dense cytoplasm, absence of filaments, and presence of microtubules.
Figure 4.
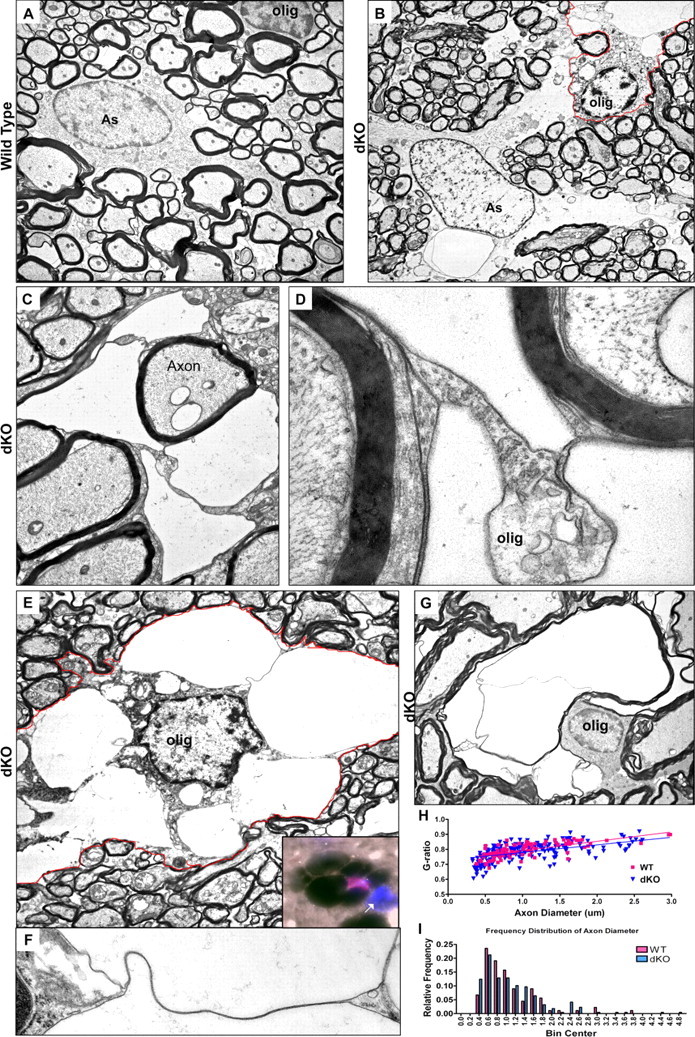
Electron micrographs depicting oligodendrocyte vacuolation within dKO white matter. A, Astrocyte (As) and oligodendrocyte (olig) in white matter of wild-type spinal cord. The astrocyte can be identified by marginal chromatin and cytoplasmic intermediate filaments. B–G, Vacuolated oligodendrocytes and astrocyte in dKO white matter. B, Vacuolated oligodendrocyte abutting edematous astrocyte in dKO cerebellar white matter. The oligodendrocyte is identified by marginal chromatin, microtubules, and relatively electron-dense cytoplasm. Note that the astrocyte also shows extensive pathology with loss of well defined cytoplasmic organelles. The oligodendrocyte is outlined in red. C, Highly compartmentalized oligodendrocyte cytoplasm. D, Higher-power view of the field in C helps identify the compartmentalized cytoplasm as oligodendrocytic due to the presence of microtubules and lack of filaments, and because its membranes are contiguous with compact myelin. E, An oligodendrocyte containing numerous membrane-bound vacuoles within its cytoplasm. The oligodendrocyte is outlined in red. Inset: immunohistochemical staining showing the nucleus of a vacuolated cell positive for both DAPI (blue) and olig2 (red). Note the unaffected, olig2-negative nucleus at right (arrow). F, Detail from E showing a continuous ribbon of oligodendrocyte cytoplasm connecting the perinuclear cytoplasm and plasma membrane between two vacuoles, demonstrating that the vacuoles are contained within the oligodendrocyte. G, Intramyelinic edema. H, I, dKO and wild-type g-ratios (H) and axon diameter (I) are not significantly different in the cerebellum. Magnification: (A), 4000×; (B), 2600×; (C), 6000×; (D), 35,000×; (E), 5000×; (F), 21,000×; (G), 3600×.
Figure 5.

Myelin pathology in dKO white matter. A–C, Toluidine blue stained 1 μm epoxy sections of the dorsal cuneate of the lumbar spinal cord of non-Cre (Cx43fl/fl Cx30−/− (A) and dKO (B, C) mice. D, Electron micrograph showing enlarged oligodendrocyte cytoplasm interpolating between an apparently normal axon and its myelin sheath. Inset depicts ectopic expression of myelin in one of several locations within the oligodendrocyte cytoplasm. E, Myelin whorls, likely representing Wallerian axonal degeneration. F, G, Edema and splitting of myelin sheaths. Scale bar: (A, B), 100 μm; (C), 10 μm. Magnification: (D), 4000×, inset: 15,000×; (E), 9000×; (F), 8000×; (G), 13,000×, inset: 4000×.
When compared with astrocytes in control tissue (Fig. 4A), astrocytes in dKO mice demonstrated loss of cytoplasmic structure and well defined organelles (Fig. 4B). These astrocytes were often in close contact with vacuolated oligodendrocytes (Fig. 4B). Extensively vacuolated oligodendrocyte cytoplasm surrounded some myelinated fibers (Fig. 4C; detail, 4D).
To determine whether the distended vacuoles were within the oligodendrocyte cytoplasm, or belonged to other cell processes that abutted the oligodendrocyte, higher-power electron micrographs were taken (Fig. 4E,F). These images showed narrow ribbons of oligodendrocyte cytoplasm connecting the perinuclear material with the cytoplasm located at the edges of the cell, indicating that the vacuoles were confined to the same oligodendrocyte cytoplasm (Fig. 4E,F). These vacuoles were typically bound by a single membrane. At the LM level, the nuclei of these vacuolated cells were immunoreactive for oligodendrocyte markers such as the transcription factor olig2 (inset, Fig. 4E). Intramyelinic edema was also observed (Fig. 4G).
We also performed g-ratio analysis to quantify myelin sheath thickness in cerebellar white matter. Figure 4, H and I, shows regression lines of these comparisons and the frequency distribution of axon sizes. The extent of myelination and the average axon size were comparable in dKO and controls. In areas of the cord lacking evidence of myelin pathology, EM images showed no differences between mice of different genotypes in myelin compaction, as determined by the periodicity of major and minor dense lines (data not shown).
Within the spinal cord, distribution of oligodendrocyte and myelin pathology showed no predilection for sensory versus motor tracts (data not shown). Myelin stripped from the axon was frequently contained within enlarged oligodendrocyte cytoplasm (Fig. 5D). Fasciculation of myelin, evidence of Wallerian degeneration, and intramyelinic edema were also noted (Figs. 4G, 5E–G). In the dorsal cuneate, pathology was observed in 14 ± 4% of myelinated axons in dKO, versus 2 ± 1% in controls, indicating that not all astrocytes, oligodendrocytes and/or myelin sheaths were affected (see Figs. 4C, 5B–D).
Fewer mature CC1 positive oligodendrocytes were present in the dKO
In wild-type mice, Cx43 GJ plaques are found within the corpus callosum in association with interfascicular arrays of oligodendrocytes (arrows, Fig. 6A). To determine how the absence of astrocyte gap junctions, and the ensuing astrocyte and oligodendrocyte pathology, might impact myelination, we performed immunohistochemical staining and Western blotting for myelin basic protein. Western blots of homogenates prepared from corpus callosum (Fig. 6B; quantification in 6C) and cerebellum (data not shown) indicated that MBP was 51% reduced in dKO mice when compared with other genotypes (p < 0.05 vs Cre).
Figure 6.
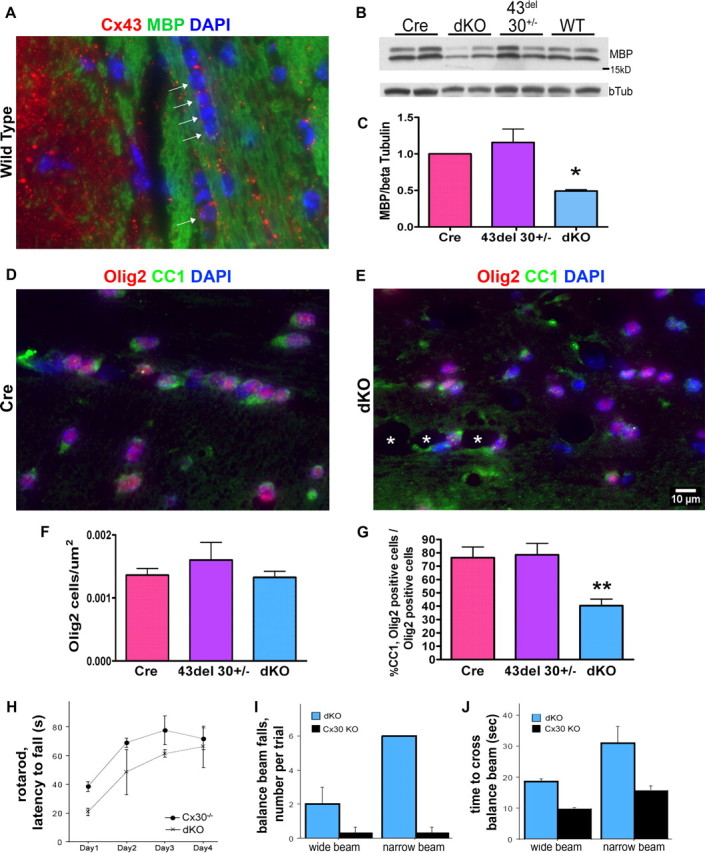
Myelin and oligodendrocyte abnormalities are associated with sensorimotor deficits in dKO mice. A, Immunohistochemistry for Cx43 (red) and myelin basic protein (MBP; green) demonstrates Cx43 puncta adjacent to oligodendrocytes in an interfascicular chain (arrows) in the corpus callosum of a wild-type mouse. Blue, DAPI nuclear stain. B, C, Representative Western blot shows 40% reduced MBP in dKO corpus callosum homogenates relative to β-tubulin (p < 0.05). D–G, Immunohistochemistry for olig2 (red) and the mature marker CC1 (green) demonstrates that although total numbers of oligodendrocytes in control and dKO corpus callosum are similar, there are fewer mature oligodendrocytes in the dKO (p < 0.01). Asterisks in E mark white matter vacuoles. Blue, DAPI. H, dKO mice (×) fall off of the rotarod more quickly than do Cx43fl/fl Cx30−/− control mice (Cx30−/−, ●) control mice. I, J, dKO mice (blue bars) have more slips (I) and are slower (J) than controls (black bars) while crossing wide and narrow balance beams. B–G, n = 5–6 mice per genotype. H–J, n = 3 mice per genotype.
To assess numbers of oligodendrocytes, unbiased morphometric criteria were used to count DAPI+, olig2+ oligodendrocyte nuclei throughout the rostral-caudal axis of the corpus callosum. Although no difference was noted in the number of olig2+ oligodendrocytes, the percentage of olig2+ cells also labeled with the mature marker CC1 was 60% decreased in dKO mice (Fig. 6D–G) (p < 0.01 vs Cre). These data indicated a selective deficiency in mature oligodendrocytes. Additionally, there was a two-fold increase in the presence of olig2− cells in the corpus callosum of dKO mice (p < 0.05; data not shown).
We next sought to determine the consequences of myelin pathology using behavioral tests sensitive to white matter lesions (Kuhn et al., 1995). In the rotarod assay, dKO mice were impaired in motor coordination and sensorimotor adaptation over repeated trials (Fig. 6H). In the balance beam task, dKO mice slipped more frequently and had greater latency to cross than did single-KO controls (Fig. 6I,J).
Gray matter pathology is comprised of edematous astrocytes and has functional consequences
Within the CA1 region of the hippocampus, EM analysis showed swollen cells with features characteristic of astrocytes, including intermediate filaments and thin marginal chromatin (Fig. 7A). Consistent with their identification as astrocytes, these cells had processes abutting normal-appearing blood vessels, neurons, and synapses (Fig. 7A–D). Other areas of hippocampus such as dentate gyrus were unaffected (Fig. 7E).
Figure 7.
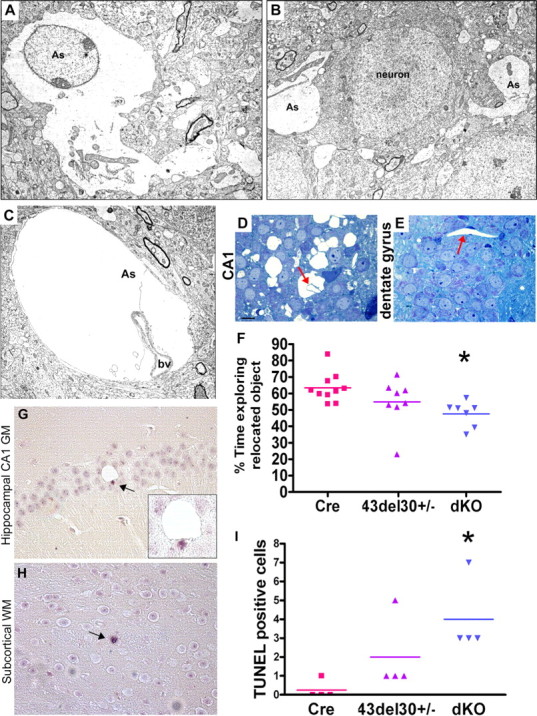
Astrocyte pathology in the CA1 pyramidal cell layer of the hippocampus and spatial memory impairment. A–C, Electron micrographs depicting edematous astrocyte (As) processes identified by the presence of glycogen granules and intermediate filaments within the CA1 region of the hippocampus of a 4-month-old dKO mouse. B, Astrocyte processes flank pyramidal neurons, identified by Nissl substance. C, Astrocyte foot processes abutting blood vessels (bv) are enlarged and depleted of most cytoplasmic organelles. D, E, Toluidine blue-stained 1 μm sections through the CA1 pyramidal cell layer (D) and dentate gyrus (E) demonstrate that pathology is not present in other regions of the hippocampus. Arrows, blood vessels. F, dKO mice fail to preferentially explore a relocated object, indicating impaired spatial memory (p < 0.05 versus mGFAP-Cre mice). Cumulative data from two experiments with 5–6 mice per genotype are shown. G–I, TUNEL assay shows a significant increase in apoptotic cells in dKO brains (p < 0.05, n = 4 brains per genotype). Some apoptotic cells were apparent in close proximity to vacuoles (G). Magnification: (A), 4000×; (B), 4000×; (C), 5000×. Scale bar: (in D) D, E, 10 μm.
We hypothesized that loss of astrocyte GJs, and the concomitant swelling of CA1 astrocytes, might lead to hippocampal dysfunction and/or neuronal losses. To test this hypothesis, we challenged dKO and control mice with spatial memory tasks designed to assay hippocampal function, and subsequently performed neuronal cell counts. The spatial memory task measures preference for an object which has been relocated to a novel location. Mice of all genotypes exhibited comparable exploratory behavior (data not shown). However, dKO mice failed to preferentially explore a relocated object, indicating impaired spatial working memory (p < 0.05) (Fig. 7F). We also determined that dKO animals and controls performed similarly in a visual recognition task thought to be less dependent on hippocampal circuitry (data not shown). These data indicate that loss of astrocyte connexins and concomitant glial edema causes marked deficiencies in some types of memory.
We next examined the number of CA1 neurons. We found no difference in the number of NeuN+ neurons present in the CA1 layer of dKO, CreCx43fl/fl Cx30+/−, and mGFAP-Cre brains (supplemental Fig. S2, available at www.jneurosci.org as supplemental material; n = 4–5 brains per genotype). Synaptic density, as assessed by immunoreactivity for the presynaptic vesicle protein synaptophysin, was unchanged (data not shown). No differences were noted between dKO and control hippocampi in immunoreactivity for other neuronal proteins including calbindin and MAP2 (data not shown).
Cell death/proliferation
To test the possibility that neurons or mature oligodendrocytes in the dKO mice might be undergoing apoptosis, we used immunostaining for the cleaved, active form of the downstream apoptotic effector caspase 3, and performed TUNEL staining for fragmented DNA. Increased apoptosis was noted in dKO brains (p < 0.05 versus mGFAP-Cre) (Fig. 7G–I and data not shown). Subcellular distribution of cathepsin D indicated no evidence of lysosomal rupture or fusion with intracytoplasmic vacuoles, arguing against necrotic/autophagic cell death (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (Sato et al., 2008). We then addressed the possibility that clearance of cellular debris would result in microglial activation; however, we observed no differences in Iba1+ microglial morphology or distribution (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). To test for cellular proliferation, we used an antibody against proliferating cell nuclear antigen (PCNA). No differences were noted in occurrence of PCNA+ cells in the hippocampus, corpus callosum, or cerebellum (data not shown).
Discussion
The glial syncytium is comprised of networks of astrocytes and oligodendrocytes which are highly interconnected via GJs. It has been hypothesized that GJ connectivity is important in the spatial buffering of ions such as K+ (Orkand et al., 1966) accompanied by water to facilitate osmotic equilibrium. Dynamic changes in glial volume are recorded as a consequence of physiologic synaptic transmission and also in pathological situations (Hansson, 1994; MacVicar et al., 2002; Risher et al., 2009).
Astrocyte Cx43 and Cx30 are known for their roles in GJ connectivity between astrocyte processes and as partners in the formation of heterotypic GJs between astrocytes and oligodendrocytes. Oligodendrocyte/astrocyte channels composed of Cx47/Cx43 predominantly localize to oligodendrocyte somata whereas oligodendrocyte/astrocyte Cx32/Cx30 channels localize to myelin lamellae and oligodendrocyte somata (Menichella et al., 2003; Altevogt and Paul, 2004; Li et al., 2004; Kamasawa et al., 2005; Orthmann-Murphy et al., 2007a). In mice, the double-deletion of oligodendrocyte Cx47 and Cx32 causes myelin vacuolation and demyelination (Menichella et al., 2003; Odermatt et al., 2003). However, whether loss of astrocyte GJs causes oligodendrocyte and myelin pathology has not been investigated.
To address this question, mice with global deletion of Cx30 (Teubner et al., 2003) and floxed Cx43 (Theis et al., 2003) were crossed with mice expressing Cre recombinase under the mouse GFAP promoter (Garcia et al., 2004; Herrmann et al., 2008). Within CNS white matter in these animals, widespread edema and vacuolation affected oligodendrocytes, myelin, and astrocytes, whereas gray matter pathology was restricted to hippocampal CA1 astrocytes.
Pathology was only noted in dKO mice. Expression of a single allele of either Cx43 or Cx30 was sufficient to protect against glial edema, indicating that these connexins are at least partially redundant. The ability of Cx30 to compensate for Cx43 indicates that protein-protein interactions mediated by the long cytoplasmic tail of Cx43 (Sorgen et al., 2004) are not essential for preventing cellular edema. Likewise, this compensation argues against a role for Cx43 hemichannels. Rather, we suggest that the function of Cx43 and Cx30 in limiting cellular edema is performed by a characteristic common to both Cx family members, such as GJ intercellular communication. The distinct pathologies observed in myelinated and nonmyelinated tissues may reflect specialized functions attributable to astrocyte:oligodendrocyte/myelin GJs versus astrocyte:astrocyte GJs. This is the first report of a structural phenotype in astrocytes and oligodendrocytes as a consequence of loss of astrocyte connexins.
Oligodendrocyte and myelin pathology in dKO mice
Affected white matter in adult dKOs included the corpus callosum, subcortical white matter, internal capsule, anterior commissure, cerebellar white matter, and corticospinal tracts. The principle finding in white matter of the dKO CNS was intracytoplasmic vacuolation of oligodendrocytes. Vacuoles were typically bound by single membranes. Although the etiology of these vacuoles is uncertain, they are unlikely to be of lysosomal origin (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Pathology was first noted at P23, a time-point coincident with the developmentally regulated expression of Cx30 (Dahl et al., 1996; Nagy et al., 1999) and with the loss of Cx43 in our dKO mice (Fig. 2). Aged animals (12 months) exhibited similar pathology, albeit slightly less severe.
The distribution and appearance of the myelin-associated vacuoles in Cx43/Cx30 dKO mice bore a striking resemblance to Cx47/Cx32 dKO mice, where vacuoles are located mostly in the periaxonal space (Menichella et al., 2003; Odermatt et al., 2003); to mice with compound haploinsufficiencies for Cx47, Cx32, and the inwardly rectifying potassium channel Kir4.1, where vacuoles develop in association with external layers of myelin (Menichella et al., 2006); and to Kir4.1−/− mice (Neusch et al., 2001). Both oligodendrocytes and astrocytes express Kir4.1 (Poopalasundaram et al., 2000; Higashi et al., 2001; Neusch et al., 2001). Furthermore, vacuolation in Cx47/Cx32 dKOs correlates with neuronal stimulation (Menichella et al., 2006). The similar pathology noted in these different animal models is consistent with a common role for oligodendrocyte/astrocyte GJs in spatial buffering of [K+] ions (and metabolites) released during neuronal activity.
Using immunostaining and Western blotting, we also detected a significant defect in oligodendrocyte maturation and hypomyelination without changes in oligodendrocyte cell number. Oligodendrocytes derived from Kir4.1−/− mice have also been noted to have a less mature phenotype (large soma, fewer oligodendrocytes elaborating myelin sheaths) without changes in total cell number (Neusch et al., 2001). Together with our findings, this raises the possibility that oligodendrocyte differentiation is influenced by astrocyte GJ communication.
Oligodendrocyte pathology might contribute to motor deficits detected on the rotarod assay, which is sensitive to pathology in the white matter (Kuhn et al., 1995). The ability to overcome initial deficits on the rotarod by dKO mice was similar to that noted in Cx43-deficient mice, indicating that upregulation of Cx30 does not compensate for the delayed motor learning (Frisch et al., 2003).
Gray matter pathology in dKO mice
In nonmyelinated tissues, edema was primarily restricted to astrocytes in the CA1 region of the hippocampus, and was not associated with loss of neurons or synaptic density. Astrocytes modify synaptic transmission by releasing bioactive nucleotides (“gliotransmitters”), neurotransmitter uptake, and synthesis of substrates for neuronal activity (Araque et al., 1999; Allen and Barres, 2005; Deitmer et al., 2006). Therefore, astrocyte dysfunction could impact efficacy of synaptic transmission in a behaviorally measurable way, without neuronal or synaptic loss. Our studies demonstrated impairment in hippocampal-dependent spatial learning, and are consistent with previous results in Cx30-null mice (Dere et al., 2003). Similar deficits have not been detected in astrocyte-targeted Cx43 KO mice (Frisch et al., 2003).
The restricted distribution of gray matter edema suggests that vulnerability of gray matter astrocytes varies depending on the microenvironment. In this regard, it is of interest to note that measurements of tetramethylammonium diffusion indicate that the CA1 region of the hippocampus has less extracellular volume than other hippocampal regions, potentially increasing local extracellular concentrations of ions such as K+ which are released during physiologic or pathologic neuronal activity (McBain et al., 1990; Pérez-Pinzón et al., 1995). We speculate that the pronounced swelling observed in dKO CA1 astrocytes represents an attempt to restore osmotic equilibrium against ionic concentrations that have exceeded a critical threshold. This would be consistent with the observation that dKO mice have increased hippocampal [K+]out and increased susceptibility to epileptiform events (Wallraff et al., 2006).
An alternative possibility is that the driving force for astrocytic edema in CA1 is the accumulation of glucose, or its metabolites, at these sites. In agreement with Rouach et al. (2008), we found immunoreactivity for Cx43 and Cx30 in the CA1 region of the hippocampus was enriched in perivascular endfeet (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material, and data not shown). Rouach et al. (2008) showed that astrocyte GJs at this site constitute the molecular basis for intercellular trafficking of blood-derived glucose and its metabolites, resulting in the release of lactate to neurons. Thus, in the absence of GJ connectivity, water could also follow an osmotic gradient formed by glucose trapped within the cytoplasm of perivascular astrocytes. The passive water channel aquaporin-4, which is the rate-limiting step in the development of intracellular edema (Yang et al., 2008), is also enriched on these astrocytic perivascular endfeet, and this did not change in the dKO mice (data not shown).
Astrocyte swelling has been well characterized in a number of diseases, including hepatic encephalopathy (Norenberg, 1977), traumatic brain injury (Bullock et al., 1991), and stroke (Kimelberg, 1995). These conditions are associated with high mortality rates, and effective therapies are limited. Intracellular edema also occurs in mouse models including water intoxication, focal cerebral ischemia (Manley et al., 2000; Feustel et al., 2004) and bacterial meningitis (Papadopoulos and Verkman, 2005). These scenarios are characterized by movement of water from the vasculature into the parenchyma across an intact blood brain barrier, leading to astrocyte swelling and decreased extracellular space. Consequences include pathologically elevated extracellular concentrations of [K+] and excitatory amino acids.
These results add information to the model of water/ion regulation in the brain, namely that astrocyte GJs are important direct or indirect contributors to water regulation. These data are in agreement with the growing consensus that astrocytes play important roles in modulating brain function. Our findings provide compelling evidence that astrocyte GJs have an essential role in maintaining astrocytic and oligodendrocytic structural and functional integrity.
Footnotes
This investigation was supported by the National Multiple Sclerosis Society (Grants RG3827A5/1 to C.F.B. and RG1001/K11 to C.S.R.) and the National Institutes of Health (Grants NS 08952 and NS 11920 to C.S.R.). C.S.R. is the Wollowick Family Foundation Professor in Multiple Sclerosis Research. We thank Michael Sofroniew for mGFAP-Cre mice and Klaus Willecke for Cx43fl/fl Cx30−/− mice. We thank Eliseo Eugenin for helpful discussion. Data in this study are from a thesis to be submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.
References
- Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15:542–548. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Brand-Schieber E, Werner P, Iacobas DA, Iacobas S, Beelitz M, Lowery SL, Spray DC, Scemes E. Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J Neurosci Res. 2005;80:798–808. doi: 10.1002/jnr.20474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock R, Maxwell WL, Graham DI, Teasdale GM, Adams JH. Glial swelling following human cerebral contusion: an ultrastructural study. J Neurol Neurosurg Psychiatry. 1991;54:427–434. doi: 10.1136/jnnp.54.5.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Manthey D, Chen Y, Schwarz HJ, Chang YS, Lalley PA, Nicholson BJ, Willecke K. Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J Biol Chem. 1996;271:17903–17910. doi: 10.1074/jbc.271.30.17903. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, McCarthy KD, Scemes E, Giaume C. Information processing and transmission in glia: calcium signaling and transmitter release. Glia. 2006;54:639–641. doi: 10.1002/glia.20428. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Frisch C, Teubner B, Söhl G, Willecke K, Huston JP. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur J Neurosci. 2003;18:629–638. doi: 10.1046/j.1460-9568.2003.02784.x. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy HS, John GR, Lee SC, Brosnan CF, Spray DC. Reciprocal regulation of the junctional proteins claudin-1 and connexin43 by interleukin-1beta in primary human fetal astrocytes. J Neurosci. 2000;20:RC114, 1–6. doi: 10.1523/JNEUROSCI.20-23-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Aggleton JP. Spontaneous recognition of object configurations in rats—effects of fornix lesions. Exp Brain Res. 1994;100:85–92. doi: 10.1007/BF00227281. [DOI] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y, Kimelberg HK. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 2004;35:1164–1168. doi: 10.1161/01.STR.0000124127.57946.a1. [DOI] [PubMed] [Google Scholar]
- Frisch C, Theis M, De Souza Silva MA, Dere E, Söhl G, Teubner B, Namestkova K, Willecke K, Huston JP. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur J Neurosci. 2003;18:2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Hansson E. Metabotropic glutamate receptor activation induces astroglial swelling. J Biol Chem. 1994;269:21955–21961. [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, Spray DC, Brosnan CF. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci U S A. 1999;96:11613–11618. doi: 10.1073/pnas.96.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Electrical coupling between astrocytes and between oligodendrocytes studied in mammalian cell cultures. Glia. 1988;1:64–73. doi: 10.1002/glia.440010108. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Current concepts of brain edema—review of laboratory investigations. J Neurosurg. 1995;83:1051–1059. doi: 10.3171/jns.1995.83.6.1051. [DOI] [PubMed] [Google Scholar]
- Kuhn PL, Petroulakis E, Zazanis GA, McKinnon RD. Motor function analysis of myelin mutant mice using a rotarod. Int J Dev Neurosci. 1995;13:715–722. doi: 10.1016/0736-5748(96)81215-9. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Hayzoun K, Selimi F, Mariani J, Strazielle C. Motor coordination in mice with hotfoot, Lurcher, and double mutations of the Grid2 gene encoding the delta-2 excitatory amino acid receptor. Physiol Behav. 2003;80:333–339. doi: 10.1016/j.physbeh.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KG, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Lou N, Kang N, Takano T, Hu F, Han X, Xu Q, Lovatt D, Torres A, Willecke K, Yang J, Kang J, Nedergaard M. A central role of connexin 43 in hypoxic preconditioning. J Neurosci. 2008;28:681–695. doi: 10.1523/JNEUROSCI.3827-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton PR. Principles and practices of unbiased stereology: an introduction for bioscientists. Baltimore: The Johns Hopkins UP; 2002. [Google Scholar]
- Mumby DG, Gaskin S, Glenn MJ, Schramek TE, Lehmann H. Hippocampal damage and exploratory preferences in rats: memory for objects, places, and contexts. Learn Mem. 2002;9:49–57. doi: 10.1101/lm.41302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Patel D, Ochalski PA, Stelmack GL. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999;88:447–468. doi: 10.1016/s0306-4522(98)00191-2. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE. Connexin26 in adult rodent central nervous system: Demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol. 2001;441:302–323. doi: 10.1002/cne.1414. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Nakase T, Söhl G, Theis M, Willecke K, Naus CC. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–2075. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford UP; 1995. pp. 717–731. [Google Scholar]
- Norenberg MD. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest. 1977;36:618–627. [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci. 2007a;34:629–641. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007b;27:13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906–13912. doi: 10.1074/jbc.M413627200. [DOI] [PubMed] [Google Scholar]
- Pérez-Pinzón MA, Tao L, Nicholson C. Extracellular potassium, volume fraction, and tortuosity in rat hippocampal CA1, CA3, and cortical slices during ischemia. J Neurophysiol. 1995;74:565–573. doi: 10.1152/jn.1995.74.2.565. [DOI] [PubMed] [Google Scholar]
- Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–372. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia. 2009;57:207–221. doi: 10.1002/glia.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe WA, Messersmith E, Meyer-Franke A, Wipke B, Karlik SJ. Connexin 43 gap junction proteins are up-regulated in remyelinating spinal cord. J Neurosci Res. 2007;85:945–953. doi: 10.1002/jnr.21194. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Sato T, Machida T, Takahashi S, Murase K, Kawano Y, Hayashi T, Iyama S, Takada K, Kuribayashi K, Sato Y, Kobune M, Takimoto R, Matsunaga T, Kato J, Niitsu Y. Apoptosis supersedes necrosis in mitochondrial DNA-depleted Jurkat cells by cleavage of receptor-interacting protein and inhibition of lysosomal cathepsin. J Immunol. 2008;181:197–207. doi: 10.4049/jimmunol.181.1.197. [DOI] [PubMed] [Google Scholar]
- Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, Frisch C, Söhl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Söhl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–195. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiencken-Barger AE, Djukic B, Casper KB, McCarthy KD. A role for Connexin43 during neurodevelopment. Glia. 2007;55:675–686. doi: 10.1002/glia.20484. [DOI] [PubMed] [Google Scholar]
- Yang B, Zador Z, Verkman AS. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem. 2008;283:15280–15286. doi: 10.1074/jbc.M801425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Rivieccio MA, Lutz S, Scemes E, Brosnan CF. The TLR3 ligand polyI: C downregulates connexin 43 expression and function in astrocytes by a mechanism involving the NF-kappaB and PI3 kinase pathways. Glia. 2006;54:775–785. doi: 10.1002/glia.20418. [DOI] [PMC free article] [PubMed] [Google Scholar]