

Abstract
ChvE is a chromosomally encoded protein in Agrobacterium tumefaciens that mediates a sugar-induced increase in virulence (vir) gene expression through the activities of the VirA/VirG two-component system and has also been suggested to be involved in sugar utilization. The ChvE protein has homology to several bacterial periplasmic sugar-binding proteins, such as the ribose-binding protein and the galactose/glucose-binding protein of Escherichia coli. In this study, we provide direct evidence that ChvE specifically binds the vir gene-inducing sugar d-glucose with high affinity. Furthermore, ChvE mutations resulting in altered vir gene expression phenotypes have been isolated and characterized. Three distinct categories of mutants have been identified. Strains expressing the first class are defective in both virulence and d-glucose utilization as a result of mutations to residues lining the sugar-binding cleft. Strains expressing a second class of mutants are not adversely affected in sugar binding but are defective in virulence, presumably due to impaired interactions with the sensor kinase VirA. A subset of this second class of mutants includes variants of ChvE that also result in defective sugar utilization. We propose that these mutations affect not only interactions with VirA but also interactions with a sugar transport system. Examination of a homology model of ChvE shows that the mutated residues associated with the latter two phenotypes lie in two overlapping solvent-exposed sites adjacent to the sugar-binding cleft where conformational changes associated with the binding of sugar might have a maximal effect on ChvE's interactions with its distinct protein partners.
Virulent strains of Agrobacterium tumefaciens contain the tumor-inducing (Ti) plasmid that carries virulence (vir) operons. Products of vir operons are involved in infecting wound sites of dicotyledonous plants and initiating tumor formation. The expression of vir genes in A. tumefaciens is activated by plant-released signals, namely, phenolic derivatives, acidic pH, and monosaccharides (for a review, see reference 6), via the combined activities of the periplasmic protein ChvE and the VirA/VirG two-component regulatory system. Upon perception of these plant signals, autophosphorylated VirA, a transmembrane histidine kinase, transfers a phosphoryl group to VirG, a response regulator, and then the phosphorylated VirG activates the expression of vir genes by binding vir boxes in their promoters (8, 19, 24, 31, 52).
Perception and transduction of the sugar signals is crucial to the virulence of A. tumefaciens: strains lacking ChvE, a chromosomally encoded putative sugar-binding protein, are significantly less virulent than wild-type strains (17, 18). Previous studies have shown that, in fact, sugar signaling is neither sufficient for nor absolutely required for vir gene expression. Rather, sugars vastly increase both the sensitivity of VirA to phenol derivatives, such as acetosyringone (AS), and the maximal levels of vir gene expression observed at saturating levels of such compounds (for a review, see reference 26). The periplasmic domain of VirA is required for transduction of the sugar and pH signals (7, 8, 16, 41), whereas the so-called “linker” domain, located in the cytoplasm between the second transmembrane domain and the kinase domain, is required for perception and transduction of the phenolic signals (8, 46, 47).
A working model for the ChvE/sugar/VirA signaling pathway suggests that monosaccharide-bound ChvE interacts with the periplasmic domain of VirA to relieve periplasmic repression, resulting in maximal sensitivity of VirA to phenolic signals (7, 11, 32, 41). However, limited evidence has been presented to reveal how ChvE recognizes monosaccharides and how it interacts with the periplasmic domain of VirA. Shimoda et al. (41) identified a mutant chvE allele [chvE(T211M)] that is able to suppress a sugar-insensitive virA allele [virA(E210V)], thereby restoring the sugar-sensing ability. The suppressing effect of chvE(T211M) was then proposed to be the result of the specific restoration of the capacity of VirAE210V to bind ChvET211M. However, ChvET211M also activated wild-type VirA in the absence of sugars (32), suggesting that this mutant may not be a site-specific suppressor of VirAE210V. Based on a homology model of ChvE, a recent study (16) does predict, though, that the residue T211 is located on the surface of the ChvE protein, consistent with the model that T211 is in a position to interact with the periplasmic domain of VirA.
Based on sequence similarity, ChvE is a member of the periplasmic sugar-binding protein (PSBP) family. The structures of some PSBPs, including two ChvE homologues in Escherichia coli, ribose-binding protein (RBP) and glucose/galactose-binding protein (GBP), have been solved. The family of PSBPs shares very similar structural features, and each of them contains two similar but distinct globular domains connected by a flexible hinge (38). A sugar-binding site is located at the cleft between the two domains. PSBPs play an important role in active sugar transport, and some of them also serve as an initial receptor for sugar chemotaxis (45). A wealth of evidence has demonstrated that some specialized regions located on the surfaces of PSBPs are important for transport and chemotactic functions. In the case of RBP, four distinct regions spanning the N-terminal and C-terminal domains are involved in interaction with its permease (a transport partner), its chemotransducer (a chemotactic partner), or both (5, 15). In GBP, one residue was identified as being specifically involved in chemotaxis but not transport (36, 49). For maltose-binding protein (MBP), which is also a member of the PSBP family, two well-defined regions located on each domain of the protein are involved in interaction with its chemotransducer (54). These regions partially overlap with the regions involved in interaction with its permease (25, 54). Structural analysis indicates that both domains of MBP have direct interactions with its transport partners (35).
ChvE also appears to be a highly versatile protein: not only does it play an important role in virulence, but as in the case of the PSPBs described above, it has been indicated to be a primary receptor for transport of and chemotaxis toward some sugars (7). This raises important biological/biochemical questions. How can ChvE interact with three presumably different periplasmic components of systems that are respectively involved in virulence, sugar utilization, and chemotaxis? How are the interactions of ChvE with these periplasmic components structurally segregated: do the interactions occur on the same or different regions of ChvE? To address these issues, we employed genetic and biophysical approaches to identify the residues of ChvE involved in sugar utilization versus the residues involved in virulence. The residues of both groups were mapped onto a homology model of ChvE based on a high-resolution crystal structure of E. coli GBP (PDB ID, 2ipn). Our results identify an extended surface spanning both the N-terminal and C-terminal domains of ChvE that is essential for interacting with VirA and that partially overlaps the surface responsible for the interaction of ChvE with a putative ABC sugar transport protein.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The E. coli TOP10 and XL1-Blue strains were used for the construction and amplification of plasmids. E. coli BL21(DE3) was used for the T7-directed expression of pET22b-derived plasmids.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZM15 ΔlacX74 deoR nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL (Strr) endA1 λ− | Invitrogen |
| BL21λ(DE3) | F−ompT gal dcm lon hsdSB (rB− mB−) with DE3, a λ prophase carrying the T7 RNA pol gene | Invitrogen |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac[F′ proAB laclqZΔM15 Tn10 (Tcr)] | Stratagene |
| A. tumefaciens | ||
| A348 | C58 background carrying pTiA6 | 17 |
| A1068 | A723 (C58 background with pTiB6806) carrying chvE::Tn5; Kmr | 18 |
| AB300 | A348 chvE deletion | This study |
| Plasmids | ||
| pBBR1MCS5 | Broad-host-range plasmid cloning vector; Gmr | 21 |
| pET22b | E. coli T7 expression vector; Apr | Novagen |
| pSW209Ω | pSM243cd derivative with a PvirB::lacZ fusion; IncP Spr | 50 |
| pK18mobsacB | Suicide plasmid in X. campestris pv. campestris; Kmr | 39 |
| pRG165 | PN25-chvE in pYW15; Apr | 16 |
| pGN102 | PN25-chvE-Strep in pBBR1MCS5 | This study |
| pFH100 | chvE-His6 in pET22b | This study |
| pFH101 | chvE(S197W)-His6 in pET22b | This study |
| pFH102 | chvE(R92C)-His6 in pET22b | This study |
| pFH103 | chvE(T72K)-His6 in pET22b | This study |
| pFH105 | chvE(D142V)-His6 in pET22b | This study |
| pLH3 | chvE(T187R)-His6 in pET22b | This study |
| pLH4 | chvE(T187P)-His6 in pET22b | This study |
The first amino acid residue on the mature ChvE protein (not including the 24-amino-acid signal peptide) is numbered as 1. Mutants with single amino acid substitutions are denoted by the combination of the original amino acid and its position in the amino acid sequence of the mature ChvE protein followed by the substituted amino acid, as described in Materials and Methods.
E. coli strains were cultured in Luria-Bertani (LB) medium at 37°C (30). A. tumefaciens strains were grown at 25°C on LB medium, AB minimal medium (9), or AB induction medium (53). Antibiotics for E. coli were used at the following concentrations (μg/ml in liquid medium/solid medium): spectinomycin (50/100), ampicillin (50/125), kanamycin (25/50), and gentamicin (12.5/25). Antibiotics for A. tumefaciens were used at the following concentrations (μg/ml in liquid medium/solid medium): spectinomycin (50/100), carbenicillin (30/100), and gentamicin (100/200).
Strain and plasmid construction.
To introduce a nonpolar chvE (Atu2348) null mutation into the chromosome of strain A348 (17), marker-exchange eviction mutagenesis (4) was carried out by sequential pair PCRs (PCRs) using the suicide plasmid pK18mobsacB (39). The first PCR amplified the upstream chvE flanking sequence and a small portion of the 5′ chvE coding sequence using primers I (5′AATGCGGCCGATGATGAAA3′) and II (5′TTATTTCAGCTGAATGGACTTCAT3′), creating an 800-bp product. The second PCR generated a 500-bp fragment containing the downstream chvE flanking sequence and internal sequence from the 3′ end of chvE using primers III (5′CAAGGAAGACCAGCTG AAATAA3′) and IV (5′AGCGTCCCGGGCGATTCCTT3′). In the third PCR, the products from the first two PCRs were used as templates to generate a 1.3-kb fragment carrying chvE flanking sequences and a small in-frame internal sequence using primers I and IV.
The 1.3-kb PCR fragment was cloned into the XmaI site of pK18mobsacB, and the resulting construct was electroporated into A348. Colonies were screened for the first recombination event on LB medium with kanamycin and for the second recombination on LB medium with 10% sucrose. The resulting isolate, containing the nonpolar chvE null mutation (confirmed by PCR using primers that were outside the region of mutagenesis), was named AB300.
For the construction of pGN102, a 1.2-kb PN25-chvE fragment was excised from pRG165 (16) as an NcoI-KpnI fragment and ligated into pBBR1MCS5 (21). The resulting construct, pGN100, was used to construct the allele encoding a C-terminally epitope-tagged version of ChvE. Two primers were used to clone this allele: the first (5′TCCGCAACAGCGGCGACGTC3′) amplified across the unique AatII restriction of chvE site at position 371 on the chvE gene, and the second (5′GGGGTACCCCTTATTATTTTTCGAACTGCGGGTGGCTCCAAGCGCTTTTCAGCTGGTCTTCCT3′) carried the sequence for StrepTagII (SAWSHPQFEK). The primer was designed to add the epitope tag sequence immediately before the stop codon of chvE and also had a KpnI site at the end to enable easy cloning. The underlined base pairs in the primer sequences denote restriction sites. The resulting 765-bp PCR product, digested with KpnI-AatII, was ligated with pGN100 (digested with the same enzymes) to result in the construct pGN102.
To express the ChvE protein with the His epitope tag at its C terminus, the open reading frame of the chvE gene was amplified from pGN102 using the oligonucleotides ChvENdeI5 (5′GGAATTCCATATGAAGTCCATTATTTCG3′) and ChvEXhoI3 (5′CCGCTCGAGTTTCAGCTGGTCTTCCTTG3′). The restriction sites are indicated by the underlined base pairs. A 1.1-kb PCR product was digested with NdeI and XhoI and cloned into those sites of pET22b, generating pFH100 which contains the coding region of full-length ChvE fused in frame to six His codons at its carboxyl terminus. The same technique was used to construct ChvE mutants with the His epitope tag at their C termini as shown in Table 1.
Mutagenesis and screening.
Random mutagenesis of the chvE allele on pGN102 was carried out using a GeneMorph II EZClone domain mutagenesis kit from Stratagene. The chvE gene fragment was randomly mutagenized by PCR using the Mutazyme II DNA polymerase according to the manufacturer's instructions. The purified PCR product was then used in the GeneMorph II EZClone reaction mixture to regenerate the pGN102 plasmid for expression of the mutated chvE. The mutagenized plasmid library was transformed into AB300 carrying pSW209Ω (50) and plated onto AB induction medium (pH 5.5) containing 40 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and 10 μM AS in the presence of either 14 mM glucose (glucose medium) or 24 mM glycerol (glycerol medium) as a sole carbon source. White or light-blue colonies on the glucose medium plates were candidates with reduced VirA activity. On the glycerol medium plates, blue colonies were putative mutants reflecting the activation of vir gene expression under noninducing conditions for wild-type ChvE. The putative mutants were isolated and further tested by vir gene induction assay. Mutant chvE alleles were sequenced after verification that their phenotypes were plasmid linked in a fresh AB300 strain carrying pSW209Ω. Some mutants carrying double mutations were used to regenerate single mutations by PCR site-directed mutagenesis using a QuikChange site-directed mutagenesis kit from Stratagene.
Mutants with single amino acid substitutions were denoted by the combination of the original amino acid, in the one-letter notation, and its position in the amino acid sequence of the mature ChvE protein (not including a 24-amino-acid signal peptide predicted using the SignalP version 3.0 software [3]), followed by the substituted amino acid. The mutant T187M from our study is equivalent to mutant T211M described in previous studies (16, 41) because the designation of the latter counted the 24-amino-acid signal peptide.
vir gene induction assays.
vir gene induction was analyzed by triplicate measurements of β-galactosidase activity produced from a strain carrying a PvirB::lacZ fusion on pSW209Ω (50). Cultures grown overnight in LB medium were used to inoculate AB induction medium buffered at pH 5.5 with 20 mM MES (morpholineethanesulfonic acid) containing 14 mM glucose (Fisher Scientific) or 28 mM glycerol (Invitrogen) at various concentrations of AS (Sigma). Samples were prepared and assayed by the method of Miller (30). The effective dose (ED50) is defined as the concentration of AS that yields 50% of maximal vir gene expression (β-galactosidase activity). It is used as a measure to quantify the sensitivity of VirA to the inducing phenolic signal (32).
Bacterial growth assays.
A. tumefaciens strains were grown to stationary phase on AB minimal medium containing 28 mM glycerol as a sole carbon source at 25°C in the presence of antibiotics and then diluted to an optical density at 600 nm of 0.06 to 0.08 in AB minimal medium (pH 5.5) supplemented with antibiotics in the presence of either glucose or glycerol as a sole carbon source. Cultures were incubated with shaking at 25°C. At intervals, measurements of the optical density at 600 nm were taken with a spectrophotometer.
Periplasmic extracts and protein purification.
For expression of the wild-type and mutant ChvE-His6 recombinant proteins, the pET22b-derived plasmids (Table 1) were transformed into E. coli BL21λ(DE3). Protein expression was induced by adding 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to mid-log-phase cultures. At 2 h postinduction, cells were harvested for periplasmic protein extraction. A modified osmotic shock procedure (34) was carried out to prepare periplasmic proteins. The harvested cell pellet was washed with 30 mM Tris (pH 8.0) and resuspended at 80 ml per gram wet weight in the same buffer with 20% sucrose. After the addition of EDTA to 1 mM, the cell suspension was incubated on ice for 10 min with gentle agitation. The cells were pelleted by centrifugation and resuspended in the same volume of ice-cold shock solution (5 mM MgSO4). After 10 min in an ice bath with shaking, the cells were pelleted by centrifugation at 8,000 × g for 20 min, and the supernatant, containing the periplasmic fluid, was decanted into a new tube.
The ChvE protein was purified under native conditions by using Ni-nitrilotriacetic acid resin according to the manufacturer's protocol (Qiagen).
Immunoblot analysis.
Each strain was grown at 25°C in AB minimal medium (pH 5.5) with glucose as a single carbon source. Equal amounts of cells were harvested, washed in phosphate-buffered saline, resuspended in Laemmli sample buffer, and heated at 100°C for 10 min. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed as described by Laemmli (22), using a 10% (wt/vol) separating gel. The gels were then transferred to a polyvinylidene difluoride membrane (Amersham). ChvE was visualized through Western blot analysis with anti-ChvE (41) primary antibody, horseradish peroxidase-conjugated anti-rabbit immunoglobulin secondary antibody (Amersham Biosciences), and ECL Plus (Amersham Biosciences).
Fluorescence spectroscopy.
Fluorescence measurements were carried out using a QuantaMaster-4/2003 spectrofluorometer (Photon Technology International). The spectral bandwidth for both excitation and emission was set to 3 nm. To determine an optimal excitation wavelength for the ChvE protein sample, excitation scanning was performed at the emission wavelength of 330 nm. Emission spectra were then measured following excitation at the optimal wavelength, 284 nm. Purified protein samples were prepared in 10 mM Tris-HCl, pH 7.3, and 50 mM NaCl. In the standard procedure, sample buffer was added up to 2 ml of the total sample volume in each cuvette, and the solution was completely mixed by stirring. The fluorescence intensity was measured after a 5-min temperature equilibration. For sugar-binding assays, 10-μl amounts of different sugar solutions at the appropriate concentrations were added to the protein samples, and readings were taken after 1 min of stirring. A control sample receiving buffer instead of sugar solution was also measured because the fluorescence intensity was affected by the increased volume (10 μl). To remove the Raman peak of water (23), a second control was used to take the reading of the buffer without protein samples. Therefore, the actual fluorescence intensities of samples (expressed as counts) were corrected by those two control readings to yield the final data. All sugars tested in the buffer alone gave zero fluorescence.
In the fluorescence-quenching experiments, the excitation wavelength was changed to 295 nm in order to reduce the absorbance by acrylamide and potassium iodide. Small volumes of 4 M quencher solutions were added to the protein and protein-ligand samples, and the fluorescence spectra from 300 to 400 nm were recorded after each addition. All measurements were performed at room temperature. The quenching data are presented as a Stern-Volmer plot, which is the plot of F0/F versus [Q], where F0 and F are the observed fluorescence in the absence and presence of quencher and [Q] is the quencher concentration. When the Stern-Volmer plot is linear, it should yield an intercept of unity on the y axis and a slope equal to the Stern-Volmer quenching constant, KSV. A change in the intrinsic lifetime of the tryptophan residues (in the absence of quencher) would affect KSV, though this is unlikely given that the emission intensity remains essentially constant upon the addition of glucose.
Determination of KD.
Fluorescence intensities were measured as described above after the addition of 10 μl of glucose solution at different concentrations to the protein samples. All the measurements were repeated at least three times. Spectral shifts of the fluorescence can be quantified by the emission fluorescence center of mass (〈vg〉), a parameter defined by the equation
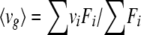 |
(1) |
where vi is the wavelength, Fi is the fluorescence intensity at vi, and the summations are over a range of appreciable values of F (43). The dissociation constant (KD) is related to 〈vg〉 by the following equation (modified from reference 1)
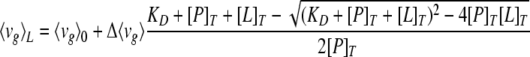 |
(2) |
where [L]T is the total concentration of ligand, 〈vg〉L is the center of mass at the given ligand concentration, 〈vg〉0 is the corresponding quantity when the ligand concentration is zero, and [P]T is the total concentration of the protein.
KD can be computed by nonlinear least-square fitting using 〈vg〉L measured at different ligand concentrations.
Model construction.
See the supplemental material for details of model construction.
RESULTS
Isolation of ChvE mutants with altered ability to activate the VirA/VirG system.
To characterize the residues of ChvE proteins involved in virulence, we screened for two types of ChvE mutants that have an altered activity in their response to sugar under vir gene induction conditions with limiting (low) concentrations of AS: (i) “down” mutants have a reduced ability to activate VirA activity in the presence of glucose, and (ii) “up” mutants are capable of inducing VirA activity even in the absence of added glucose. For this purpose, we created a chvE mutant library by random mutagenic PCR (see Materials and Methods) and transformed a pool of plasmid DNA carrying the chvE mutant library into a chvE in-frame deletion strain, AB300, carrying pSW209Ω (PvirB-lacZ) for use as a reporter of vir gene expression (50). The resulting transformants were plated on AB minimal medium (pH 5.5) containing a low level (10 μM) of AS and either glucose (glucose medium) or glycerol (glycerol medium) as a sole carbon source. In glucose medium, the wild-type colonies were blue; thus, colonies harboring mutants of ChvE with reduced color were candidates for down mutants that are incapable of glucose-dependent response to low levels of AS. In glycerol medium, the wild-type colonies were white; thus, blue colonies identified under these conditions harbored up mutants of ChvE that activated vir gene expression at low AS concentrations even in the absence of exogenously added glucose. After the confirmation that the phenotypes of isolated mutants were indeed plasmid linked, the expression levels of the mutant ChvE proteins were examined by immunoblot analysis. Only the mutants that had expression levels similar to the level in the wild type (data not shown) were chosen for sequence analysis and characterization.
ChvE down mutants with impaired ability to activate the VirA/VirG system in the presence of glucose.
We identified 10 down mutants carrying single mutations in the chvE coding sequence. These alleles showed ∼17- to 1,184-fold reductions in vir gene expression at a low AS concentration as determined by the expression of a PvirB-lacZ fusion (Table 2). Strains expressing three different ChvE down mutant proteins (ChvEL181M, ChvET72K, and ChvET187R) were characterized via examination of their dose response to AS in the presence of 14 mM glucose (Fig. 1A). The ED50 for AS (32) in the strain with wild-type ChvE was about 3 μM AS, while the ED50 values for the strains expressing ChvEL181M,ChvET72K, and ChvET187R were approximately 50, 80, and 85 μM AS, respectively. The ED50 values quoted for ChvET72K and ChvET187R are lower limits, because the activity of β-galactosidase did not reach a maximum at the tested AS concentrations. We conclude that these ChvE mutant proteins greatly decreased the sensitivity of VirA to AS when the inducing sugar glucose is present. Moreover, at saturating levels of AS, none of the mutants reached the maximum activity of vir gene expression exhibited by the wild-type strain, a phenotype also observed in the case of the wild-type strain tested in the absence of inducing sugar (Fig. 1B).
TABLE 2.
Effects of ChvE mutant protein expression in AB300 strain (ΔchvE) on induction of PvirB-lacZ by 10 μM AS at pH 5.5 in the presence or absence of d-glucose
| Growth condition, type of mutant tested | Protein expressed | β-Galactosidase activity (Miller units) | Fold decrease (down mutant) or increase (up mutant) in β-galactosidase activity | Sugar utilizationa |
|---|---|---|---|---|
| Presence of d-glucose, down mutant | Wild-type ChvE | 2,978 | 1 | Normal |
| ChvET180M | 178 | 17 | Slow | |
| ChvEA190T | 74 | 40 | Normal | |
| ChvER92C | 71 | 42 | Slow | |
| ChvEL181 M | 69 | 43 | Normal | |
| ChvER92H | 29 | 104 | Slow | |
| ChvER191H | 19 | 160 | Normal | |
| ChvET72K | 10 | 298 | Slow | |
| ChvET187R | 7 | 436 | Normal | |
| ChvER182H | 6 | 467 | Normal | |
| ChvED142V | 3 | 1,184 | Slow | |
| None (vector) | 8 | 372 | Slow | |
| Absence of d-glucose, up mutant | Wild-type ChvE | 24 | 0 | Normal |
| ChvEE53K | 378 | 16 | Normal | |
| ChvEA186V | 700 | 30 | Normal | |
| ChvES197W | 1,579 | 67 | Slow | |
| ChvET187 M | 1,718 | 73 | Normal | |
| ChvET187P | 4,018 | 173 | Normal |
Growth assay was carried out in AB minimal medium with glucose as sole carbon source as described in Materials and Methods. Slow, cells grew ≥20% more slowly than the wild type; Normal, cells grew at the wild-type rate.
FIG. 1.

AS dose responses of ChvE mutants in vir gene expression. (A) Cells (AB300/pSW209Ω) carrying one of the chvE down mutants [chvE(T72K), chvE(L181M), and chvE(T187R)], wild-type chvE (pGN102), or the vector control (pBBR1MCS5) were grown in AB induction medium containing glucose as a sole carbon source. (B) Cells (AB300/pSW209Ω) carrying one of the chvE up mutants [chvE(T187P), chvE(T187M), and chvE(S197W)] or wild-type chvE (pGN102) were grown in AB induction medium with glycerol as the sole carbon source. Data shown are the means of the results of three independent experiments performed in duplicate. Error bars represent standard deviations. wt, wild type.
ChvE up mutants that activate the VirA/VirG system in the absence of glucose.
When tested in medium with a low level of AS in the absence of inducing sugars, the strains expressing five different mutant ChvE proteins (ChvES197W, ChvEA186V, ChvEE53K, ChvET187P, and ChvET187M) showed ∼16- to 173-fold increases in vir gene induction compared to the level in the strain with wild-type ChvE (Table 2) even though they all exhibited a wild-type level of vir gene induction in the presence of glucose (data not shown). Note that residue T187 corresponds to residue T211 described by others (16, 41) who included the 24-amino-acid signal peptide in their designation. The ED50 for AS in strains with three of the mutant proteins (ChvET187P, ChvET187M, and ChvES197W) tested in glycerol-containing medium were 2, 18, and 7 μM, respectively, values considerably lower than that for the strain expressing wild-type ChvE (about 100 μM) (Fig. 1B). Moreover, each of the three mutant strains tested reached much higher levels of maximal induction than the wild type. These data indicate that the up mutant proteins do not require inducing sugar to enhance the sensitivity of VirA to AS or to promote maximal vir induction. Indeed, the addition of glucose (14 mM) to the medium for these up mutant strains failed to give rise to an additional enhancement in maximal vir induction over that observed for the corresponding mutant in glycerol medium (data not shown).
Effect of ChvE mutation on sugar-dependent growth.
An earlier report by Cangelosi et al. (7) demonstrated that a chvE mutant strain of A. tumefaciens had a slow growth phenotype when cultured in the presence of certain sugars as sole carbon source. Presumably, this impaired ability to grow on sugars reflects ChvE's role in sugar transport rather than a vir-dependent process. We therefore determined whether the chvE mutant strains isolated on the basis of changes in vir gene expression might also have altered growth phenotypes in liquid medium containing d-glucose as a sole carbon source. Cells harboring wild-type ChvE have a shorter lag phase and doubling time than those that do not express this protein (Fig. 2A). Furthermore, there was no growth difference between the mutant and wild-type cells in liquid medium with glycerol as a sole carbon source (data not shown). These observations clearly confirm the earlier report (7) that ChvE plays an important role in glucose utilization in A. tumefaciens. Growth curves were obtained for three down mutant strains (T72K, T187R, and L181M) and one up mutant strain (S197W) over 51 h in minimal medium with glucose as a sole carbon source (Fig. 2A). Strains expressing ChvET187R and ChvEL181M showed the wild-type growth phenotype, suggesting that neither substitution affects the sugar-binding or transport activity of ChvE. In contrast, the strain with ChvET72K behaved more like the chvE deletion strain in terms of lag phase, suggesting a defective function related to sugar utilization. The strain with the up mutant protein ChvES197W also showed a growth defect similar to that of ChvET72K, implying that the S197W mutation impaired the ability of ChvE to utilize sugar while at the same time allowing vir induction to proceed in the absence of sugar.
FIG. 2.

Effect of ChvE mutations on growth of A. tumefaciens strains. (A) Cells (AB300/pSW209Ω) carrying chvE mutants, wild-type chvE (pGN102), or vector control (pBBR1MCS5), were grown in AB minimal medium containing glucose as a sole carbon source. (B) Cells (AB300/pSW209Ω) carrying chvE mutants, wild-type chvE plasmid (pGN102), or the vector control (pBBR1MCS5) were grown for 24 h in AB minimal medium with glucose as the sole carbon source. Blank bars, controls; black bars, ChvE down mutants; gray bars, ChvE up mutants. Data shown are the means of the results of three independent experiments performed in duplicate. Error bars represent standard deviations. O.D.600, optical density at 600 nm; wt, wild type.
Because there is a large difference after 24 h in the glucose-dependent growth of the chvE deletion strain (AB300) carrying an empty vector versus that of the same strain carrying wild-type chvE (Fig. 2A), the growth phenotypes of all mutant strains were examined at this time point. The strains expressing five down mutant ChvE proteins (ChvER92H, ChvED142V, ChvER92C, ChvET180M, and ChvET72K) and one up mutant protein (ChvES197W) showed at least 20% slower growth than the strain expressing wild-type ChvE (Fig. 2B). The fact that these mutants grew normally in glycerol medium (data not shown) rules out the possibility that these mutants have a general growth defect. Therefore, the residues altered in these ChvE mutants affect both VirA activity and glucose-dependent growth, suggesting that the mutations may affect sugar-binding activity and/or interaction with putative glucose transport components. The other five down mutants (A190T, L181M, R191H, R182H, and T187R) and the remaining up mutants (A186V, E53K, T187P, and T187M) affect VirA activity but not sugar-dependent growth. Thus, these mutations may affect interactions with VirA without having an appreciable effect on ChvE's ability to bind glucose and to pass it on to proteins involved in glucose transport.
Cangelosi et al. (7) also showed that ChvE plays a role in chemotaxis toward some vir-inducing sugars, including glucose. We intended to utilize a swarm plate assay to characterize the chemotaxis phenotype of the chvE mutants described above. However, we were unable to confirm that ChvE is involved in chemotaxis toward glucose in A. tumefaciens (see the supplemental material) and, therefore, did not examine the chemotactic activity of the isolated chvE mutants.
Direct binding of d-glucose to ChvE detected by tryptophan fluorescence.
Heretofore, physical measurement of the sugar-binding capacity of ChvE has not been obtained. Intrinsic Trp fluorescence is a convenient method for monitoring the binding of ligands, particularly when Trp residues are proximal to the binding site (51). This method has been extensively used to examine the effects of ligands on GBP (27, 29, 42). GBP and some other PSBPs have Trp residues in their sugar-binding clefts which contribute to binding via CH-π interactions between the sugar and the aromatic ring (28, 49; for a review, see reference 38). Upon binding of substrates, the Trp residues are generally shielded from solvent due to a variety of effects that include (i) physical occlusion by the substrate, (ii) a decrease in solvent accessibility arising from the formation of a more-compact conformation with greater interactions between PSBP's two domains, and (iii) a decrease in the overall dynamics of the structure (10, 23, 27, 29, 48). ChvE has only two Trp residues, W18 and W183. To examine their likely locations relative to the binding site, we constructed a homology model of the protein based on the structure of GBP (Fig. 3; also see the supplemental material). Based on sequence alignment, both of these residues are predicted to lie within the putative sugar-binding pocket (Fig. 3): W183 is conserved in GBP, where it interacts extensively with d-glucose, whereas W18 of ChvE is conserved as a hydrophobic Met in GBP or an aromatic Phe residue in a related RBP (see Table S1 in the supplemental material). Thus, W18 and W183 are likely to be ideally situated to report the binding of d-glucose or any concomitant conformational changes that are likely to occur as a result of binding.
FIG. 3.

Homology model for ChvE, based on the crystal structure of the ligand-bound form of GBP (PDB ID, 2ipn). The side chains of W18 and W183 are shown in ball-and-stick representation in green. Although the level of sequence homology between ChvE and GBP is not great enough to define the fine details of the interaction between ChvE and d-glucose, d-glucose is expected to bind in the cleft between the two domains of ChvE. The sugar (purple) has been placed in the binding cleft based on the known structure of GBP. The Trp residues are both located in the binding cleft, proximal to the bound ligand.
To characterize sugar binding, ChvE-His6 was purified via Ni-nitrilotriacetic acid resin and tested for its fluorescence properties in the presence or absence of sugars. The addition of d-glucose at a high concentration (0.1 mM) to the solution containing the wild-type ChvE protein resulted in a blue shift of the maximum-intensity wavelength of fluorescence from 353 nm to 343 nm (Fig. 4A), which is consistent with the expected decrease in the polarity and an increase in the rigidity of the environment sampled by the Trp residues. In contrast, ChvE failed to show any spectral changes upon the addition of 0.1 mM d-ribose (Fig. 4B), which is a noninducing sugar for virulence (2). To determine the stoichiometry and affinity of the interaction of ChvE with d-glucose, the spectrum was measured as a function of increasing ligand concentration. The resulting titration curve conformed well to a classical binding isotherm, with a stoichiometry of one sugar/ChvE and a KD of 0.6 μM for wild-type ChvE (Fig. 5A), which is the same order of magnitude as that of E. coli GBP for d-glucose or d-galactose (42).
FIG. 4.

Tryptophan fluorescence properties of wild-type ChvE. Emission spectra of wild-type ChvE in the presence and absence of 0.1 mM d-glucose (A) and 0.1 mM d-ribose (B). Purified ChvE protein (0.18 μM) samples were prepared in 10 mM Tris-HCl, 50 mM NaCl, pH 7.3. The excitation wavelength was 284 nm, and both the excitation and emission slits were 3 nm. Fluorescence intensity was determined as described in Materials and Methods.
FIG. 5.

Determination of KDs of d-glucose for wild-type and mutant ChvE proteins. Protein concentration was 0.2 μM. The solid lines through the data represent the best fit obtained using equation 2 in Materials and Methods. (A) Wild-type ChvE. KD = (6.30 ± 0.77) × 10−7 M. (B) ChvER92C. KD = (5.80 ± 0.36) × 10−3 M.
To further probe the molecular basis for the ligand-induced fluorescence changes, we determined the extent to which the binding of d-glucose was able to shield the Trp residues from ionic and small-molecule fluorescence-quenching agents, specifically, iodide ion and acrylamide (13, 14). In the absence of d-glucose, acrylamide quenched the fluorescence emission of the ChvE protein: a roughly linear decrease in fluorescence intensity was obtained with increasing concentrations of acrylamide (Fig. 6). A Stern-Volmer plot of the data is linear and predicts a quenching constant (KSV) of 6.3 M−1, which is consistent with a largely exposed location on the surface of the protein. The value of KSV decreased dramatically, to 1.0 M−1, in the presence of 0.1 mM d-glucose, consistent with a decrease in accessibility for the quencher. Similar patterns were obtained with potassium iodide (KI) as a quencher (data not shown). Taken together, these data are in excellent agreement with the predicted location of the Trp residues in the homology model of ChvE.
FIG. 6.

Quenching of protein fluorescence emission by acrylamide. Acrylamide with different concentrations was added to ChvE protein samples (0.2 μΜ in 10 mM Tris-HCl, 50 mM NaCl, pH 7.3) in the presence and absence of 0.1 mM d-glucose. Emission wavelength was monitored at 352 nm for ChvE without sugar and at 342 nm for ChvE with d-glucose. Excitation wavelength was 295 nm in both cases. F0, fluorescence intensity before adding the quencher; F, fluorescence intensity after adding the quencher.
Tryptophan residue mutations.
The results of the tryptophan fluorescence experiments strongly suggest that the W18 and/or W183 residue plays an important role in sugar binding, and the ChvE model suggests that these residues might contribute to the structural stability of the protein by contributing to and consolidating the hydrophobic cores of the protein's domains. To test this prediction, we generated W18F, W18A, W183F, and W183A mutants by site-directed mutagenesis. Only the ChvEW18F protein was expressed at a level comparable to that of the wild-type ChvE protein, whereas the other three mutant proteins were expressed at very low or undetectable levels as assayed by immunoblots (Fig. 7A). This result suggests that the two Trp residues are critical to maintaining the kinetic stability of the natively folded structure of the protein, probably by contributing to the structural integrity of the pocket.
FIG. 7.

Characterization of ChvE mutants containing Trp residue mutations. (A) Expression levels of mutant proteins. Cells (AB300) carrying Trp residue mutations, wild-type chvE (pGN102), or vector control (pBBR1MCS5) as indicated in each lane were grown in LB medium. Equivalent amounts of cell extracts were electrophoresed through sodium dodecyl sulfate-polyacrylamide gels. The ChvE protein was visualized by immunoblotting using an anti-ChvE antibody. These proteins were transferred onto three separate polyvinylidene difluoride membranes. (B) Effect of W18F mutation on growth of A. tumefaciens. Cells (AB300) carrying chvE(W18F), wild-type chvE (pGN102), or the vector control (pBBR1MCS5) were grown for 24 h in AB minimal medium with glucose as a sole carbon source. O.D.600, optical density at 600 nm. (C) Effect of W18F mutation on induction of PvirB-lacZ in the presence of 10 μM AS, pH 5.5, and 14 mM glucose. Data shown are the means of the results of three independent experiments performed in duplicate. Error bars represent standard deviations. wt, wild-type.
The W18F mutation has major deleterious effects on the ability of ChvE to function in vivo, as well as to bind d-glucose in vitro (Fig. 7 and Table 3). The growth of the W18F mutant strain was significantly slower than that of the strain carrying wild-type chvE at 24 h of culture in AB minimal medium containing glucose as a sole carbon source (Fig. 7B), while no significant growth difference was observed when glycerol was used as a sole carbon source (data not shown). The capacity of ChvEW18F to induce vir gene expression was also tested. Under inducing conditions (in the presence of glucose and low concentration of AS with pH 5.5), the strain with ChvEW18F yielded significantly less expression from a PvirB-lacZ fusion than the strain with wild-type ChvE (Fig. 7C). Moreover, the KD of ChvEW18F for d-glucose is about 4,000-fold higher than that of wild-type ChvE (Table 3). As predicted, these results show that W18 is an important residue for the sugar-binding activity of ChvE.
TABLE 3.
Characteristics of ChvE mutants
| ChvE proteins | vir induction | Glucose-mediated growtha | KD (M)b for d-glucose |
|---|---|---|---|
| Wild-type ChvE | Normal | Normal | (6.30 ± 0.77) × 10−7 |
| ChvER92C | Down | Slow | (5.80 ± 0.36) × 10−3 |
| ChvET72K | Down | Slow | (4.37 ± 0.10) × 10−7 |
| ChvED142V | Down | Slow | (3.12 ± 0.41) × 10−7 |
| ChvET187R | Down | Normal | (5.20 ± 0.35) × 10−7 |
| ChvET187P | Up | Normal | (5.27 ± 0.44) × 10−7 |
| ChvES197W | Up | Slow | (4.74 ± 1.12) × 10−7 |
| ChvEW18F | Down | Slow | (2.80 ± 0.22) × 10−3 |
Growth assay was carried out in AB minimal medium with glucose as sole carbon source as described in Materials and Methods. Slow, cells grew ≥20% more slowly than the wild type; Normal, cells grew at the wild-type rate.
Data shown are the means of the results of three independent experiments performed in duplicate. Standard deviations are indicated.
Sugar-binding properties of the ChvE mutant proteins.
An examination of the locations of the up and down ChvE mutations on the model of ChvE indicated that all but one were expected to lie along the surface of the protein (see below), where they might affect interactions with ChvE's protein partners rather than perturbing binding to d-glucose. The only mutation observed in the proposed sugar-binding cleft was Arg 92 to His or Cys. This residue is largely conserved in RBP and GBP, contacting the ligand (see Table S1 in the supplemental material). We therefore expected that the R92C mutation would disrupt glucose binding. To assess this prediction, the KDs of six purified mutant proteins were determined by Trp fluorescence titrations: four of them (ChvER92C, ChvET72K, ChvED142V, and ChvES197W) impaired glucose-dependent growth, while the other two (ChvET187R and ChvET187P) did not. As predicted, ChvER92C decreased the affinity for d-glucose by approximately 10,000-fold compared to that of the wild type (Fig. 5B). In contrast, the remaining altered proteins all had slightly higher affinities for d-glucose (up to a factor of 2) (Table 3), irrespective of their growth phenotypes. This result strongly indicates that the R92 residue is critical for glucose binding, whereas the other residues tested likely define surfaces important for interaction with ChvE's partner protein(s) involved in sugar utilization and virulence.
In addition to glucose, a number of other monosaccharides can enhance the sensitivity of VirA to phenolic signals in a ChvE-dependent pathway (2). To better understand the sugar-binding properties of the ChvE protein, we determined the binding affinities of ChvE for four of the other vir gene-inducing sugars (Table 4). The sugars are listed here in decreasing order of affinity: d-galactose > l-arabinose > d-glucose > d-fucose > d-xylose. This order does not agree completely with the results obtained from the earlier vir gene induction studies (2) in which d-xylose and d-glucose were found to be more active in the induction assays. One explanation for this discrepancy is that the effective amount of the monosaccharide available for serving as inducing signal could be different from that in the culture medium. This is because despite the addition of glycerol as carbon source in the induction assay, inducing sugars can be also consumed as carbon source to various degrees (44). The KDs of ChvER92C and ChvEW18F for d-galactose and l-arabinose are approximately 1,000-fold higher than the comparable KDs of wild-type ChvE (Table 4), suggesting that residues R92 and W18 are also important for binding those two vir gene-inducing sugars.
TABLE 4.
KDs of wild-type ChvE, ChvER92C, and ChvEW18F for different sugars
| Protein |
KD (M)a
|
|||
|---|---|---|---|---|
| d-Galactose | l-Arabinose | d-Xylose | d-Fucose | |
| Wild-type ChvE | 1.30 × 10−7 | 4.70 × 10−7 | 1.73 × 10−5 | 2.24 × 10−6 |
| ChvER92C | 3.32 × 10−4 | 1.22 × 10−3 | NA | NA |
| ChvEW18F | 1.52 × 10−3 | 9.10 × 10−4 | NA | NA |
NA, not available.
In order to examine the biological significance of the increased KD value in R92C, we measured the growth of a strain carrying this mutant protein in different concentrations of glucose at 24 h. Specifically, the R92C mutant strain should exhibit the wild-type growth phenotype in response to saturating concentrations of glucose. Indeed, when the glucose concentration in the medium was lower than the KD of ChvER92C for glucose (5.8 mM, Table 3), the R92C mutant strain grew as poorly as the chvE deletion mutant (Fig. 8). However, the R92C-containing strain grew significantly faster than the chvE deletion strain in 10 mM glucose, and the growth phenotype of R92C was close to that of the wild-type strain when the glucose concentration was increased to 100 mM. This result supports our conclusion that the R92 residue is involved in sugar-binding activity and that defects in growth under our standard conditions caused by the R92C mutation can be overcome by saturating sugar concentrations. The same result was observed when this mutant was tested for vir induction at various glucose concentrations. The deleterious effects of this mutation on both AS sensitivity and maximal levels of vir induction observed at 10 mM glucose were greatly reduced when the same strain was tested at 100 mM glucose (data not shown).
FIG. 8.

Growth of ChvE mutant R92C in different concentrations of glucose. Cells (AB300/pSW209Ω) carrying chvE(R92C), wild-type chvE (pGN102), or vector control (pBBR1MCS5) were grown for 24 h in AB minimal medium containing glucose as a sole carbon source. Data shown are the means of the results of three independent experiments performed in duplicate. Error bars represent standard deviations. O.D.600, optical density at 600 nm; wt, wild type.
DISCUSSION
ChvE plays a critical role in the virulence of A. tumefaciens via its effect(s) on VirA, rendering this histidine kinase both more sensitive to inducing phenolic derivatives, such as AS, and capable of greater levels of vir gene expression at saturating levels of such compounds. The genetic and biophysical studies reported here demonstrate the following. (i) ChvE displays high-affinity and specific binding to the vir gene-inducing sugar, d-glucose, but not the noninducing sugar d-ribose. (ii) Residues in ChvE that were identified as essential for binding d-glucose on the basis of a homology model built from GBP have been confirmed to be required for binding ligand. (iii) ChvE has an extended surface spanning both the C-terminal and N-terminal domains that is likely to be important for interaction with VirA. (iv) Some of the residues that are proposed to interact with VirA also interfere with growth on d-glucose, strongly suggesting that these residues are also essential for interaction with a permease involved in sugar transport.
Random mutagenesis of chvE was performed to identify residues of ChvE that could be involved in interaction between ChvE and the periplasmic domain of VirA. Strains carrying either down or up mutations could exhibit either normal or slow growth in medium containing glucose as a sole carbon source. Therefore, ChvE mutants can be categorized into two classes. Strains expressing the first class of mutants are affected in both VirA activity and sugar-dependent growth. Residues mutated in this class may be involved in either sugar-binding activity or interaction of the sugar-bound form of ChvE with the periplasmic region of VirA, as well as interaction with additional sugar transport components. In strains expressing the second class of mutants, VirA activity but not sugar-dependent growth is affected. Thus, altered residues in the second class may be residues that specifically affect the interaction between ChvE and VirA. Consistent with this hypothesis, we found that in strains expressing comparable levels of both wild-type ChvE and the up mutant ChvET187P, the mutant phenotype is dominant (data not shown), suggesting an enhanced affinity of ChvET187P for the periplasmic domain of VirA.
The PSBP family belongs to the periplasmic-binding protein (PBP) superfamily. In this superfamily, proteins such as ChvE often have dual activities associated with their abilities to interact with signaling systems (e.g., chemotransducers or sensor kinases) and with ATP-dependent transporters. In each case, the binding of ligand induces an allosteric transition in the PBP, resulting in a change in its ability to interact with and potentially mediate specific conformational changes in signaling or transport proteins (for a review, see reference 38). Thus, mutations identified in this work are good candidates for sites involved in direct protein-protein interactions with VirA and/or the putative sugar transporter, GguB (20) (see below). Indeed, when mapped onto the predicted structure of ChvE, the mutated sites define an extended surface spanning the C-terminal and N-terminal lobes (Fig. 9B and D). Furthermore, the locations of some residues identified in ChvE are very similar to those in RBP and GBP. Residues T72 and D142 of ChvE, which are suggested to be involved in both virulence and sugar utilization, overlap approximately with residues A70 and G134 of RBP (5, 15). The latter represents a region involved in interaction with both its transporter and chemotransducer. In contrast, residue G74 of GBP, found in the a-helix of the protein that is equivalent to the location of T72 of ChvE, was identified as specific for interaction of GBP with its chemotactic partner (36).
FIG. 9.

Bacterial sensing proteins in complex with their cognate periplasmic binding proteins. (A) Quorum-sensing protein LuxP in complex with the periplasmic protein LuxQ (33). Green, LuxP; gray, the periplasmic domain of LuxQ. (B) Structural model of the ChvE protein showing up and down mutations. (C) MBP in complex with the periplasmic maltose transporter protein (35). Green, MBP; gray, MalF and MalG. (D) Model of the ChvE protein showing glucose utilization mutations responsible for down phenotypes.
Recently published structures of PBPs complexed with sensing and transporter proteins help explain how the mutations in ChvE affect binding to its protein partners, VirA and its permease. The structure of the PBP LuxP in complex with the periplasmic domain of the signaling protein LuxQ has provided a first glimpse of a PBP engaged with a two-component sensor kinase (33). LuxP is a member of the type I PBP family, of which GBP, RBP, and ChvE are also members (12), allowing a three-dimensional comparison of the binding surfaces. Superimposition of our model of ChvE onto a model of LuxP suggests a geometry by which ChvE might interact with VirA. Significantly, the up and down mutations identified in this work map to approximately the same region used by LuxP to bind LuxQ. However, it is important to not overinterpret this result, because the degree of structural similarity of VirA's periplasmic domain to that of LuxQ is not yet known. Nevertheless, it is interesting to note that the mutationally defined surface of ChvE spans both of its domains; similarly, LuxP interacts with LuxQ over both domains. This arrangement allows ligand-induced changes in the orientation of the two domains to drive conformational changes in the binding partners of PBPs (33). Furthermore, Neiditch et al. (33) have examined variations of residues in the N-terminal lobe of LuxP which are in direct contact with LuxQ. Four mutants were identified that disrupt function. These mutations overlap extensively with the locations of mutations in the N-terminal lobe of ChvE.
The residues implicated in the interaction of ChvE with VirA are shown in Fig. 9B, and the majority of these surround the binding cleft. Residues 53 and 72 are located within the N-terminal lobe. Within the C-terminal lobe, a sequence spanning from residue 180 to residue 191 comprises a functional hot spot that forms a continuous surface defined by the side chains of residues 180 to 182, 186, 187, 190, and 191; the remaining residues within this segment project away from this surface. D142 extends the surface, while S197 lies at the periphery of the apparent binding surface. In both domains, the residues that decrease VirA signaling tend to cluster close to the cleft. Conversely, the activating mutations tend to lie peripheral to the cleft. We hypothesize that the activating mutations serve as auxiliaries to the primary determinants (identified in the down mutations). The auxiliaries might serve to enhance the affinity of ChvE for the activated form of VirA through electrostatic (E53K) or hydrophobic (A186V, T187M, T187P, and S197W) interactions. T187 is particularly interesting because at T187, replacements with Pro or Met enhanced VirA activity in the absence of an inducing sugar, while placement of the very polar residue Arg at that position drastically impaired the activity of VirA in response to the sugar signal. This indicates that T187 is a critical site for interaction with VirA.
It is interesting to note that although all of the mutants identified in this work were selected based on screens for vir gene activation, nearly half of the strains with these mutations also showed a phenotype of slow growth on glucose but normal growth on glycerol. The slow-growth phenotype could be the result either of disruption of the sugar-binding capacity of ChvE or an impaired interaction between ChvE and a putative ABC transporter, presumably GguB (20). Though earlier studies indicated that GguB is not involved in sugar-dependent growth (20), our preliminary studies indicate that it does function in this role (He et al., unpublished data). Tryptophan fluorescence assays were used to examine the binding of sugars to the wild-type and mutant versions of ChvE. Consistent with our model, we found that residues R92 and W18 are both required for high-affinity binding of d-glucose to ChvE. In contrast, other mutations that resulted in defects in glucose-dependent growth did not affect the binding affinity for glucose and thus represent candidate residues for interaction with the proposed transporter. The structure of MBP complexed with the transmembrane region of the maltose transporter is shown in Fig. 9C, providing a model for a PBP interacting with an ABC transporter. MBP is a type II PBP, so it is not possible to provide a precise comparison of the three-dimensional mapping, although the general location of the cleft and the two domains remains conserved between type I and II proteins (38). Significantly, the residues important for growth on glucose span both domains and lie along the same face of ChvE as that used by MBP to interact with the maltose transporter. In this orientation, glucose bound to ChvE would be well positioned to move into the ABC transporter.
The finding that about half of the mutants identified by a screen for VirA activation also lead to impaired putative interactions with a transporter indicates that this protein uses an overlapping but nonidentical set of residues to mediate interactions with the two types of protein partners. This is also consistent with the structures modeled in Fig. 9A and C, which show that similar surface-spanning dual-domain architecture of two distantly related PBPs is used to interact with signaling versus transport proteins.
Taken together, the genetic and biophysical data presented here provide direct evidence for sugar-binding activity of ChvE and identify functional surfaces of ChvE involved in sugar utilization and virulence. Our observations suggest three distinct sets of ChvE interaction sites—a set of sugar-binding sites, sites for interacting with other putative sugar utilization components, and sites for interacting with the periplasmic domain of VirA—and lay a groundwork for ongoing biochemical studies of these systems.
Supplementary Material
Acknowledgments
We are grateful to the Goulian laboratory for the spectrofluorometer, plasmids, and helpful discussions. We thank Greg Caputo in the DeGrado laboratory for help with the analysis of tryptophan-quenching data and Yuanqing Lin in the GRASP Laboratory for help with computing KD. We also thank Christian Baron for providing anti-ChvE antibodies, which were raised in Yasunori Machida's laboratory. Many thanks to Arlene Wise for helpful discussions and critical reading of the manuscript.
This work was supported by National Science Foundation grants MCB 0421885 and IOS-0818613 (to A.N.B.), National Institutes of Health grant RO1 GM47369 (to A.N.B.), and National Institutes of Health grant AI74866 (to W.F.D.).
Footnotes
Published ahead of print on 24 July 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Andujar-Sanchez, M., V. Jara-Perez, and A. Camara-Artigas. 2007. Thermodynamic determination of the binding constants of angiotensin-converting enzyme inhibitors by a displacement method. FEBS Lett. 5813449-3454. [DOI] [PubMed] [Google Scholar]
- 2.Ankenbauer, R. G., and E. W. Nester. 1990. Sugar-mediated induction of Agrobacterium tumefaciens virulence genes: structural specificity and activities of monosaccharides. J. Bacteriol. 1726442-6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340783-795. [DOI] [PubMed] [Google Scholar]
- 4.Berger, B. R., and P. J. Christie. 1993. The Agrobacterium tumefaciens virB4 gene product is an essential virulence protein requiring an intact nucleoside triphosphate-binding domain. J. Bacteriol. 1751723-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binnie, R. A., H. Zhang, S. Mowbray, and M. A. Hermodson. 1992. Functional mapping of the surface of Escherichia coli ribose-binding protein: mutations that affect chemotaxis and transport. Protein Sci. 11642-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brencic, A., and S. C. Winans. 2005. Detection of and response to signals involved in host-microbe interactions by plant-associated bacteria. Microbiol. Mol. Biol. Rev. 69155-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cangelosi, G. A., R. G. Ankenbauer, and E. W. Nester. 1990. Sugars induce the Agrobacterium virulence genes through a periplasmic binding protein and a transmembrane signal protein. Proc. Natl. Acad. Sci. USA 876708-6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, C. H., and S. C. Winans. 1992. Functional roles assigned to the periplasmic, linker, and receiver domains of the Agrobacterium tumefaciens VirA protein. J. Bacteriol. 1747033-7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chilton, M. D., T. C. Currier, S. K. Farrand, A. J. Bendich, M. P. Gordon, and E. W. Nester. 1974. Agrobacterium tumefaciens DNA and PS8 bacteriophage DNA not detected in crown gall tumors. Proc. Natl. Acad. Sci. USA 713672-3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Auria, S., A. Ausili, A. Marabotti, A. Varriale, V. Scognamiglio, M. Staiano, E. Bertoli, M. Rossi, and F. Tanfani. 2006. Binding of glucose to the D-galactose/D-glucose-binding protein from Escherichia coli restores the native protein secondary structure and thermostability that are lost upon calcium depletion. J. Biochem. 139213-221. [DOI] [PubMed] [Google Scholar]
- 11.Doty, S. L., M. C. Yu, J. I. Lundin, J. D. Heath, and E. W. Nester. 1996. Mutational analysis of the input domain of the VirA protein of Agrobacterium tumefaciens. J. Bacteriol. 178961-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dwyer, M. A., and H. W. Hellinga. 2004. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 14495-504. [DOI] [PubMed] [Google Scholar]
- 13.Eftink, M. R., and C. A. Ghiron. 1976. Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry 15672-680. [DOI] [PubMed] [Google Scholar]
- 14.Eftink, M. R., and C. A. Ghiron. 1981. Fluorescence quenching studies with proteins. Anal. Biochem. 114199-227. [DOI] [PubMed] [Google Scholar]
- 15.Eym, Y., Y. Park, and C. Park. 1996. Genetically probing the regions of ribose-binding protein involved in permease interaction. Mol. Microbiol. 21695-702. [DOI] [PubMed] [Google Scholar]
- 16.Gao, R., and D. G. Lynn. 2005. Environmental pH sensing: resolving the VirA/VirG two-component system inputs for Agrobacterium pathogenesis. J. Bacteriol. 1872182-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garfinkel, D. J., and E. W. Nester. 1980. Agrobacterium tumefaciens mutants affected in crown gall tumorigenesis and octopine catabolism. J. Bacteriol. 144732-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, M. L., G. A. Cangelosi, W. Halperin, and E. W. Nester. 1990. A chromosomal Agrobacterium tumefaciens gene required for effective plant signal transduction. J. Bacteriol. 1721814-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin, S., T. Roitsch, R. G. Ankenbauer, M. P. Gordon, and E. W. Nester. 1990. The VirA protein of Agrobacterium tumefaciens is autophosphorylated and is essential for vir gene regulation. J. Bacteriol. 172525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kemner, J. M., X. Liang, and E. W. Nester. 1997. The Agrobacterium tumefaciens virulence gene chvE is part of a putative ABC-type sugar transport operon. J. Bacteriol. 1792452-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovach, M. E., P. H. Elzer, D. S. Hill, G. T. Robertson, M. A. Farris, R. M. Roop II, and K. M. Peterson. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166175-176. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 23.Lakowicz, J. R. 1999. Principles of fluorescence spectroscopy. Plenum Press, New York, NY.
- 24.Leroux, B., M. F. Yanofsky, S. C. Winans, J. E. Ward, S. F. Ziegler, and E. W. Nester. 1987. Characterization of the virA locus of Agrobacterium tumefaciens: a transcriptional regulator and host range determinant. EMBO J. 6849-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martineau, P., W. Saurin, M. Hofnung, J. C. Spurlino, and F. A. Quiocho. 1990. Progress in the identification of interaction sites on the periplasmic maltose binding protein from E coli. Biochimie 72397-402. [DOI] [PubMed] [Google Scholar]
- 26.McCullen, C. A., and A. N. Binns. 2006. Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annu. Rev. Cell Dev. Biol. 22101-127. [DOI] [PubMed] [Google Scholar]
- 27.McGowan, E. B., T. J. Silhavy, and W. Boos. 1974. Involvement of a tryptophan residue in the binding site of Escherichia coli galactose-binding protein. Biochemistry 13993-999. [DOI] [PubMed] [Google Scholar]
- 28.Miller, D. M., III, J. S. Olson, J. W. Pflugrath, and F. A. Quiocho. 1983. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J. Biol. Chem. 25813665-13672. [PubMed] [Google Scholar]
- 29.Miller, D. M., III, J. S. Olson, and F. A. Quiocho. 1980. The mechanism of sugar binding to the periplasmic receptor for galactose chemotaxis and transport in Escherichia coli. J. Biol. Chem. 2552465-2471. [PubMed] [Google Scholar]
- 30.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.Mukhopadhyay, A., R. Gao, and D. G. Lynn. 2004. Integrating input from multiple signals: the VirA/VirG two-component system of Agrobacterium tumefaciens. ChemBioChem 51535-1542. [DOI] [PubMed] [Google Scholar]
- 32.Nair, G. R. 2006. Novel insights into vir gene regulation in Agrobacterium tumefaciens. Ph.D. thesis. University of Pennsylvania, Philadelphia.
- 33.Neiditch, M. B., M. J. Federle, A. J. Pompeani, R. C. Kelly, D. L. Swem, P. D. Jeffrey, B. L. Bassler, and F. M. Hughson. 2006. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 1261095-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neu, H. C., and L. A. Heppel. 1964. On the surface localization of enzymes in E. coli. Biochem. Biophys. Res. Commun. 17215-219. [DOI] [PubMed] [Google Scholar]
- 35.Oldham, M. L., D. Khare, F. A. Quiocho, A. L. Davidson, and J. Chen. 2007. Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450515-521. [DOI] [PubMed] [Google Scholar]
- 36.Ordal, G. W., and J. Adler. 1974. Properties of mutants in galactose taxis and transport. J. Bacteriol. 117517-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reference deleted.
- 38.Quiocho, F. A., and P. S. Ledvina. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol. 2017-25. [DOI] [PubMed] [Google Scholar]
- 39.Schafer, A., A. Tauch, W. Jager, J. Kalinowski, G. Thierbach, and A. Puhler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 14569-73. [DOI] [PubMed] [Google Scholar]
- 40.Reference deleted.
- 41.Shimoda, N., A. Toyoda-Yamamoto, S. Aoki, and Y. Machida. 1993. Genetic evidence for an interaction between the VirA sensor protein and the ChvE sugar-binding protein of Agrobacterium. J. Biol. Chem. 26826552-26558. [PubMed] [Google Scholar]
- 42.Silhavy, T. J., and W. Boos. 1975. The “hidden ligand” of the galactose-binding protein. Eur. J. Biochem. 54163-167. [DOI] [PubMed] [Google Scholar]
- 43.Silva, J. L., E. W. Miles, and G. Weber. 1986. Pressure dissociation and conformational drift of the beta dimer of tryptophan synthase. Biochemistry 255780-5786. [DOI] [PubMed] [Google Scholar]
- 44.Stulke, J., and W. Hillen. 1999. Carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 2195-201. [DOI] [PubMed] [Google Scholar]
- 45.Tam, R., and M. H. Saier, Jr. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57320-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toyoda-Yamamoto, A., N. Shimoda, and Y. Machida. 2000. Genetic analysis of the signal-sensing region of the histidine protein kinase VirA of Agrobacterium tumefaciens. Mol. Gen. Genet. 263939-947. [DOI] [PubMed] [Google Scholar]
- 47.Turk, S. C., R. P. van Lange, T. J. Regensburg-Tuink, and P. J. Hooykaas. 1994. Localization of the VirA domain involved in acetosyringone-mediated vir gene induction in Agrobacterium tumefaciens. Plant Mol. Biol. 25899-907. [DOI] [PubMed] [Google Scholar]
- 48.Vivian, J. T., and P. R. Callis. 2001. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 802093-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vyas, N. K., M. N. Vyas, and F. A. Quiocho. 1988. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 2421290-1295. [DOI] [PubMed] [Google Scholar]
- 50.Wang, Y., A. Mukhopadhyay, V. R. Howitz, A. N. Binns, and D. G. Lynn. 2000. Construction of an efficient expression system for Agrobacterium tumefaciens based on the coliphage T5 promoter. Gene 242105-114. [DOI] [PubMed] [Google Scholar]
- 51.Ward, L. D. 1985. Measurement of ligand binding to proteins by fluorescence spectroscopy. Methods Enzymol. 117400-414. [DOI] [PubMed] [Google Scholar]
- 52.Winans, S. C. 1990. Transcriptional induction of an Agrobacterium regulatory gene at tandem promoters by plant-released phenolic compounds, phosphate starvation, and acidic growth media. J. Bacteriol. 1722433-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winans, S. C., R. A. Kerstetter, and E. W. Nester. 1988. Transcriptional regulation of the virA and virG genes of Agrobacterium tumefaciens. J. Bacteriol. 1704047-4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, Y., C. Conway, M. Rosato, Y. Suh, and M. D. Manson. 1992. Maltose chemotaxis involves residues in the N-terminal and C-terminal domains on the same face of maltose-binding protein. J. Biol. Chem. 26722813-22820. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.