

Abstract
The native product of open reading frame 112 (orf112) and a recombinant variant of the RstB protein, encoded by Vibrio cholerae pathogen-specific bacteriophages VGJφ and CTXφ, respectively, were purified to more than 90% homogeneity. Orf112 protein was shown to specifically bind single-stranded genomic DNA of VGJφ; however, RstB protein unexpectedly bound double-stranded DNA in addition to the single-stranded genomic DNA. The DNA binding properties of these proteins may explain their requirement for the rolling circle replication of the respective phages and RstB's requirement for single-stranded-DNA chromosomal integration of CTXφ phage dependent on XerCD recombinases.
Vibrio cholerae, the etiologic agent of cholera, is a gram-negative bacterium which hosts several specific filamentous phages (1, 7, 8, 9, 10, 11, 13). CTXφ phage has been the most studied due to its role in pathogenicity and horizontal gene transfer (6). This phage is usually integrated into the genomes of toxigenic strains of V. cholerae, but it is also able to replicate directly from the bacterial chromosome (6) and to produce infective phage particles with potential for transducing the cholera toxin genes into nonpathogenic environmental strains (6, 13). Another filamentous phage important for its role in horizontal gene transfer is VGJφ, which is able to recombine with the CTXφ genome to originate a hybrid phage endowed with the full potential for virulence conversion. The hybrid phage shows an increased infectivity due to its specificity for the receptor mannose-sensitive hemagglutinin (receptor mannose-sensitive hemagglutinin pilus), which is ubiquitous among environmental strains (1, 2). Therefore, elucidating the biology of these phages is crucial for understanding the evolution of bacterial pathogenesis.
The genomes of CTXφ and VGJφ carry the putative homologous rstB and open reading frame 112 (orf112) genes, respectively. The requirement of rstB for the integration of CTXφ into the bacterial chromosome has been described (14). However, the biochemical function of the gene product has not been elucidated. Genes rstB and orf112 are positional and size homologues of genes encoding single-stranded DNA (ssDNA)-binding proteins (SSB) in other filamentous phages (1). It is expected that the proteins encoded by rstB and orf112 exert similar functions in the biology of their respective phages (1). Thus, we wanted to evaluate the ssDNA-binding activity of these ORF products.
To asses whether the Orf112 product and RstB have SSB activity, sufficient amounts of pure proteins are required. This paper describes quick purification protocols used to obtain both protein species and the evaluation of their DNA binding activities. The Orf112 protein was obtained from V. cholerae strain 569B (serogroup O1, Inaba classical biotype) infected with VGJφ, which expresses high levels of the protein. The infected bacteria were inoculated into 300 ml of LB broth and were cultured with shaking overnight at 200 rpm and 37°C. Parallel uninfected batches of 569B were also processed. Cells were collected by centrifugation for 15 min at 9,000 × g and at 4°C and stored at −20°C until processed.
A recombinant rstB gene with a hexahistidine tag coding region fused to the C terminus of the respective protein product (RstB-His) was constructed by cloning the gene into the expression vector pBAD/Myc-HisC (Invitrogen). The rstB gene was PCR amplified from the CTXφ genome using the oligonucleotides CNC06-171 (5′-AGTTCCATGGGGAAATTATGGGTGATAAT-3′) and CNC06-173 (5′-CATCAAGCTTTAATGGGT-3′), which introduce restriction sites for NcoI and HindIII at the amplicon ends. The amplified fragment was digested with both enzymes and cloned into the same sites of pBAD/Myc-HisC. In the resultant construction, named pBAD/Myc-HisC-rstB 9, expression of the recombinant protein is inducible by arabinose.
Plasmid pBAD/Myc-HisC-rstB 9 was electroporated into Escherichia coli Top 10. A 1-ml sample of an overnight, 5-ml, ampicillin-supplemented LB broth culture of transformed Top 10 was inoculated into 300 ml of fresh broth. The culture was incubated with orbital shaking at 200 rpm and 37°C until it reached an optical density at 600 nm of 0.5. To induce expression of the RstB-His protein, 0.002% (wt/vol) arabinose was added and the culture was reincubated for three additional hours. Parallel batches of pBAD/Myc-HisC-transformed E. coli Top 10 were processed as a negative control. Cells were sedimented by centrifugation for 15 min at 9,000 × g and 4°C and stored at −20°C until processed.
The expression of the Orf112 and RstB-His proteins was monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Cell extracts of VGJφ-infected 569B and E. coli Top 10 transformed with pBAD/Myc-HisC-rstB 9 contained proteins with apparent molecular sizes of 12.7 kDa (Fig. 1) and 16 kDa (Fig. 2B), respectively, which are not observed in cells from control cultures. The sizes match those predicted from orf112 (12.72 kDa) (see reference 1) and recombinant rstB-his (16.8 kDa).
FIG. 1.
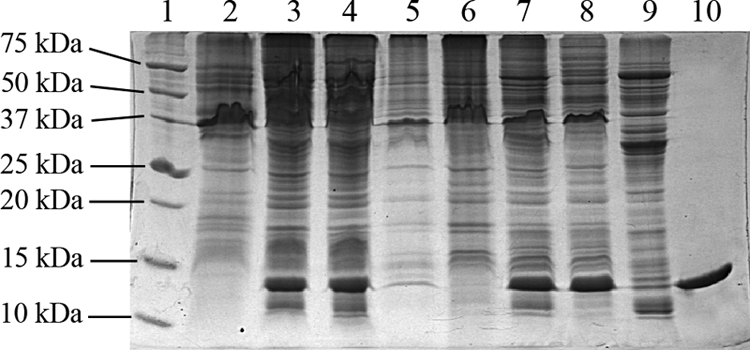
SDS-PAGE monitoring of the purification process of Orf112 protein. Lane 1, broad-range protein molecular mass markers (Promega); lane 2, cell extract of non-VGJφ-infected V. cholerae 569B; lane 3, cell extract of VGJφ-infected V. cholerae 569B; lane 4, soluble fraction of the sonicate; lane 5, insoluble fraction of the sonicate; lane 6, precipitate at 30% (NH3)2SO4; lane 7, supernatant at 30% (NH3)2SO4; lane 8, precipitate at 50% (NH3)2SO4; lane 9, supernatant at 50% (NH3)2SO4; lane 10, Orf112 protein electro-eluted after preparative SDS-PAGE.
FIG. 2.

Isolation and purification of RstB-His. (A) Chromatogram on a Ni-CAM HC His tag affinity column. (B) SDS-PAGE monitoring of the purification process of RstB-His. Lane 1, broad-range protein molecular mass markers (Promega); lane 2, cell extract of uninduced cultures; lane 3, cell extract of expression-induced cultures; lane 4, soluble fraction of the sonicate from expression-induced cultures; lane 5, insoluble fraction of the sonicate from expression-induced cultures; lane 6, soluble fraction of the 8 M urea extract; lane 7, RstB-His eluted from the column.
These proteins are not secreted into the growth medium (not shown); thus, they were released from the cells by ultrasonic disruption as previously described (4). V. cholerae was suspended in 15 ml of 20 mM Tris-HCl buffer, pH 7.5, while E. coli cells were suspended in 15 ml of 20 mM sodium phosphate, 0.5 M NaCl, 10 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride, pH 8.0. Cell lysates were cleared by centrifugation for 40 min at 9,000 × g and 4°C. SDS-PAGE detected Orf112 protein in the soluble fraction, while RstB-His remained in the insoluble fractions of cell extracts (Fig. 1). Subsequently, 569B cell lysate supernatants containing Orf112 protein were fractionated with ammonium sulfate. At 30% ammonium sulfate, several contaminants precipitated but Orf112 protein remained in solution, while at 50% ammonium sulfate, Orf112 precipitated and was recovered by centrifugation. The pellet was washed twice with 50% ammonium sulfate and finally resuspended into 3 ml of 20 mM Tris-HCl buffer, pH 7.5. Removal of excess salt was achieved by gel filtration before the extract was applied to a preparative SDS-PAGE gel. Briefly, a 15% polyacrylamide gel (17 by 19 by 0.5 cm) was run at a constant current intensity of 100 mA and with free voltage at 4°C, until the bromophenol blue dye migrated off the gel. The gels were negatively stained with imidazole-zinc (3), and the Orf112 protein band was identified by comparing bands with the bands of a negative control applied in a neighboring lane of the same gel, where this protein was not visible. The ORF112 protein band was cut from the gel, and the slice was fragmented and introduced into a dialysis bag with a 6- to 8-kDa molecular mass cutoff in 10 ml of 24 mM Tris-HCl-250 mM glycine-0.5% (wt/vol) SDS buffer. The protein was electro-eluted for 5 h at a current intensity of 70 mA and with free voltage at 4°C. Reverse current was applied for 5 min to release membrane-bound proteins, and gel fragments were discarded. The same sample was dialyzed against 1 liter of 0.5 M Tris-HCl, 0.25 M glycine buffer, pH 7.5, for 24 h at 4°C with constant stirring. The dialysis was repeated with 20 mM Tris-HCl, 0.5 M NaCl buffer, pH 7.5, for 24 h. No contaminants were seen when 25 μg of this Orf112 protein-dialyzed extract was checked by SDS-PAGE and Coomassie brilliant blue staining (Fig. 1, lane 10).
RstB-His protein was recovered from the insoluble fraction of the E. coli lysate by dissolving the lysate in 15 ml of a buffer containing 8 M urea, 20 mM sodium phosphate, 0.5 M NaCl, and 10 mM imidazole, pH 8.0. The mixture was stirred overnight at 4°C and cleared by centrifugation at 9,000 × g for 40 min. The supernatant was applied to a Ni-CAM HC matrix (Sigma), and urea was removed using a linear gradient from 8 to 0 M urea as previously described (5). The presence of 10 mM imidazole in the sample and binding buffer was intended to reduce the level of contaminants bound to the column. Protein was eluted using a gradient of imidazole (10 to 250 mM) in 20 mM sodium phosphate, 0.5 M NaCl buffer, pH 8.0. Fractions were assayed by SDS-PAGE, and those containing the RstB-His protein were pooled according to purity rather than yield. RstB-His protein was obtained with 90% purity (Fig. 2B, lane 7), according to a densitometry scan of Coomassie brilliant blue-stained gels, using a Gene Genius gel documentation system (Syngene Synoptics Ltd., Cambridge, United Kingdom). The gradient-based removal of urea allowed effective solubilization of RstB-His without significant precipitation of protein in the column, as described before (5).
Biological activity was assayed by retardation assays of VGJφ genomic ssDNA by 0.5% agarose gel electrophoresis conducted with 20 mM EDTA, 40 mM Tris-acetate buffer. Various amounts of each protein and ssDNA from VGJφ (0.5 μg) mixed in 20% glycerol, 0.25 mM EDTA, 0.3 μM bovine serum albumin, 20 mM Tris-HCl up to a total volume of 40 μl were incubated at room temperature for 30 min and loaded into the gel for analysis. The electrophoresis was run at 100 V and 4°C until colorant exit. Ethidium bromide (1 μg/ml) was used for 30 min to stain DNA bands, which were documented in a Gene Genius gel system (Syngene Synoptics Ltd., Cambridge, United Kingdom).
Orf112 protein exhibited DNA retardation activity, showing a high specificity of binding for the circular ssDNA of VGJφ, but was unable to bind double-stranded DNA (dsDNA) (Fig. 3A). However, RstB was able to bind ssDNA as well as dsDNA substrates (Fig. 3B). No retardation was observed with protein preparations from negative controls or when the DNA-protein mixture was inactivated with 1:1 (vol/vol) phenol-chloroform, indicating that the binding activity is intrinsic to the purified proteins.
FIG. 3.

Gel retardation assays of VGJφ-ssDNA by Orf112 protein or RstB-His binding, as measured by 0.5% agarose gel electrophoresis. (A) Binding of Orf112 protein. Lane 1, control of 500 ng of genomic ssDNA of VGJφ; lanes 2 to 6; same as for lane 1 plus 1.25, 2.50, 5.00, 10.0, and 20.0 μg of Orf112 protein, respectively; lane 7, same as for lane 6 but treated with phenol-chloroform; lane 8, linearized replicative-form dsDNA of VGJφ; lane 9, same as for lane 8 plus 20.0 μg of Orf112; lane 10, linearized pUC19; lane 11, same as for lane 10 plus 20.0 μg of Orf112. (B) Binding of RstB-His. Lane 1, control of 500 ng of genomic ssDNA of VGJφ; lane 2, same as for lane 1 plus 20 μg of RstB treated with phenol-chloroform; lanes 3 to 8, same as for lane 1 plus 0.62, 1.25, 2.50, 5.00, 10.0, and 20.0 μg of RstB-His, respectively; lane 9, linearized replicative-form dsDNA of VGJφ; lane 10, same as for lane 9 plus 20.0 μg of RstB-His; lane 11, linearized pUC19; lane 12, same as for lane 11 plus 20.0 μg of RstB-His.
In the case of RstB, which binds to ssDNA and dsDNA substrates, we wanted to rule out the possibility that this effect was caused by the His tail fused to the recombinant protein. For this, we recloned RstB in the same vector as RstB-His but without the His tail. We used the same procedure described above for RstB-His but used oligonucleotides CNC06-171 (see above) and CNC06-172 (5′-TACTGCAGTCAAGATTTAATGGGTTG-3′) for RstB amplification. In this case, CNC06-172 introduced a restriction site for PstI, which was used for cloning into pBAD/Myc-HisC.
For purification of this RstB variant, E. coli growth and protein expression induction was done as described for RstB-His. Again, RstB was recovered in the insoluble fraction after cell disruption by sonication. Inclusion bodies were resuspended in 15 ml of 50 mM phosphate buffer, pH 7.7, containing 8 M urea, and after overnight stirring at 4°C, the suspension was cleared by centrifugation (9,000 × g, 40 min). The supernatant was applied to an SP Sepharose fast-flow column (Amersham, United Kingdom), and urea was removed as described above for RstB-His. The RstB that bound to the matrix was eluted using a gradient of 0 to 2 M NaCl and was obtained with about 92% purity (data not shown). RstB without the His tail also showed binding activity toward ssDNA and dsDNA substrates (data not shown), ruling out the possibility that the His hexamer is responsible for the nonspecific binding of RstB-His.
Since RstB has affinity for both ssDNA and dsDNA, the possibility exists that this protein simply binds any DNA nonspecifically due to an effect of a charge interaction with the phosphate backbone of DNA. In order to study the effect of the charge in the DNA-binding activity of RstB, NaCl was included in the reaction mixture at a concentration from 0 to 500 mM (Fig. 4). As can be seen in Fig. 4, the retardation activity of ssDNA was only partially inhibited from a starting concentration of 400 mM (Fig. 4A), while the retardation of dsDNA started to be partially inhibited at 300 mM NaCl (Fig. 4B). Since RstB continues to bind at high salt concentrations, which should equilibrate the charge effect, these results indicate that the DNA binding activity is not due to the presence of positively charged amino acids in the protein backbone but rather to the presence of domains that specifically recognize the ssDNA or dsDNA.
FIG. 4.

Effect of salt concentration and nonrelated dsDNA competition on the binding of RstB. (A) RstB binding to phage ssDNA in the presence of 0 to 500 mM NaCl as detected by 0.5% agarose gel electrophoresis. Lane 1, 400 ng of genomic ssDNA of VGJφ (control); lane 2, same as for lane 1 plus 15.6 μg of RstB-His; lanes 3 to 7, same as for lane 2 plus 100, 200, 300, 400, and 500 mM NaCl, respectively. (B) RstB binding to replicative-form phage dsDNA in the presence of 0 to 500 mM NaCl. Lane 1, 400 ng of dsDNA of VGJφ (control); lane 2, same as for lane 1 plus 15.6 μg of RstB-His; lanes 3 to 7, same as for lane 2 plus 100, 200, 300, 400, and 500 mM NaCl. (C) DNA-binding competition by RstB. Lane 1, 400 ng of genomic ssDNA of VGJφ (control); lane 2, same as for lane 1 plus 15.6 μg of RstB-His; lanes 3 and 4, same as for lane 2 plus 500 and 1,000 ng of sheared calf thymus dsDNA, respectively; lane 5, 500 ng of calf thymus DNA (control).
We also investigated whether RstB-His has more affinity for the phage ssDNA than for a nonrelated dsDNA; a DNA-binding competition experiment in which the protein was incubated with a constant amount of ssDNA of VGJφ phage and increasing amounts of calf thymus DNA was performed (Fig. 4C). The ssDNA of VGJφ was retarded by RstB-His even in the presence of 500 and 1,000 ng of calf thymus dsDNA (Fig. 4C, lanes 3 and 4). These results indicate that RstB protein has more affinity for the phage ssDNA than for a non-phage-related dsDNA.
Until we know more, RstB is the first protein of a filamentous phage which shows affinity for both ss- and dsDNA, at least in vitro. It is possible that RstB needs another protein from the host or the phage itself to recognize the ssDNA in a specific manner, or perhaps the affinity of RstB for both ss- and dsDNA is an intrinsic property of the protein, which is needed on one hand for binding to genomic ssDNA during the rolling circle replication of the phage and on the other hand for holding the hairpin dsDNA secondary structure formed by the phage genome that functions as the site for integration into the bacterial chromosome (12). This hairpin structure is used by XerCD recombinases as a substrate for recombining the phage genome with the bacterial chromosomal dif site (12), and RstB may act jointly with XerCD to achieve integration. This could explain the requirement of RstB for the integration of CTXφ.
It is concluded that Orf112 and RstB proteins purified by the protocols described in this paper were biologically active and obtained at a high degree of purity, which paves the way for further characterization of these proteins. The SSB activities of these two proteins are shown for the first time. Consequently, we propose to rename their respective genes gVVGJφ and gVCTXφ and their proteins pVVGJφ and pVCTXφ, to follow the denomination of genes of canonical phages of the Inovirus genus. A biochemical and chemical-physical characterization of both proteins is in progress and will be published elsewhere. Should the in vitro role demonstrated for these proteins operate in vivo as well, it might explain their role for rolling circle replication and why rstB is required for CTXφ integration.
Footnotes
Published ahead of print on 17 July 2009.
REFERENCES
- 1.Campos, J., E. Martinez, E. Suzarte, B. L. Rodriguez, K. Marrero, Y. Silva, T. Ledón, R. del Sol, and R. Fando. 2003. VGJφ, a novel filamentous phage of Vibrio cholerae, integrates into the same chromosomal site as CTXφ. J. Bacteriol. 1855685-5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campos, J., E. Martinez, K. Marrero, Y. Silva, B. L. Rodriguez, E. Suzarte, T. Ledon, and R. Fando. 2003. Novel type of specialized transduction for CTXφ or its satellite phage RS1 mediated by filamentous phage VGJφ in Vibrio cholerae. J. Bacteriol. 1857231-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castellanos-Serra, L., and E. Hardy. 2001. Detection of biomolecules in electrophoresis gels with salts of imidazole and zinc II: a decade of research. Electrophoresis 22864-873. [DOI] [PubMed] [Google Scholar]
- 4.Chakrabarti, S. R., K. Chaudhuri, K. Sen, and J. Das. 1996. Porins of Vibrio cholerae: purification and characterization of OmpU. J. Bacteriol. 178524-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colangeli, R., A. Heijbel, A. M. Williams, C. Manca, J. Chan, K. Lyashchenko, and M. L. Gennaro. 1998. Three-step purification of lipopolysaccharide-free, polyhistidine-tagged recombinant antigens of Mycobacterium tuberculosis. J. Chromatogr. B 714223-235. [DOI] [PubMed] [Google Scholar]
- 6.Davis, B. M., and M. K. Waldor. 2003. Filamentous phages linked to virulence of Vibrio cholerae. Curr. Opin. Microbiol. 635-42. [DOI] [PubMed] [Google Scholar]
- 7.Faruque, S. M., I. Bin Naser, K. Fujihara, P. Diraphat, N. Chowdhury, M. Kamruzzaman, F. Qadri, S. Yamasaki, A. N. Ghosh, and J. J. Mekalanos. 2005. Genomic sequence and receptor for the Vibrio cholerae phage KSF-1Φ: evolutionary divergence among filamentous vibriophages mediating lateral gene transfer. J. Bacteriol. 1874095-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honma, Y., M. Ikema, C. Toma, M. Ehara, and M. Iwanaga. 1997. Molecular analysis of a filamentous phage (fs1) of Vibrio cholerae O139. Biochim. Biophys. Acta 1362109-115. [DOI] [PubMed] [Google Scholar]
- 9.Ikema, M., and Y. Honma. 1998. A novel filamentous phage, fs2, of Vibrio cholerae O139. Microbiology 1441901-1906. [DOI] [PubMed] [Google Scholar]
- 10.Jouravleva, E. A., G. A. McDonald, C. F. Garon, M. B. Finkelstein, and R. A. Finkelstein. 1998. Characterization and possible function of a new filamentous bacteriophage from Vibrio cholerae. Microbiology 144315-324. [DOI] [PubMed] [Google Scholar]
- 11.Kar, S., R. K. Ghosh, A. N. Ghosh, and A. Ghosh. 1996. Integration of the DNA of a novel filamentous bacteriophage VSK from Vibrio cholerae O139 into the host chromosomal DNA. FEMS Microbiol. Lett. 14517-22. [DOI] [PubMed] [Google Scholar]
- 12.Val, M.-E., M. Bouvier, J. Campos, D. Sherratt, F. Cornet, D. Mazel, and F. Barre. 2005. A new mechanism for bacteriophage integration using single strand DNA. Mol. Cell 19559-566. [DOI] [PubMed] [Google Scholar]
- 13.Waldor, M. K., and J. J. Mekalanos. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 2721910-1914. [DOI] [PubMed] [Google Scholar]
- 14.Waldor, M. K., E. J. Rubin, G. D. Pearson, H. Kimsey, and J. J. Mekalanos. 1997. Regulation, replication, and integration functions of the Vibrio cholerae CTXφ are encoded by region RS2. Mol. Microbiol. 24917-926. [DOI] [PubMed] [Google Scholar]