

Abstract
It was previously demonstrated that microbial communities of pig manure were composed of both bacteria and archaea. Recent studies have shown that bacteria are aerosolized from pig manure, but none have ever focused on the airborne archaeal burden. We sought here to develop and apply molecular ecology approaches to thoroughly characterize airborne archaea from swine confinement buildings (SCBs). Eight swine operations were visited, twice in winter and once during summer. Institute of Occupational Medicine cassettes loaded with 25-mm gelatin filters were used to capture the inhalable microbial biomass. The total genomic DNA was extracted and used as a template for PCR amplification of the archaeal 16S rRNA gene. High concentrations of archaea were found in SCB bioaerosols, being as high as 108 16S rRNA gene copies per cubic meter of air. Construction and sequencing of 16S rRNA gene libraries revealed that all sequences were closely related to methanogenic archaea, such as Methanosphaera stadtmanae (94.7% of the archaeal biodiversity). Archaeal community profiles were compared by 16S rRNA gene denaturing gradient gel electrophoresis. This analysis showed similar fingerprints in each SCB and confirmed the predominance of methanogenic archaea in the bioaerosols. This study sheds new light on the nature of bioaerosols in SCBs and suggests that archaea are also aerosolized from pig manure.
Over the last 30 years, swine production in Canada evolved from small family farms to industrial facilities. Pig producers have increased animal density, building mechanization, and confinement in order to decrease working and feeding time and to optimize space, leading to an increased contamination of air by bioaerosols.
Even though the swine confinement building (SCB) environment has been studied for several years, little is known about the real concentration and nature of airborne microorganisms. Moreover, increasing confinement level in modern barns has raised bioaerosol levels, modifying the health risk of exposed workers. Thus far, using culture-dependent methods was the only strategy developed and used to describe SCB bioaerosol content and levels (6, 7). However, it is well known that culture-independent approaches are more likely to reveal the presence of microorganisms never suspected in most environments (2). In aerobiology, there are only a few reports using culture-independent methods (4, 15). Nehme et al. (20) applied molecular approaches to quantify and describe the bacterial aerosols in SCB and reported as much as 108 bacteria per cubic meter of air, with significantly higher concentrations during winter, when the confinement is maximal. The data obtained were also compared to recent biodiversity studies of swine manures (13, 22, 25). Anaerobic gram-positive bacteria, being the greater part of the microbiological aerosols, appeared to originate from the swine manure. Those manure biodiversity studies revealed the presence of methanogenic archaea in hog wastes (22, 25). Since bacteria observed in the aerosols seem to originate from the manure, it is plausible that archaea from pig slurries are also aerosolized.
We report here the characterization of the archaeal community of SCB bioaerosols by using cultivation-independent approaches. The phylogeny of airborne archaea was assessed using 16S rRNA gene sequences. Archaeal biodiversity profiles were determined with PCR-denaturing gradient gel electrophoresis (DGGE), and the concentration of aerosolized archaea was evaluated by real-time PCR by quantifying archaeal 16S rRNA gene copies in the air samples.
MATERIALS AND METHODS
Sampling sites.
Eight SCBs from the Quebec City area (Eastern Canada) were visited on three occasions: twice in winter and once in summer. Samples were all collected in growing and finishing rooms, occupied by weight-market pigs (∼100 kg). Wastes were collected as a liquid mixture in a pit located under a slatted floor (66 to 100% slatted). All pigs were fed with feeds from the COOP Fédérée (Agricultural Field, Montréal, Quebec, Canada) or from on-farm mills.
Sample collection and preparation.
Airborne samples were collected over 4 h, in triplicate or quadruplicate, using Institute of Occupational Medicine (IOM) (SKC, Ancaster, Ontario, Canada) cassettes loaded with gelatin membranes 25 mm in diameter (SKC). They were taken 1 m above the floor, as far away as possible from doors, windows, or other ventilation sources. The IOM-gelatin membrane system was plugged into a Gilair-5 pump (Levitt-Sécurité Limitée, Dorval, Quebec, Canada) calibrated at 2 liters min−1 using a DryCal 2 flow meter (SKC). After sampling, cassettes were put on ice and brought to the laboratory. Replicate gelatin membranes were pooled in 0.9% NaCl solution (5 ml/membrane) and dissolved by vortexing at maximum speed for 30 min at room temperature with a Multi-Pulse Vortexer (Glas-Col, Terre Haute, IN). The resulting solution was divided into aliquots to obtain an equivalent of 144 liters of air in each tube and then centrifuged (10 min, 21,000 × g, room temperature). Pellets were kept at −20°C until their use for total DNA extraction.
Total DNA extraction.
Isolation of total genomic DNA from pellets was performed by using a QIAamp DNA minikit (Qiagen, Mississauga, Ontario, Canada) according to the manufacturer's instructions for tissue with the required modifications for bacteria. Total DNA was eluted in 200 μl of DNase/RNase-free water (Sigma, Oakville, Ontario, Canada).
Quantitative real-time PCR for total archaea.
PCR amplifications were carried out with the DNA Engine Opticon 2 (Bio-Rad, Mississauga, Ontario, Canada). Quantification of archaea was performed with the ARC787F and ARC1059R primers and the TaqMan probe ARC915F (Table 1). The components for the 16S rRNA gene quantification assay per 25 μl were as follows: 1 to 100 ng of DNA template, 0.5 μM concentrations of each primer, 0.15 μM concentrations of TaqMan probe, and 12.5 μl of a 2× reaction mix obtained from a QuantiTech Probe PCR probe kit (Qiagen). The PCR program to amplify a 272-bp product was as follows: one hold at 37°C for 10 min and one hold at 95°C for 15 min, followed by 45 cycles of 95°C for 15 s and 60°C for 60 s. A standard curve was built using serial dilutions of the Methanosarcina mazei (ATCC BAA-159D) 16S rRNA gene cloned in a pCR4-TOPO plasmid (Invitrogen, Carlsbad, CA). The PCR efficiencies and detection sensitivities of the Methanosarcina mazei 16S rRNA gene preparation and the environmental samples were determined by using serial 10-fold dilutions. The data were acquired with the computer software Opticon Monitor (version 2.02.24; Bio-Rad) and analyzed by linear regression of the log10 (target copy number) = f (threshold cycle) function. PCR efficiencies were determined as follows: E = 10−slope − 1. Negative controls and field blanks were also added to each PCR run to evaluate the PCR reagent contamination.
TABLE 1.
Primers and probe sequences used in this studya
| Method and primer or probe | Nucleotide sequence (5′-3′)b | Reference |
|---|---|---|
| Quantitative PCR | ||
| ARC787F | ATT AGA TAC CCS BGT AGT CC | 27 |
| ARC915F | (FAM)-AGG AAT TGG CGG GGG AGC AC-(TAMRA) | 27 |
| ARC1059R | GCC ATG CAC CWC CTC T | 27 |
| Nested PCR (DGGE and cloning) | ||
| Ar3F | TTC CGG TTG ATC CTG CCG GA | 10 |
| Ar9R | CCC GCC AAT TCC TTT AAG TTT C | 10 |
| 1492R | GGT TAC CTT GTT ACG ACT T | 12 |
| Parch340F-GCc | CCC TAC GGG GYG CAS CAG | 21 |
| 519R | TTA CCG CGG CKG CTG | 21 |
All of the primers and DNA probes used in this study were purchased from IDT (Coralville, IA).
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxy-tetramethylrhodamine.
For DGGE, this primer has the following GC-clamp at the 5′ end: CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG (19).
DGGE.
A nested PCR was used to amplify the variable V3 region of 16S rRNA gene sequences from archaea. A first step consisted of using Ar3F and 1492R primers to amplify the archaeal community, followed by an amplification of diluted PCR products using PARCH340-GC and 519r primers (Table 1). For both steps, the components for the 16S rRNA gene amplification per 25 μl were as follows: 1 to 100 ng of DNA template, 0.25 μM concentrations of each primer, 1.5 mM MgCl2, 2% dimethyl sulfoxide, 0.2 mM deoxynucleoside triphosphate, 1× TaKaRa PCR buffer (10 mM Tris-HCl [pH 8.3], 50 mM KCl), and 1.25 U of TaKaRa Taq polymerase (Fisher Scientific, Ottawa, Ontario, Canada). PCR was performed with a DNA Engine DYAD thermocycler (Bio-Rad). The first step of the nested PCR protocol was 5 min at 95°C, followed by 30 cycles of 60 s at 95°C, 60 s at 55°C, and 60 s at 72°C, with a final extension step of 5 min at 72°C. The second step protocol was 5 min at 95°C, followed by 30 cycles of 60 s at 95°C, 60 s at 53.5°C, and 120 s at 72°C, with a final extension step of 5 min at 72°C. After gel electrophoresis (1.5% [wt/vol] agarose gel) of 5-μl subsamples of the PCR products, the amount of amplified DNA was quantified by comparing band intensities to standard curves obtained by using an EZ Load Precision molecular mass ruler (Bio-Rad). Band intensities were measured with GeneTools analysis software (SynGen, Cambridge, England).
Profiles of the amplified 16S rRNA gene sequences were produced by DGGE as described by Muyzer (18) using the DCode apparatus (Bio-Rad). The PCR products were loaded (60 ng) onto an 8% polyacrylamide gel in 0.5× TAE buffer (20 mM Tris [pH 8.0], 10 mM acetic acid, 0.5 mM EDTA; Bio-Rad) with a denaturant gradient (100% denaturant was 7 M urea and 40% [vol/vol] deionized formamide). The electrophoresis was carried out in 0.5× TAE buffer at 60 V for 16 h at 60°C. The DNA fragments were stained for 15 min in 0.5× TAE buffer with SYBR Gold (Molecular Probes, Eugene, OR). The gels were washed in distilled water for 15 min. Images of the gels were obtained using an imaging system ChemiGenius 2 (SynGen) and the imaging software GeneSnap (SynGen).
DNA bands from the polyacrylamide gels were excised and incubated in 40 μl of sterile DNase/RNase-free water (Sigma) overnight at 4°C. A 5-μl portion of the DNA extract was used as the template in a PCR amplification with the PARCH340 (no GC clamp) and 519r primers as described above. The PCR products were cloned with the TOPO-TA cloning kit (Invitrogen). Three clones from each DGGE band were randomly chosen and digested with restriction enzyme HaeIII, HhaI, or MspI (New England Biolabs, Pickering, Ontario, Canada) in order to confirm the purity of the reamplified DNA from the DGGE bands.
16S rRNA gene library construction.
PCR runs were carried out on samples collected during winter 1 from barns D, G, and H using the Ar3F and 1492R primers as described above. Nested PCR was then performed using the primers Ar3F and Ar9R, corresponding to nucleotides 7 to 927 in Escherichia coli (Table 1). The PCR mixture was as described above, and the PCR conditions were as follows: one hold at 95°C for 5 min, followed by 30 cycles at 95°C for 60 s, 55°C for 60 s, and 72°C for 60 s, followed in turn by a final extension step of 5 min at 72°C. The PCR products were cloned into E. coli using a TOPO-TA cloning kit (Invitrogen) to generate 16S rRNA gene libraries. Approximately 200 clones were randomly picked from each library and grown overnight in LB medium with 100 mg of ampicillin (Sigma) ml−1 at 37°C. The insert from each clone was directly amplified from E. coli culture using the M13 primers (TOPO-TA cloning kit). The final PCR products were then digested with restriction enzymes HaeIII and HhaI, according to the manufacturer's specifications, and separated on a 2% agarose gel that was electrophoresed at 100 V for at least 30 min. Restriction fragment length polymorphisms were grouped according to their riboprint patterns. The PCR products of all different riboprint patterns were sequenced on both strands (Plateforme de Séquençage et de Génotypage, CHUL Research Center, Québec, Quebec, Canada). When two or more clones had the same riboprint pattern, up to 10 clones were sequenced for confirmation.
Phylogenetic analysis.
Each cloned DNA sequence was compared to sequences available in databases, using BLASTN (1) from the National Center of Biotechnology Information (http://www.ncbi.nlm.nih.gov/BLAST/). Potential DNA chimeric structures were searched with the CHECK_CHIMERA program (RDP-II) (5). All of the sequences and their closest relatives were then aligned using CLUSTAL W software (23). The sequences were screened for manual alignment correction with the ARB package (14). Finally, the phylogenetic tree was constructed with ARB using the maximum-likelihood method, with pairwise gap removal and a conservation filter of 50%.
Statistical analysis.
The statistical method used to perform comparisons of airborne archaeal concentrations between summer and winter was a one-way analysis of variance. Relationships between parameters were expressed using the Pearson correlation coefficient as the linearity between these observed parameters and statistically analyzed to determine whether it was significant using a Student t test. The results were considered significant if the P value was ≤0.05. The data were analyzed by using the statistical package program SAS (SAS Institute, Cary, NC).
Nucleotide sequence accession numbers.
The nucleotide sequence data reported in the present study appear in the GenBank nucleotide sequence database under the accession numbers FJ001234 to FJ001326.
RESULTS
Total archaeal counts in the SCB.
The primer set ARC787F-ARC1059R and the probe ARC915F were successfully used to quantify the archaeal community in the air (efficiency = 88.8%, R2 = 0.996), with a standard curve being linear from 101 to 106 gene copies per reaction. The quantitative PCR results expressed in 16S rRNA gene copy number were considered representative of the airborne archaeal community concentrations. No PCR inhibition effect was observed in diluted reaction mixtures of the environmental samples (data not shown).
The real-time PCR counts revealed a high number of 16S rRNA gene copies (106 to 108) per cubic meter of air. There was no significant difference between the two winter visits for the concentration of archaea in the air (Fig. 1A). However, there was a significant decrease of 16S rRNA gene copy number of archaea during the summer period (3.5-fold less than in winter, P < 0.01) (Fig. 1B).
FIG. 1.
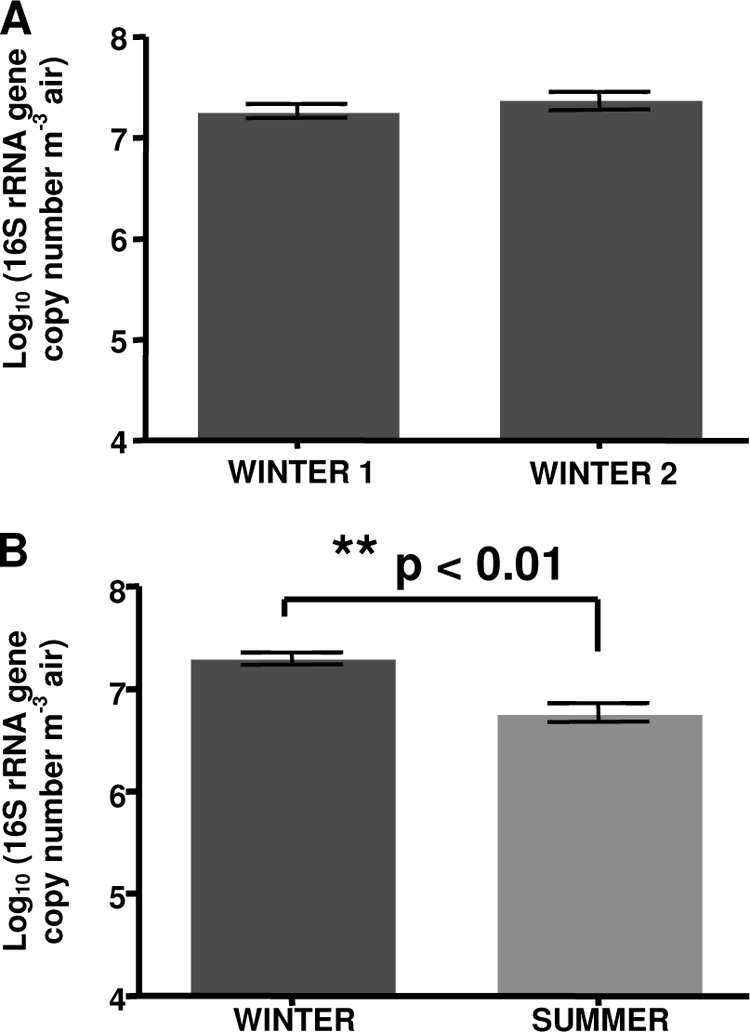
Number of 16S rRNA gene copies from archaea as determined by real-time PCR. (A) Comparison between the two winter samples (n = 8 each). (B) Comparison between the winter and summer samples (winter, n = 16; summer, n = 8). The error bars represent the standard deviation.
DGGE profiles analysis.
Diversity within the SCB archaeal bioaerosol populations was determined by 16S rRNA gene-targeted PCR-DGGE analysis. A 20-to-50% denaturing gradient analysis (Fig. 2) was performed on each sample.
FIG. 2.

DGGE profiles of the 16S rRNA gene V3 region obtained from SCB bioaerosols during the winter and the summer seasons. Lanes A to H: sampled SCB. Numbers 1 to 6: DNA bands that were sequenced for microbial identification.
Six different bands were observed among all DGGE profiles. The presence of two similarly migrating bands (bands 1 and 2) suggests that some species were present in all samples. Even though each sampled SCB aerosol had a similar DGGE profile, some slight differences were observed since a number of DNA bands were not always detected (bands 3 to 6). A comparison between winter 1 and winter 2 DGGE profiles showed similar patterns for each SCB, while the analysis of the summer profiles also showed that they were highly similar to those obtained for winter samples. Bands 5 and 6 were observed only during the summer, in barn C, D, and E air samples, whereas band 3 was retrieved only from barn B during the winter.
Six major bands were extracted from the gel, reamplified, cloned, and sequenced (∼150 nucleotides). Five of six sequences obtained from these bands showed high identity (95.4 to 100%) to sequences affiliated with the family Methanobacteriaceae. All of these sequences (bands 1 to 4 and band 6) retrieved from the DGGE profiles were related to Methanosphaera stadtmanae (95.4 to 96.7% sequence identity) and uncultured archaeon clones, respectively, obtained from human intestinal samples (16) and bovine rumen fluid (26) (Fig. 3). The band 5 sequence is part of the CA11 group (9) and more closely related to the uncultured archaeon clone EcoP2-45F (98.7% sequence identity), retrieved from pig feces samples (M. Lange et al., unpublished data).
FIG. 3.

Phylogenetic tree derived from three 16S rRNA gene clone libraries constructed from bioaerosol DNA. The evolutionary distance among 16S rRNA sequences and representative members of related families is illustrated by an unrooted phylogenetic tree. The tree was inferred using the maximum-likelihood method with aligned sequences containing 900 to 950 positions. The scale bar indicates the nucleotide substitutions per position. GenBank accession numbers are given next to each species names. Experimental sequences are named as follows. In “4A_1 FJ001275 (522),” for example, the first three characters correspond to the ribotype, the numbers beside the ribotype are its GenBank accession number, and the numbers in parentheses indicate the number of clones obtained under this particular ribotype for the entire clone library.
16S rRNA gene clone library analysis.
As shown in Fig. 2, DGGE profiles were highly similar; three winter environmental samples from SCB aerosols were then randomly used for the phylogenetic analysis (barns D, G, and H using winter 1 samples). Three different 16S rRNA gene libraries were derived from these samples. A total of 566 clones contained an insert that could be sequenced. A preliminary screening using restriction fragment length polymorphism revealed at least 16 ribotypes. All sequences recovered from the SCB bioaerosols were related to uncultured methanogenic archaeal sequences in public databases (Fig. 3). The average determined length of the 16S rRNA gene was ∼900 bp, and phylogenetic analysis was based on 750 to 900 aligned homologous nucleotides. No sequence was identified as a possible chimera. Phylotypes were named based on a representative clone of its group.
Two phylotypes (11A1 and 4A1), represented by a total of 535 clones and whose closest known relative was Methanosphaera stadtmanae (96.1 to 96.4% sequences identity), constituted the main sequences retrieved from SCB bioaerosol samples (94.5% of the clones). These clones were grouped in the phylogenetic tree among Methanosphaera stadtmanae identified from human intestinal samples and other unidentified methanogens obtained from bovine rumen fluid.
Approximately 2.8% of the clones were phylogenetically classified into the CA11 group, being more closely related to unidentified archaea (9). Eleven phylotypes (15 clones) were obtained that showed similarities to uncultured archaeon clones collected from pig feces samples (GenBank accession number AY911629), chimpanzee and human intestinal communities (GenBank accession numbers AY911614 and AY907247), and goat rumen (GenBank accession number DQ402017).
More than 2.6% of the clones were classified among Methanosarcinales. One phylotype (1 clone) was only 92.6% similar to Methanosarcina siciliae, obtained from marine canyon sediments (8), and one phylotype (14 clones) was 92.8% similar to the unidentified methanogen ARC 63, obtained from bovine rumen fluid (36).
DISCUSSION
In addition to archaeal quantification using real-time PCR, archaeal biodiversity in SCB bioaerosols was characterized by analysis of 16S rRNA gene fragments using PCR-DGGE and 16S rRNA gene library construction. Previous studies about SCB bioaerosol analysis measured only the culturable concentration of bacteria in the air (7, 11). Duchaine et al. (7) observed a bacterial concentration in the air of up to 105 CFU m−3 and found that this contamination level significantly decreased in summer. They hypothesized that increased ventilation rates in summer could explain this diminution. More recently, Nehme et al. (20) showed, using culture-independent methods, that total airborne bacterial concentration was more important than previously evaluated using culture, being up to 1,000 times higher than the concentration of airborne culturable bacteria. They also confirmed that bacterial concentrations in SCB aerosols were significantly lower in summer than values obtained in winter.
There are no data in the literature about airborne archaea, either culturable or total, to compare to the results obtained here. No study has ever focused on the biodiversity or the concentration of archaea in air, whatever the environment, either by using culture-dependent methods or culture-independent ones. SCBs were an ideal environment for testing molecular methods for airborne-archaeon analysis, since hog wastes are known to contain these types of microorganisms (22, 25). Snell-Castro et al. (22) showed that pig manure was mainly dominated by two kinds of archaea: a group of uncultured archaea affiliated with clone CA11 (9), of unknown activity and only distantly related to Thermoplasmatales, and Methanobacteriales affiliated with Methanobrevibacter smithii and Methanosphaera stadtmanae. These authors also observed very few clones affiliated with Methanomicrobiales archaea.
Archaeal sequences could be found in all bioaerosol samples of the swine confinement buildings by DGGE and cloning. All sequences retrieved were related to methanogenic archaea, particularly Methanosphaera stadtmanae, suggesting that analyzed bioaerosol samples had a low biodiversity of archaea. Some DGGE profiles showed slight differences but were not season related. These differences could result from bias in air sampling and DNA extraction steps. In addition, other sequences obtained during the present study were almost all related to uncultured archaeal clones from pig feces, primate intestinal samples, or rumen fluid samples. It is probable that airborne archaea observed in the present study were aerosolized from the swine manure, as described for the bacterial community (20).
Data obtained in our study revealed the presence of the same two main archaeal groups described by Snell-Castro et al. from swine manure (22). However, no Methanomicrobiales were observed, either in DGGE profiles or in clone libraries. Two hypotheses were raised to explain this result: a preferential aerosolization or “universal” archaeal primers biased toward a group of microorganisms, as suggested by Baker et al. (3). These authors analyzed the specificity of archaeon-specific PCR primers and showed that most universal archaeal primers are biased, with poor specificity toward Korarchaeotes and Nanoarcheae. Moletta et al. (17), while studying anaerobic digestors and landfill biogas biodiversity, found that some bacteria were preferentially aerosolized in this kind of environment. Archaeal microorganisms were not aerosolized.
High concentrations of archaea (up to 108 16S rRNA copies per cubic meter of air) were found in the bioaerosols from SCBs. These values are considered high since more usual places, such as schools, offices, and houses, show less than 103 CFU m−3 (24). A comparison of these quantitative data with those of Nehmé et al. (20), who used the same samples that we used to analyze airborne bacterial concentration in SCBs, showed that the airborne archaeal concentration was in the same order of magnitude as total bacteria. Moreover, a correlation (n = 8, R2 = 0.58, P < 0.0001) of the total archaeal concentration in air and the total airborne bacteria has been observed (Fig. 4). These results contradict those of Moletta et al. (17), who observed that archaea were not aerosolized from anaerobic digesters, in contrast to some groups of bacteria (e.g., Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria) that were observed in the biogas. As observed by Moletta et al. (17) for bacteria, aerosolization of archaea could be group dependent. However, under the conditions described here, little is known about bacterial/archaeal ratios, but co-aerosolization was demonstrated.
FIG. 4.
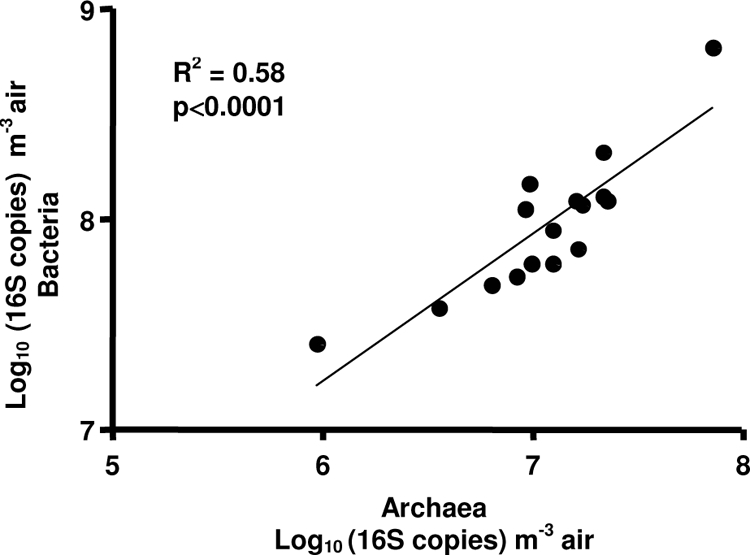
Correlation between the number of bacterial and archaeal 16S rRNA gene copies in SCBs.
Although archaeal concentrations in bioaerosols decreased significantly during the summer, similar diversities characterized the summer and winter bioaerosols. These results show the same trend observed previously for total bacteria (20). Nehme et al. (20) explained this concentration diminution by an increase of building ventilation during the summer in order to maintain a stable and comfortable temperature for animals. However, Kim et al. (11) found that the ventilation rates could not explain the decrease in particulate and microorganism concentrations in the air observed in this environment. It is possible that the animal's activity diminishes during the summer due to higher temperatures inside the barns, thus creating fewer bioaerosols. Nevertheless, even if the number of archaea diminished significantly, their concentration remained high, ∼106 archaeal 16S rRNA copies per cubic meter of air.
Thus far, the presence of anaerobic archaea was never suspected in bioaerosols, but we demonstrated here high airborne archaeal concentrations. The large quantity of airborne archaea observed in this kind of environment raises questions about the health of pigs and farmers. However, no data about the effect of airborne archaea on human and animal respiratory health are currently available. The study of the role of archaea in the human response to SCB bioaerosols constitutes a new challenge.
Acknowledgments
B.N. and V.L. are CIHR Strategic Training Fellows in PHARE, research funded by the CIHR Strategic Training Program and Partner Institutes. C.D. acknowledges CIHR/IRSST (2001-2006) and FRSQ (2006-2010) scholarships and Canadian Center for Health and Safety in Agriculture time release support (2006-2008). This project was funded by the Natural Sciences and Engineering Research Council of Canada (288122-04), IRSST, and FQRNT.
C.D. is a member of the FRSQ Respiratory Health Network.
Footnotes
Published ahead of print on 26 June 2009.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann, R. I., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker, G. C., J. J. Smith, and D. A. Cowan. 2003. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 55:541-555. [DOI] [PubMed] [Google Scholar]
- 4.Brodie, E. L., T. Z. DeSantis, J. P. Parker, I. X. Zubietta, Y. M. Piceno, and G. L. Andersen. 2007. Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. USA 104:299-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole, J. R., B. Chai, T. L. Marsh, R. J. Farris, Q. Wang, S. A. Kulam, S. Chandra, D. M. McGarrell, T. M. Schmidt, G. M. Garrity, and J. M. Tiedje. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crook, B., J. F. Robertson, S. A. Glass, E. M. Botheroyd, J. Lacey, and M. D. Topping. 1991. Airborne dust, ammonia, microorganisms, and antigens in pig confinement houses and the respiratory health of exposed farm workers. Am. Ind. Hyg. Assoc. J. 52:271-279. [DOI] [PubMed] [Google Scholar]
- 7.Duchaine, C., Y. Grimard, and C. Cormier. 2000. Influence of building maintenance, environmental factors, and seasons on airborne contaminants of swine confinement buildings. Am. Ind. Hyg. Assoc. J. 61:56. [PubMed] [Google Scholar]
- 8.Elberson, M. A., and K. R. Sowers. 1997. Isolation of an aceticlastic strain of Methanosarcina siciliae from marine canyon sediments and emendation of the species description for Methanosarcina siciliae. Int. J. Syst. Bacteriol. 47:1258-1261. [DOI] [PubMed] [Google Scholar]
- 9.Godon, J.-J., E. Zumstein, P. Dabert, F. Habouzit, and R. Moletta. 1997. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63:2802-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurgens, G., K. Lindstrom, and A. Saano. 1997. Novel group within the kingdom Crenarchaeota from boreal forest soil. Appl. Environ. Microbiol. 63:803-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim, K. Y., H. J. Ko, H. T. Kim, Y. S. Kim, Y. M. Roh, and C. N. Kim. 2007. Effect of ventilation rate on gradient of aerial contaminants in the confinement pig building. Environ. Res. 103:352-357. [DOI] [PubMed] [Google Scholar]
- 12.Lane, D. J. 1991. 16S/23S rRNA sequencing, p. 115-175. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. Wiley, Chichester, United Kingdom.
- 13.Leser, T. D., J. Z. Amenuvor, T. K. Jensen, R. H. Lindecrona, M. Boye, and K. Moller. 2002. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 68:673-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier, Yadhukumar, A. Buchner, T. Lai, S. Steppi, G. Jobb, W. Förster, I. Brettske, S. Gerber, A. W. Ginhart, O. Gross, S. Grumann, M. May, B. Nonhoff, B. Reichel, R. Strehlow, A. Stamatakis, N. Stuckmann, A. Vilbig, M. Lenke, T. Ludwig, A. Bode, and K.-H. Schleifer. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maron, P. A., D. P. H. Lejon, E. Carvalho, K. Bizet, P. Lemanceau, L. Ranjard, and C. Mougel. 2005. Assessing genetic structure and diversity of airborne bacterial communities by DNA fingerprinting and 16S rDNA clone library. Atmos. Environ. 39:3687-3695. [Google Scholar]
- 16.Miller, T. L., and M. J. Wolin. 1985. Methanosphaera stadtmanae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 141:116-122. [DOI] [PubMed] [Google Scholar]
- 17.Moletta, M., J. P. Delgenes, and J. J. Godon. 2007. Differences in the aerosolization behavior of microorganisms as revealed through their transport by biogas. Sci. Total Environ. 379:75-88. [DOI] [PubMed] [Google Scholar]
- 18.Muyzer, G. 1999. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 2:317-322. [DOI] [PubMed] [Google Scholar]
- 19.Muyzer, G., E. C. De Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nehme, B., V. Létourneau, R. J. Forster, M. Veillette, and C. Duchaine. 2008. Culture-independent approach of the bacterial bioaerosol diversity in the standard swine confinement buildings, and assessment of the seasonal effect. Environ. Microbiol. 10:665-675. [DOI] [PubMed] [Google Scholar]
- 21.Ovreas, L., L. Forney, F. L. Daae, and V. Torsvik. 1997. Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl. Environ. Microbiol. 63:3367-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snell-Castro, R., J. J. Godon, J. P. Delgenes, and P. Dabert. 2005. Characterisation of the microbial diversity in a pig manure storage pit using small subunit rDNA sequence analysis. FEMS Microbiol. Ecol. 52:229-242. [DOI] [PubMed] [Google Scholar]
- 23.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorne, P. S., and C. Duchaine. 2007. Airborne bacteria and endotoxin, p. 989-1004. In C. J. Hurst, R. M. Crawford, J. L. Garland, D. A. Lipson, A. L. Mills, and L. D. Stetzenbach (ed.), Manual of environmental microbiology, 3rd ed. ASM Press, Washington, DC.
- 25.Whitehead, T. R., and M. A. Cotta. 1999. Phylogenetic diversity of methanogenic archaea in swine waste storage pits. FEMS Microbiol. Lett. 179:223-226. [DOI] [PubMed] [Google Scholar]
- 26.Whitford, M., R. Teather, and R. Forster. 2001. Phylogenetic analysis of methanogens from the bovine rumen. BMC Microbiol. 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu, Y., C. Lee, J. Kim, and S. Hwang. 2005. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89:670-679. [DOI] [PubMed] [Google Scholar]