

Abstract
We studied meropenem in 23 pre-term (gestational age, 29 to 36 weeks) and 15 full-term (gestational age, 37 to 42 weeks) neonates. Meropenem doses of 10, 20, and 40 mg/kg were administered as single doses (30-min intravenous infusion) on a random basis. Blood was obtained for determining the meropenem concentration nine times. Each child required other antimicrobials for proven/suspected bacterial infections. Samples were assayed by high-performance liquid chromatography analysis. Population pharmacokinetic parameter values were obtained by employing the BigNPAG program. Model building was performed by the likelihood ratio test. The final model included estimated creatinine clearance (CLcr) (Schwartz formula) and weight (Wt) in the calculation of clearance (meropenem clearance = 0.00112 × CLcr + 0.0925 × Wt + 0.156 liter/hr). The overall fit of the model to the data was good (observed = 1.037 × predicted − 0.096; r2 = 0.977). Given the distributions of estimated creatinine clearance and weight between pre-term and full-term neonates, meropenem clearance was substantially higher in the full-term group. A Monte Carlo simulation was performed using the creatinine clearance and weight distributions for pre-term and full-term populations separately, examining 20- and 40-mg/kg doses, 8- and 12-h dosing intervals, and 0.5-h and 4-h infusion times. The 8-h interval produced robust target attainments (both populations). If more resistant organisms were to be treated (MIC of 4 to 8 mg/liter), the 40-mg/kg dose and a prolonged infusion was favored. Treating clinicians need to balance dose choices for optimizing target attainment against potential toxicity. These findings require validation in clinical circumstances.
In many countries, pre-term infants are frequently admitted to the neonatal intensive care unit. Because of this, they frequently suffer infections with nosocomially acquired organisms (15, 24, 25). Meropenem, because of its broad-spectrum activity, which includes the majority of nosocomially acquired pathogens, would be an agent of great utility in these circumstances.
Unfortunately, little information is available regarding the disposition of antimicrobial agents in the neonatal patient population (22). This information is critical if the clinician is to be able to prescribe the correct dose and schedule of an antimicrobial agent that will minimize toxicity and maximize the probability of a good outcome. For β-lactam antibiotics, the work of a number of laboratories has indicated that the time that plasma concentrations exceed the MIC of the pathogen is the pharmacodynamic variable most closely linked to clinical outcome (10). Therefore, the dosing interval which is selected for antimicrobial administration will have a major impact on the adequacy of the dose chosen.
Newborns are well known to have clearances of drugs which differ from those seen in children and adults (2). Much of this difference is attributable to the maturation of both renal and nonrenal clearance pathways. Furthermore, volumes of distribution (V) tend to be larger, when adjusted for weight, in newborns than those seen in older children and adults (2). Consequently, any evaluation of a new agent which does not take into account physiological differences of newborn infants relative to older children or adults will not be able to provide optimal information to the clinician that will allow the choice of the proper dosing interval to maximize the time above the MIC (Time>MIC).
Because meropenem is such a potentially valuable agent for the neonatally infected group, we decided to examine both full-term and pre-term infants for their dispositions towards meropenem. We report here the single-dose pharmacokinetic parameters seen after doses of 10, 20, and 40 mg/kg of body weight of meropenem in both pre- and full-term newborns.
MATERIALS AND METHODS
Patients and study design.
Thirty-eight newborn infants were studied at two centers: the Neonatal Intensive Care Units of the Erasmus MC-Sophia Children's Hospital, Rotterdam, The Netherlands, and Charles University, Prague, Czech Republic. For inclusion in this clinical investigation, potential participants were required to meet the following criteria. (i) Participants had to be hospitalized pre-term and full-term neonates in their first 4 weeks of life who had received a minimum of 48 h of antibiotic therapy for a known or presumed bacterial infection. (ii) The subject's overall condition needed to be good or fair. (iii) Parents or legal guardians needed to have given written informed consent. (iv) The participants were not to meet any of the following exclusion criteria: (a) so severely ill that they were not likely to survive the duration of the trial; (b) born to mothers who are known or suspected to be positive for human immunodeficiency virus or have hepatitis; (c) born to mothers who are addicted to drugs or alcohol; (d) having a major congenital abnormality; (e) having a known history of immediate hypersensitivity to any β-lactam antibiotic; (f) having seizures within the previous 3 days; (g) having unconjugated bilirubin concentrations sufficiently high enough to warrant exchange transfusion; (h) having received ceftriaxone or cefotoxin (known to interfere with the meropenem assay) within the previous 3 days; (i) having plasma creatinine values of >140 μmol/liter; (j) having received a systemic investigational drug; or (k) having any condition which in the opinion of the investigator made the subject unsuitable for enrollment. All studied infants continued to receive their standard antibiotic regimens. In addition, a single dose of meropenem was administered as a 30-min intravenous infusion. The neonates were divided into a pre-term group (29 to 36 weeks of gestational age; n = 23) and a group of full-term infants (37 to 42 weeks of gestational age; n = 15). Pre-term and full-term infants were further randomly subdivided into three subgroups each. Each of the three groups was administered one of the following doses: 10 mg/kg, 20 mg/kg, or 40 mg/kg of meropenem.
Informed consent.
Written informed consent was obtained from the parents of the infants according to institutional guidelines.
Sampling scheme and sample handling.
Blood samples (200 μl) were collected immediately before and at the following time points after the start of the infusion: 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, and 24 h. Blood samples were collected into NH4-heparinized tubes and shaken slightly. Blood samples were kept cold (4°C) for at least 3 but no more than 15 min under centrifugation (4°C for 10 min at 3,500 rpm). The plasma was split and transferred into two plastic tubes. Samples were quickly frozen on dry ice and maintained on dry ice until stored at −70°C.
Assay of meropenem in plasma and urine.
The concentration of meropenem in human plasma was determined by a precise high-performance liquid chromatography procedure. The validation procedure followed international guidelines.
Briefly, an aliquot of plasma (30 μl) was stabilized by an addition of 150 μl of 0.05 M sodium phosphate buffer (pH 7.09) containing the internal standard (cefpodoxime) and was deproteinated by adding 200 μl of acetonitrile. After vigorously mixing and centrifuging the samples, the acetonitrile was removed by extraction with 500 μl of dichloromethane. After centrifugation, 100 μl of the aqueous phase of each sample was injected onto the high-performance liquid chromatography system. The chromatographic separation of meropenem from endogenous material was performed and an internal standard was determined on a Spherisorb ODS2 column (5 μm, 250 × 4.6 mm), by using an isocratic solvent system consisting of 0.05 M sodium dihydrogen phosphate buffer (pH 5.08) and acetonitrile (90:10, vol/vol).
The retention times of meropenem and cefpodoxime were 9.0 and 12.8 min, respectively. The eluent was monitored by UV absorption at 296 nm. The Turbochrom 3 (version 3.2, 1991; PE Nelson, Cupertino, CA) software was used for evaluation of the chromatograms. Calibration was performed by weighted (1/concentration) linear regression. The flow rate was 0.6 ml/min.
The meropenem concentrations of 390 plasma samples from the study were measured in a total of 15 sequences. Standard curves were prepared in blank human plasma. The standard curve was linear between 0.0455 and 203 μg/ml. The limit of quantitation was 0.0455 μg/ml. Fifty-one of the 390 samples were below the limit of quantitation. The between-day coefficient of variation of the spiked quality control samples ranged from 6.2 to 9.9%.
Pharmacokinetic methods.
Population pharmacokinetic parameter estimation for meropenem was performed using the NPAG (non-parametric adaptive grid with adaptive γ) program package developed by Leary et al. (20). This program provides maximum-likelihood estimates of the population mean pharmacokinetic parameter values and their distributions without making assumptions as to the shape of the underlying distributions. One- and two-compartment open models with zero-order input and first-order elimination from the central compartment were evaluated. For observation weighting, the inverse of the assay variance was employed as an approximation of the total observation variance. This initial estimate was multiplied by a scalar (γ), which was iteratively estimated to provide the best estimate of overall observation variance. The system was parameterized as the V of the central compartment, the plasma clearance, and the intercompartmental transfer rate constants.
Population analyses were performed for the population as a whole. Bayesian posterior estimates of pharmacokinetic parameter values were derived for each patient using that patient's plasma samples and the probability density function derived from the full population analysis.
We examined the dependence of total meropenem plasma clearance and V on a number of demographic population descriptors. These descriptors included sex, gestational age, weight (kg), heel-to-crown length (cm), creatinine clearance (calculated according to the method of Schwartz et al. [23]), dose, and pre-term versus full-term status. This analysis was performed using the generalized linear module of the SYSTAT for Windows program (v. 11.0). Analyses were performed both stepwise forward and stepwise backward. The level of significance was set at an alpha equal to 0.05. This allowed us to examine the covariates to be evaluated within the population model.
These analyses were performed by expanding the base pharmacokinetic model. The significance of model expansion was determined by the likelihood ratio test (twice the log-likelihood difference between the base and the expanded model, examined against a χ2 distribution with the appropriate number of degrees of freedom).
In order to evaluate possible doses and schedules for recommendations for pre-term and full-term infants, we employed the technique of Monte Carlo simulation and evaluation of target attainment rates by MIC, as described previously (13). The ADAPT II package of programs of D'Argenio and Schumitzky was employed for Monte Carlo simulation (6). Both normal and log-normal distributions were evaluated. The choice between distributions was made by determining the fidelity with which the starting parameter values and their dispersions were recapitulated by the 9,999-subject simulation.
The simulations were carried out with the parameter values and creatinine clearance and weight distributions for the full-term and pre-term infant populations considered separately.
RESULTS
Study population and clinical observations.
All 38 newborn infants, both pre-term and full-term, tolerated the single-dose infusion of meropenem well. No drug-related adverse events were identified. The demographic data of the 38 infants studied are presented in Tables 1 and 2. There were 23 pre-term and 15 full-term newborn infants studied. The pre-term infants consisted of 13 males and 10 females. Gestational ages ranged from 29 to 36 weeks with weights ranging from 952 to 2,830 g and the crown-heel length ranging from 36 to 47.5 cm. Postnatal ages ranged from 2 to 28 days of life. Creatinine clearance, calculated according to the method of Schwartz et al. (30), ranged from 13.3 to 34.2 ml/min/1.73 m2. Nine pre-term infants received 10 mg/kg of meropenem, while 8 received 20 mg/kg and 6 received 40 mg/kg of meropenem. The full-term infants consisted of 10 males and 5 females. Their gestational ages, weights, and crown-heel lengths ranged from 37 to 42 weeks, 2,340 to 4,050 g, and 43 to 53 cm, respectively. Postnatal ages ranged from 2 to 14 days of life. The creatinine clearances in this group, calculated according to the method of Schwartz et al. (30), ranged from 20.1 to 60.3 ml/min/1.73 m2. In this group, five infants received 10 mg/kg of meropenem, while five received 20 mg/kg and five received 40 mg/kg of meropenem. The antibiotics which were coadministered to the infants on the day they received meropenem are as follows: amoxicillin, ampicillin, azlocillin, cefuroxime, cefotaxime, ceftazidime, oxacillin, and ticarcillin (β-lactams); amikacin, gentamicin, and tobramycin (aminoglycosides); and lincomycin and vancomycin (others). Other medications that were coadministered include caffeine, dobutamine, furosemide, hydrocortisone, indomethacin, and phenobarbital. While there are a number of drugs which could potentially interfere with the tubular secretion of meropenem, the administration of meropenem was separated from the administration of potentially interfering drugs as much as possible. Further, it should be recognized that this is a particularly difficult patient population to study, and they were studied while undergoing treatment for an infection. The agents that we felt to be at the highest likelihood of interfering with meropenem pharmacokinetics are as follows: amoxicillin, ampicillin, azlocillin, cefuroxime, cefotaxime, ceftazidime, oxacillin, ticarcillin, and furosemide.
TABLE 1.
Demographic data and sequence of treatments of hospitalized pre-term neonates
| Center/patient no. | Sex | Gestational age (wk) | Wt (kg) | Crown-heel length (cm) | CLcra (ml/min/ 1.73 m2) | Dosing (mg/kg) |
|---|---|---|---|---|---|---|
| 0001/0001 | M | 34 | 2.83 | 47.0 | 28.6 | 40 |
| 0001/0003 | F | 36 | 2.50 | 47.0 | 21.1 | 10 |
| 0001/0004 | M | 35 | 2.10 | 47.5 | 24.3 | 40 |
| 0001/0005 | F | 31 | 1.62 | 42.0 | 22.3 | 20 |
| 0001/0006 | M | 34 | 2.44 | 47.0 | 24.5 | 10 |
| 0001/0007 | F | 30 | 1.23 | 37.0 | 24.5 | 20 |
| 0001/0009 | F | 31 | 1.72 | 39.0 | 24.7 | 10 |
| 0001/0010 | M | 30 | 1.69 | 43.0 | 13.3 | 20 |
| 0001/0011 | F | 34 | 1.86 | 42.0 | 26.6 | 40 |
| 0001/0012 | M | 30 | 1.51 | 39.0 | 24.2 | 10 |
| 0001/0013 | F | 30 | 1.64 | 39.0 | 24.7 | 10 |
| 0001/0014 | F | 31 | 1.87 | 41.0 | 34.2 | 20 |
| 0001/0015 | M | 36 | 2.47 | 45.0 | 31.3 | 40 |
| 0001/0016 | F | 35 | 1.96 | 43.0 | 19.6 | 10 |
| 0001/0017 | M | 35 | 2.52 | 45.0 | 22.3 | 20 |
| 0002/0001 | M | 31 | 1.10 | 40.5 | 18.5 | 20 |
| 0002/0002 | M | 31 | 1.55 | 42.5 | 15.1 | 40 |
| 0002/0003 | M | 35 | 2.73 | 46.0 | 29.8 | 10 |
| 0002/0004 | M | 32 | 1.79 | 45.0 | 29.2 | 20 |
| 0002/0005 | M | 30 | 1.20 | 39.0 | 15.4 | 10 |
| 0002/0006 | F | 31 | 1.50 | 46.0 | 16.2 | 40 |
| 0002/0007 | F | 29 | 0.952 | 36.0 | 17.2 | 10 |
| 0002/0008 | M | 32 | 2.20 | 44.0 | 25.7 | 20 |
| Mean | 32 | 1.87 | 42.7 | 23.2 | ||
| SD | 2 | 0.53 | 3.43 | 5.53 | ||
| Min | 29 | 0.952 | 36.0 | 13.3 | ||
| Max | 36 | 2.83 | 47.5 | 34.2 |
The creatinine clearance was calculated as follows: CLcr = 0.45·crown-heel length/plasma creatinine (2).
TABLE 2.
Demographic data and sequence of treatments of hospitalized full-term neonates
| Center/patient no. | Sex | Gestational age (wk) | Wt (kg) | Crown-heel length (cm) | CLcra (ml/min/ 1.73 m2) | Dosing (mg/kg) |
|---|---|---|---|---|---|---|
| 0001/0101 | M | 40 | 3.26 | 52.0 | 51.7 | 40 |
| 0001/0102 | F | 39 | 3.02 | 47.0 | 60.3 | 20 |
| 0001/0103 | M | 38 | 3.42 | 53.0 | 39.8 | 10 |
| 0001/0104 | M | 39 | 3.22 | 53.0 | 39.8 | 20 |
| 0001/0105 | M | 38 | 3.02 | 47.0 | 41.6 | 40 |
| 0001/0106 | M | 38 | 2.82 | 49.0 | 40.6 | 10 |
| 0001/0107 | F | 41 | 3.56 | 51.0 | 56.4 | 20 |
| 0001/0108 | M | 38 | 3.19 | 49.0 | 41.5 | 10 |
| 0001/0109 | F | 39 | 3.15 | 49.0 | 48.7 | 40 |
| 0001/0110 | F | 40 | 4.00 | 52.0 | 48.1 | 10 |
| 0001/0111 | M | 39 | 2.95 | 48.0 | 49.0 | 40 |
| 0001/0112 | M | 39 | 4.05 | 50.0 | 34.3 | 20 |
| 0002/0101 | M | 42 | 2.47 | 49.0 | 55.7 | 40 |
| 0002/0102 | F | 37 | 2.34 | 43.0 | 20.1 | 20 |
| 0002/0103 | M | 38 | 3.12 | 50.0 | 31.6 | 10 |
| Mean | 39 | 3.17 | 49.5 | 43.9 | ||
| SD | 1 | 0.47 | 3 | 10.5 | ||
| Min | 37 | 2.34 | 43 | 20.1 | ||
| Max | 42 | 4.05 | 53 | 60.3 |
The creatinine clearance was calculated as follows: CLcr = 0.45·crown-heel length/plasma creatinine (2).
The mean plasma concentration-time curves for meropenem for the 10-, 20-, and 40-mg/kg doses for pre-term infants are displayed in Fig. 1A and data for the full-term infants are in Fig. 1B. The peak concentrations are dose-proportional for the pre-term and full-term newborns at concentrations of 19.3, 42.9, and 73.8 mg/liter for the 10-, 20-, and 40-mg/kg doses in the pre-term infants. For the full-term infants, these values were 21.7, 40.6, and 59.2 mg/liter.
FIG. 1.

(A) Mean (for 10 mg/kg, n = 9; for 20 mg/kg, n = 8; for 40 mg/kg, n = 6) plasma concentrations of meropenem of hospitalized pre-term neonates after a 30-min infusion of 10, 20, or 40 mg/kg of meropenem. (B) Mean (for 10 mg/kg, n = 5; for 20 mg/kg, n = 5; for 40 mg/kg, n = 4) plasma concentrations of meropenem of hospitalized full-term neonates after a 30-min infusion of 10, 20, or 40 mg/kg of meropenem.
Demographic modeling of pharmacokinetic parameter values.
The base model without descriptors was employed to calculate Bayesian estimates for each of the children studied. We then explored different demographic covariates to help us in the generation of the final population pharmacokinetic model with demographic covariates. The general linear model module of the SYSTAT package was used to model some appropriate demographic variables to determine their influence on plasma clearance and V.
For meropenem plasma clearance, the demographic variables chosen as descriptors were as follows:
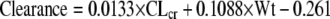 |
(1) |
The resulting meropenem plasma clearance estimate is in liters/h. The creatinine clearance (CLcr) estimate is in units of ml/min/1.73 m2 (estimated by the Schwartz formula [23]), and weight (Wt) is in kg. It should be noted that weight along with height are included in the Schwartz formula. Nevertheless, the estimator was able to find weight as affecting clearance in addition to the Schwartz formula-estimated creatinine clearance. Overall, the regression was highly statistically significant. The P value for CLcr was 0.00017 and for Wt was 0.036. The regression relationship explained 67.9% of the variance (i.e., r2 = 0.679). The relationship was the same when performed stepwise forward or stepwise backward.
For V, the physiological/demographic variable chosen was CLcr.
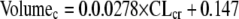 |
(2) |
In the equation, volume is in liters and CLcr is in units of ml/min/1.73 m2. Again, the relationship was highly statistically significant, with a P of <0.0001. The relationship explained 35.2% of the variance (r2 = 0.352). The general linear model procedure identified a larger model (including weight) stepwise backward but only CLcr stepwise forward. To be conservative, we employed the stepwise forward procedure.
Population modeling.
The population model for all infants (n = 38), performed without covariates, had a maximum-likelihood score of −655.7. A general linear model was developed, relating both volume of the central compartment and clearance to covariates (see above). Volume was related to estimated creatinine clearance. Clearance was related to weight and estimated creatinine clearance.
Population analyses were performed with the volume and clearance related to the covariates as indicated below:
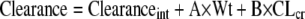 |
(3) |
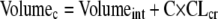 |
(4) |
The likelihood scores for the base model and the expanded models are shown in Table 3. The fit of the full model to the data was good. After the Bayesian step, the r2 was 0.977; the observed meropenem concentrations were calculated as follows: 1.037 × predicted meropenem concentrations − 0.096. Bias was −0.154 mg/liter; precision was 1.003 (mg/liter)2. This is displayed in Fig. 2.
TABLE 3.
Likelihood scores of competing modelsa
| Model | Likelihood score | 2× likelihood difference | df | P value |
|---|---|---|---|---|
| Base model | −655.7 | |||
| Wt and CLcr (in clearance) | −648.8 | 13.8 | 2 | <0.001 |
| CLcr (in vol) | −648.4 | 0.8 | 1 | NS |
df, degrees of freedom; NS, not significant.
FIG. 2.

Predicted observed plot for meropenem concentrations after the Bayesian step for the total population, using the final model (CLcr and weight in meropenem clearance—see Table 3).
The fit of the full model prior to the Bayesian step was also quite acceptable. The r2 was 0.742. Observed meropenem concentrations were calculated as follows: 0.917 × predicted meropenem concentrations + 3.748. Bias was −0.259 mg/liter; precision was 18.152 (mg/liter)2.
The final parameter values for the integrated analysis of all 38 neonates are presented in Table 4.
TABLE 4.
Final model pharmacokinetic parameter values of meropenem in hospitalized pre-term and full-term neonatesa
| Characteristic | Vol (liter) | Kcp (h−1) | Kpc (h−1) | CLSLPcr (liter/h/ml/min/1.73 m2) | CLSLPWt (liter/h/kg) | CL-INT (liter/h) |
|---|---|---|---|---|---|---|
| Mean | 0.969 | 7.71 | 30.0 | 0.00112 | 0.0925 | 0.156 |
| Median | 0.883 | 5.862 | 41.6 | 0.00510 | 0.0918 | 0.172 |
| Mode | 0.896 | 0.100 | 44.8 | 0.00510 | 0.0460 | 0.0248 |
| SD | 0.527 | 6.67 | 17.9 | 0.00151 | 0.0606 | 0.122 |
The parameter values are for the analysis of both pre- and full-term neonates. Simulations for the two subpopulations employ the separate weight and estimated creatinine clearance distributions which are shown in Tables 1 and 2. Vol, volume of the central compartment; Kcp and Kpc, first-order intercompartmental transfer rate constants; CLSLPcr, the clearance slope term with estimated creatinine clearance; CLSLPWt, the clearance slope term with weight; CL-INT, the clearance intercept term. The estimate for meropenem clearance from the mean values is given by the following equation: CL = 0.156 + 0.00112·CLcr + 0.0925·Wt.
More information regarding the V and plasma clearance can be obtained by examining the ranges observed for these parameters after the Bayesian step for the two populations. For the V, the 20% to 80% bounds on volume for the pre-term and full-term groups are 0.44 to 0.98 and 0.82 to 1.54 liters, showing considerable overlap. However, when one examines the distributions for clearance, the full-term children have a 20% to 80% range of 0.414 to 0.753 liters/h, whereas for pre-term children, this range is 0.253 to 0.398 liters/h. It is clear that the two subpopulations differ significantly in their clearances of meropenem. Full-term infants have a substantially larger plasma clearance for meropenem than do pre-term infants, as could be predicted from the increased renal clearing capacity of the full-term newborn.
Monte Carlo simulation and target attainment rates by MIC.
The log-normal distribution best regenerated the point estimates of the mean parameter values and the estimates of dispersion. For carbapenem antibiotics, organism maximal cell kill occurs when the Time>MIC of free drug meets or exceeds 40% of the dosing interval (11). Thus, 40% Time>MIC of free drug served as the target in our simulation. The target attainment rates for a dose of 20 or 40 mg/kg intravenously every 8 or 12 h with constant-rate infusions of 0.5 h and 4 h are presented in Fig. 3A (20 mg/kg) and B (40 mg/kg) for the pre-term infants and in Fig. 3C (20 mg/kg) and D (40 mg/kg) for the full-term infants. As can be seen in the figure, maximal cell kill is clearly improved by the use of a prolonged (4-h) infusion. The combination of a 40-mg/kg dose and a prolonged infusion produces target attainments in excess of 90% out to an MIC of 8 mg/liter for both the pre-term and full-term groups. More recently, Tam et al. (26) have demonstrated that carbapenem trough concentrations need to be 1.7 times the MIC to minimize resistant mutant amplification in Pseudomonas aeruginosa when meropenem is administered along with an aminoglycoside and 6.2 times the MIC to suppress resistance when administered alone. Using these as a metric, we present the likelihood of attaining a resistance suppression profile with meropenem at a dose of 40 mg/kg/day every 8 h with a 4-h infusion. This clearly demonstrates the difference between 12- and 8-h dosing intervals and also between pre-term and full-term populations.
FIG. 3.


Meropenem target (40% Time>MIC − maximal kill) attainment for a 20-mg/kg dose (A) or 40-mg/kg dose (B) every 8 or 12 h in pre-term infants. These doses are displayed for full-term infants in panels C and D.
DISCUSSION
Neonatal sepsis remains one of the main causes of mortality and morbidity of newborn infants admitted to a neonatal intensive care unit (1, 14, 16, 18). Furthermore, invasive infections such as pneumonia, meningitis, and necrotizing enterocolitis threaten the newborn infant.
Meropenem is distributed in extracellular water and is excreted mainly by glomerular filtration. Therefore, changes in body water and development of renal function influence the disposition of meropenem. Meropenem has a larger V and lower clearance in premature neonates, and even more so compared to adults, and dosing regimens thus have to be adjusted accordingly. Improvement of clearance follows the gestational age- and postnatal age-dependent increases in the glomerular filtration rate (9). Other factors in the neonatal intensive care unit that directly influence V or renal function, such as extracorporeal membrane oxygenation or exposure to indomethacin, were shown to significantly alter the neonatal pharmacokinetics of other primarily renally excreted drugs (7, 8, 9, 27).
Meropenem pharmacokinetics have been reported in seven pre-term neonates, showing a V of 0.74 (range, 0.24 to 1.2) liters/kg (30). The half-life was circa 3.4 h, which is substantially longer than the 1-h rate in adults (21). From the previous study, those authors concluded that, because of the increased half-life, a two-times-daily meropenem dose of 15 mg/kg would suffice. Most recently, Bradley and colleagues (4) studied 37 neonates and administered single doses of 10 and 20 mg/kg of meropenem as a 30-min infusion. These authors demonstrated that an 8-h interval may be more appropriate for organisms with higher MICs. They found that a 20-mg/kg, 8-h dose would provide robust coverage for their patients but that 40 mg/kg may be necessary for some infections with more resistant pathogens, like Pseudomonas aeruginosa. It should be noted, however, that these data were based on very sparse sampling, where any one patient had three blood samples obtained on one of two schedules. In contrast, the patients reported here had seven to nine samples obtained for analysis.
Here we report the results of a single-dose pharmacokinetic study of 38 newborn infants (23 pre-term and 15 full-term) using three different doses (10, 20, and 40 mg/kg) of meropenem intravenously. Because of the limits on blood withdrawal and because of ethical considerations, it was only possible to perform this study on newborns that were already infected and required antimicrobial therapy. Because of this, it is clear that there may have been some competition, particularly at the renal tubular level between drugs being administered at approximately the same period. We attempted to minimize such an interaction by not administering the meropenem at a time when concentrations of any other competing agent might be high. Nevertheless, it must be understood that some interference may have taken place. However, it should also be understood that the pharmacokinetics of meropenem were determined in a clinically realistic setting.
The population modeling that we undertook demonstrated clearly that meropenem V values were in the range of those reported previously for other agents of the β-lactam class. Examination of the probability distribution of the volume for the pre-term versus the full-term groups of newborns showed that the means were similar and that the full-term infants, somewhat surprisingly, were more variable in their V.
The clearances were significantly different in the two groups. As can be seen by examining Tables 1 and 2, pre-term infants had calculated creatinine clearances which were clearly less than those seen in the full-term neonatal group. This is to be expected on the basis of their gestational age (19, 27, 28, 29). Indeed, when one compares the probability distribution of clearance for the full population versus each of the two subpopulations, it is clear that there is at most a 15 to 20% overlap between the pre-term and full-term groups. We hypothesized that this nonoverlap of meropenem plasma clearance was due, mainly, to the differences in estimated creatinine clearances in the two patient populations.
These between-group differences need to be taken into account when designing dosing regimens that would have a high likelihood of being efficacious in the empirical therapy setting. Multiple investigators (5, 13) have demonstrated that Time>MIC is the pharmacodynamic variable most closely linked to outcome for β-lactam antibiotics. Consequently, it is important to be able to make dosing recommendations which the clinician can employ at the bedside to generate concentrations of meropenem in the plasma which remain above the MIC of clinically important pathogens for a relatively high percentage of the dosing interval.
Monte Carlo simulation combined with target attainment rate analysis has been introduced as a way of rationally evaluating dose and schedule as well as setting MIC breakpoints (3), and it has been reviewed for its applicability to children (17). This approach has been prospectively validated in a number of circumstances, both for bacteria and viruses (3, 5, 12). We evaluated meropenem at a prospective dose and schedule of 20 and 40 mg/kg every 8 and 12 h using the pre-term and full-term population pharmacokinetics (Fig. 3). Maintaining free-drug concentrations (meropenem is approximately 2% bound) of >MIC for 40% of a dosing interval achieves a maximal cell kill (11). Examination of Fig. 3 demonstrates that a dose of 40 mg/kg with an 8-h dosing interval produces target attainment rates of >90% for achieving maximal cell kill at 8 mg/liter for both pre-term and full-term infants. Use of a prolonged (4-h) infusion markedly improves target attainment rates for the 20-mg/kg dose. Obviously, antimicrobial chemotherapy is a balance between efficacy and toxicity. Meropenem has little in the way of concentration-dependent toxicity, but the balance should be explicitly judged when the decision for a 20-mg/kg versus a 40-mg/kg dose is to be made.
As indicated above, previous work in a smaller number of neonates (30) resulted in a recommendation for a 12-h dosing interval. It is likely that such a recommendation would be successful for infections where non-pseudomonal or non-Acinetobacter isolates were being treated, where MICs would be highly likely to be ≤2 mg/liter. The circumstance of the infection becomes clear after pathogen identification. It may be prudent, then, to choose an 8-h interval until the pathogen is identified and an MIC is obtained.
These data indicate that 20 to 40 mg/kg with an 8-h dosing interval should provide robust coverage for the vast majority of nosocomially acquired pathogens seen in this population. If the 20-mg/kg dose is chosen, consideration should be given to the prolonged (4-h) infusion. Care needs to be exercised for clinicians in settings where there is a high likelihood of methicillin-resistant Staphylococcus aureus, as meropenem would not be adequate empirical therapy for this pathogen. In addition, meropenem has been given to children at doses of 40 mg/kg every 8 h and has been well tolerated (empirical treatment of meningitis). The clinician's choice of dose, schedule, and interval should be weighted upon the probability of target attainment versus the probability of toxicity, modulated by the MIC distribution of likely pathogens present in their specific institution.
Finally, given the rate of loss of antibiotics from the physician's armamentarium due to resistance, regimens that help suppress this resistance should be considered, if the toxicity price is not great. Figure 4 shows the target attainment for achieving a trough value of free drug 1.7 times a baseline MIC (combination therapy with an aminoglycoside) and 6.2 times a baseline MIC (monotherapy with meropenem), a target proposed by Tam et al. (26) for resistance suppression for Pseudomonas aeruginosa. For an 8-h interval and a 4-h infusion with a 40-mg/kg dose, this target is met >80% of the time for both pre-term and full-term neonates out to an MIC of 2 mg/liter when combined with an aminoglycoside, which encompasses the vast majority of the wild-type population sensitivity of Pseudomonas aeruginosa to meropenem. For monotherapy, the resistance suppression goal falls below 79% after an MIC of 1.0 mg/liter. This provides another reason to favor the large dose every 8 h with a 4-h infusion, at least empirically.
FIG. 4.

Ability of a prolonged infusion of 40 mg/kg to attain a trough free-drug concentration >1.7 times the baseline MIC, a value associated with resistance emergence suppression when meropenem is administered with an aminoglycoside, or to attain a trough free-drug concentration >6.2 times the baseline MIC, a value associated with resistance emergence suppression when meropenem is administered alone (33).
These findings need to be prospectively validated in the clinic. Furthermore, we would like to stress that the aforementioned recommendations are primarily derived from pre-term infants with gestational ages of more than 30 weeks, because only one infant was studied with a gestational age of less than 30 weeks.
Data on the pharmacokinetics of meropenem in pre-term infants with gestational ages of less than 30 weeks are still needed before doses and schedules of meropenem can be calculated at the bedside for this very young population.
Acknowledgments
We have no conflicts to declare. The work was supported by AstraZeneca Pharmaceuticals.
Footnotes
Published ahead of print on 6 July 2009.
REFERENCES
- 1.Baker, C. J., M. E. Melish, R. T. Hall, D. T. Casto, U. Vasan, and L. B. Givner. 1992. Intravenous immunoglobulin for the prevention of nosocomial infection in low-birth-weight neonates. N. Engl. J. Med. 327:213-219. [DOI] [PubMed] [Google Scholar]
- 2.Besunder, J. B., M. D. Reed, and J. L. Blumer. 1988. Principles of drug biodisposition in the neonate. A critical evaluation of the pharmacokinetic-pharmacodynamic interface (part 1). Clin. Pharmacokinet. 14:189-216. [DOI] [PubMed] [Google Scholar]
- 3.Bradley, J. S., M. N. Dudley, and G. L. Drusano. 2003. Predicting efficacy of antiinfectives with pharmacodynamics and Monte Carlo simulation. Pediatr. Infect. Dis. J. 22:982-992. [DOI] [PubMed] [Google Scholar]
- 4.Bradley, J. S., J. B. Sauberan, P. G. Ambrose, S. M. Bhavnani, M. R. Rasmussen, and E. V. Caparelli. 2008. Meropenem pharmacokinetics, pharmacodynamics, and Monte Carlo simulation in the neonate. Pediatr. Infect. Dis. J. 27:794-799. [DOI] [PubMed] [Google Scholar]
- 5.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibiotic dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 6.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II user's guide: pharmacokinetic/pharmacodynamic systems analysis software. University of Southern California, Los Angeles, CA.
- 7.de Hoog, M., J. N. van den Anker, and J. W. Mouton. 2004. Vancomycin: pharmacokinetics and administration regimens in neonates. Clin. Pharmacokinet. 43:417-440. [DOI] [PubMed] [Google Scholar]
- 8.de Hoog, M., J. W. Mouton, and J. N. van den Anker. 2003. The use of aminoglycosides in newborn infants, p. 117-140. In I. Choonara, A. J. Nunn, and G. Kearns (ed.), Introduction to paediatric and perinatal drug therapy. Nottingham University Press, Nottingham, United Kingdom.
- 9.Drukker, A., and J. P. Guignard. 2002. Renal aspects of the term and preterm infant: a selective update. Curr. Opin. Pediatr. 14:175-182. [DOI] [PubMed] [Google Scholar]
- 10.Drusano, G. L. 2004. Antimicrobial pharmacodynamics: the interactions between bug and drug. Nat. Rev. Microbiol. 2:289-300. [DOI] [PubMed] [Google Scholar]
- 11.Drusano, G. L. 2003. Prevention of resistance: a goal for dose selection for antimicrobial agents. Clin. Infect. Dis. 36:S42-S50. [DOI] [PubMed] [Google Scholar]
- 12.Drusano, G. L., K. H. P. Moore, J. P. Kleim, W. Prince, and A. Bye. 2002. Rational dose selection for a nonnucleoside reverse transcriptase inhibitor through the use of population pharmacokinetic modeling and Monte Carlo simulation. Antimicrob. Agents Chemother. 46:913-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vesga, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escobar, G. J., T. Zukin, M. S. Usatin, J. Lemesurier, D. Honeychurch, M. A. Armstrong, and B. Folck. 1994. Early discontinuation of antibiotic treatment in newborns admitted to rule out sepsis: a decision rule. Pediatr. Infect. Dis. J. 13:860-866. [DOI] [PubMed] [Google Scholar]
- 15.Fanaroff, A. A., S. B. Korones, L. L. Wright, E. C. Wright, R. L. Poland, C. B. Bauer, J. E. Tyson, J. B. Philipis III, W. Edwards, and J. F. Lucey. 1994. A controlled trial of intravenous immunoglobulin to reduce nosocomial infections in very-low-birth-weight infants. N. Engl. J. Med. 330:1107-1113. [DOI] [PubMed] [Google Scholar]
- 16.Gladstone, I. M., R. A. Ehrenkranz, S. C. Erdberg, and R. S. Baltimore. 1990. A ten-year review of neonatal sepsis and comparison with the previous fifty-year experience. Pediatr. Infect. Dis. J. 9:819-825. [DOI] [PubMed] [Google Scholar]
- 17.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H. Tam, R. Bachhawat, C. Freeman, J. B. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein, J. O., and S. Michael Marcy. 1995. Bacterial sepsis and meningitis, p. 836. In J. S. Remington and J. O. Klein (ed.), Infectious diseases of the fetus & newborn infant. WB Saunders & Company, Philadelphia, PA.
- 19.Leake, R. D., C. W. Trygstad, and W. Oh. 1976. Inulin clearance in the newborn infant: relationship to gestational and postnatal age. Pediatr. Res. 10:759-762. [DOI] [PubMed] [Google Scholar]
- 20.Leary, R., R. Jelliffe, A. Schumitzky, and M. van Guilder. 2001. An adaptive grid non-parametric approach to pharmacokinetic and dynamic (PK/PD). 14th IEEE Symposium on Computer-Based Medical Systems. IEEE Computer Society, Bethesda, MD.
- 21.Mouton, J. W., D. J. Touw, A. M. Horrevorts, and A. A. Vinks. 2000. Comparative pharmacokinetics of the carbapenems: clinical implications. Clin. Pharmacokinet. 39:185-201. [DOI] [PubMed] [Google Scholar]
- 22.Pacifici, G. M., J. Labatia, H. Mulla, and I. Choonara. 2009. Clinical pharmacokinetics of penicillins in the neonate: a review of the literature. Eur. J. Clin. Pharmacol. 65:191-198. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz, G. J., G. B. Haycock, C. M. Edelman, Jr., and A. Spitzer. 1976. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics 58:259-263. [PubMed] [Google Scholar]
- 24.Shah, S. S., R. A. Ehrenkranz, and P. G. Gallagher. 1999. Increasing incidence of gram-negative rod bacteremia in a newborn intensive care unit. Pediatr. Infect. Dis. J. 18:591-595. [DOI] [PubMed] [Google Scholar]
- 25.Stoll, B. J., T. Gordon, S. B. Korones, S. Shankaran, J. E. Tyson, C. R. Bauer, A. A. Fanaroff, J. A. Lemons, E. F. Donovan, W. Oh, D. K. Stevenson, R. A. Ehrenkranz, L. A. Papile, J. Verter, and L. L. Wright. 1996. Early onset sepsis in very low birth weight neonates: a report from the National Institute of Child Health and Human Development Neonatal Research Network. J. Pediatr. 129:72-80. [DOI] [PubMed] [Google Scholar]
- 26.Tam, V. H., A. N. Schilling, S. Neshat, K. Poole, D. A. Melnick, and E. A. Coyle. 2005. Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:4920-4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van den Anker, J. N., W. C. Hop, R. De Groot, B. J. van der Heijden, H. M. Broerse, J. Lindemans, and P. J. Sauer. 1994. Effect of prenatal exposure to betamethasone and indomethacin on the glomerular filtration rate in the preterm infant. Pediatr. Res. 36:578-581. [DOI] [PubMed] [Google Scholar]
- 28.van den Anker, J. N., W. C. Hop, R. C. Schoemaker, B. J. van der Heijden, H. J. Neijens, and R. de Groot. 1995. Ceftazidime pharmacokinetics in preterm infants: effect of postnatal age and postnatal exposure to indomethacin. Br. J. Clin. Pharmacol. 40:439-443. [PMC free article] [PubMed] [Google Scholar]
- 29.van der Heijden, H. J., W. F. Grose, J. J. Ambagtsheer, A. P. Provoost, E. D. Wolff, and J. J. Sauer. 1988. Glomerular filtration rate in the preterm infant: the relation to gestational and postnatal age. Eur. J. Pediatr. 148:24-28. [DOI] [PubMed] [Google Scholar]
- 30.Van Enk, J. G., D. Touw, and H. N. Lafeber. 2001. Pharmacokinetics of meropenem in preterm neonates. Ther. Drug Monitor. 23:198-201. [DOI] [PubMed] [Google Scholar]