

Abstract
Toxoplasma gondii mutants identified as defective in the establishment of chronic infection were screened to isolate those specifically impaired in their ability to replicate within activated macrophages. One of the identified mutants contains an insertion in the hypothetical gene TGME49_111670. Genetic complementation restores the ability of the mutant to replicate in immune cells and produce cysts in the brains of mice. While the mutant is more sensitive to nitric oxide than is its parental strain, it is not defective in its ability to suppress nitric oxide. The disrupted protein has no significant homology to proteins with known functions, but is predicted to have one transmembrane domain. Immunofluorescence shows the protein on the parasite surface, even in activated macrophages, colocalizing with a tachyzoite surface antigen, SAG1, and oriented with its C-terminal end external. Western analysis reveals that the protein is downregulated in bradyzoites. Despite the tachyzoite specificity of this protein, mice infected with the mutant succumb to acute infection similarly to those infected with the parent strain. Serum samples from mice with chronic T. gondii infection react to a polypeptide from TGME49_11670, indicating that the protein is seen by the immune system during infection. This study is the first to characterize a T. gondii surface protein that contains a transmembrane domain and show that the protein contributes to parasite replication in activated immune cells and the establishment of chronic infection.
The apicomplexan parasite Toxoplasma gondii is an obligate intracellular organism capable of infecting any nucleated cell of warm-blooded mammals. T. gondii is able to reproduce sexually and asexually. The asexual cycle contains two stages, a fast-growing, disseminating form called a tachyzoite and a slow-growing, encysted form called a bradyzoite (10, 38). Due to uncharacterized host/parasite signaling processes, tachyzoites decrease their growth rate and change into bradyzoites. The presence of these cysts in the central nervous system and muscle tissue is a hallmark of chronic infection (10). While infection with T. gondii is generally asymptomatic in immunocompetent individuals, infections can be fatal for fetuses and those who are immunocompromised due to immune suppression therapies or human immunodeficiency virus (9, 22, 29, 30).
T. gondii elicits responses from innate and acquired immunity. The innate response involves a type 1 immune reaction involving secretion of gamma interferon (IFN-γ) by Th1 cells, CD8+ T lymphocytes, and natural killer cells, leading to the circulation of multiple proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-8 (IL-8) (1, 7, 39). The production of IFN-γ and TNF-α has been linked to resistance to acute T. gondii infections (32, 36). Differentiation of IFN-γ-secreting cells is triggered by secretion of IL-12 by T. gondii-activated macrophages, inflammatory monocytes, neutrophils, and dendritic cells (3, 12). While dendritic cells are essential for control of infection due to their cytokine production (20), they are also important for parasite dissemination (18). Macrophages activated in vitro by IFN-γ and TNF-α produce nitric oxide (NO) via inducible nitric oxide synthase (iNOS), which is effective at eliminating tachyzoites (33). However, T. gondii is able to impede NO production within infected macrophages, aiding parasite survival and replication (21, 34). The parasite pathways involved in this regulation are largely unknown. A patatin-like protein that contributes to the suppression of NO was discovered (26).
Surface antigens of the parasite facilitate the initial interactions with host cells, both immune and nonimmune. These interactions are crucial and will dictate the fate of the parasite in the host; therefore, identifying structures on the surface of T. gondii has been an active area of research. Several families of glycosylphosphatidylinositol (GPI)-anchored surface antigens have been characterized, including surface antigen 1 (SAG1), SAG1-related sequence (SRS), and SAG-unrelated surface antigen (SUSA) (28; reviewed in references 4, 16, and 19). The roles of these GPI-anchored proteins decorating the surface of the parasite have not been fully elucidated. Some have been linked to parasite attachment to and invasion of host cells, and others have been shown to elicit a strong host immune response (14, 17, 25). While the exact role of each protein in the families is unknown, many are developmentally regulated. Tachyzoite-specific surface antigens elicit an immune response during acute infection that is likely not effective against bradyzoites during chronic infection.
A modified signature-tagged mutagenesis library was used to identify T. gondii virulence mutants (11). For the present study, these signature-tagged mutagenesis mutants were secondarily screened to identify mutants unable to withstand activation of infected macrophages. One of the four mutants identified, 41E2, has an insertion within the annotation TGME49_111670 (www.toxodb.org). The TGME49_111670 protein has a predicted transmembrane domain and is shown to be a tachyzoite-specific surface antigen involved in replication in activated immune cells and the establishment of a chronic infection.
MATERIALS AND METHODS
Cell culture and parasite strains.
Strains were maintained as tachyzoites by serial passage on monolayers of human foreskin fibroblasts (HFFs) at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA), and 2 mM l-glutamine (Invitrogen). Bradyzoite conditions were RPMI 1640 supplemented with 1% fetal bovine serum and 1% penicillin-streptomycin, buffered with 50 mM HEPES to pH 8 and incubated at 37°C with ambient CO2. PRUΔHPT is the base strain for creation of the parental and mutant strains (26).
Screen for parasite replication in bone marrow-derived macrophages.
Bone marrow-derived macrophages were isolated and cultured as previously described (26). Macrophages were used for experiments between 7 and 10 days after isolation. For screening parasites, macrophages were grown on glass chamber slides (BD Biosciences, San Jose, CA) overnight at a concentration of 2 × 105 cells per ml in 250 μl of supplemented DMEM. Macrophages were challenged with 2 × 104 T. gondii insertional mutants for 3 hours, rinsed with fresh medium to remove nonadherent parasites, and either left unstimulated or activated with 50 ng/ml lipopolysaccharide (LPS; List Biologicals, Campbell, CA) and 80 U/ml gamma interferon (IFN-γ; PeproTech, Rocky Hill, NJ) for an additional 25 h. After culture, cells were fixed for 20 min in 4% buffered formalin, permeabilized with 0.2% Triton X-100, and localized by a polyclonal rabbit anti-T. gondii antibody (Fitzgerald Industries, Concord, MA) followed by an Alexa 488 anti-rabbit secondary antibody (Invitrogen, Carlsbad, CA). To evaluate whether vacuoles fused with lysosomes, cells were stained 25 h after activation with a rat anti-mouse monoclonal antibody to lysosome-associated membrane protein (LAMP1) (ID4B; Developmental Studies Hybridoma Bank, University of Iowa) and a rabbit polyclonal antibody to T. gondii. Samples were mounted using VectaShield mounting media with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA). Phase-contrast and fluorescence microscopy was used to examine their morphology and determine the number of parasites per vacuole ± standard deviation. At least 100 parasite-containing vacuoles were examined per experiment, and each experiment was repeated at least three times. Samples were visualized using a Zeiss inverted Axiovert 200 motorized microscope with a 100× objective (PlanApo 1.4-numerical-aperture oil PH3 objective); Zeiss filter sets 31, 34, 38, and 50; and Axiovision 4.3 software. Pictures were obtained using a Zeiss Axiocam MRm.
For complementation studies, parasites were allowed to invade for 3 hours before activation with 100 ng/ml LPS (Escherichia coli-derived; LIST Biological Laboratories, Campbell, CA) and 200 U/ml IFN-γ (eBioscience, San Diego, CA) and then were incubated for an additional 21 or 33 h. For NO synthase inhibition, 1 mM of aminoguanidine (Sigma-Aldrich, St. Louis, MO) was added 3 hours after parasite invasion as previously described (27). Parasites were visualized using chronic infection serum (serum collected from CBA/J mice infected with 2 × 104 PRU tachyzoites for 22 days) and Alexa Fluor 488 goat anti-mouse antibody (Molecular Probes, Seattle, WA). The number of parasites per vacuole was counted in 25 random fields, and degraded parasites in activated macrophages were noted. Student's t test was used to determine significance of the data.
Detection of reactive nitrogen intermediates.
The level of nitrite, a downstream product of NO, present in infected bone marrow-derived macrophage cultures was determined using the Griess reaction as previously described (26, 27). Samples were measured in triplicate, and the average nitrite level (μM) was determined. Student's t test was used to determine significance of the data.
Characterization of the TgTSA1 transcript.
Parasites were harvested for RNA isolation by syringing with a 27-gauge needle to release parasites. RNA was isolated using Ultraspec RNA (Biotecx Laboratories, Inc., Houston, TX) according to manufacturer's protocol. 5′ and 3′ untranslated regions (UTRs) were determined using rapid amplification of cDNA ends (Ambion, Austin, TX). cDNA was made from 5 μg of RNA using Invitrogen's SuperScript III according to the manufacturer's protocol. Regions between the defined UTRs were amplified using primers 41E2_EST178-198FW (5′-GCTTCAAAACGACATCCG-3′), 41E2_3221R (5′-CGGATAATGCAGAGTCAG), 41E2_3221F (5′-CTGACTCTGCATTATCCG-3′), and 41E2_seqR (5′-ACAAAAAAGGAACCGCCCAGC-3′). A cDNA reaction lacking reverse transcriptase was used in PCRs to control for DNA contamination. PCR products were cloned into pCR 2.1 (Invitrogen) according to the manufacturer's protocols.
Generation of 41E2 complementation constructs and electroporation into T. gondii.
Primers 41E2SpeIFW (5′-ACTAGTCGAGTTTTCGGTGAGATT-3′) and 41E2SpeIREV (5′-ACTAGTTTCTGAATCCTTTCTTGGGT-3′) were used to amplify a 4.7-kb genomic segment spanning 1.3 kb upstream of the 5′ UTR and 413 bp downstream of the poly(A) site. The genomic piece was cloned into pBC SK+ (Stratagene, La Jolla, CA) with dihydrofolate reductase (DHFR)-thymidylate synthase (8) at the SpeI site, creating pBC-DHFR-41E2comp. A hemagglutinin (HA) tag was added 222 bp upstream of the predicted translational stop site by amplifying a region surrounding EcoRV and AvrII sites, using primers 41E2HAF (5′-GATATCTTACCCATACGACGTCCCAGACTACGCGGAGCAGAGTGCTCCGACG-3′) and 41E2_seqR. pBC-DHFR-41E2HA with a C-terminal HA tag was constructed by removing an 831-bp piece of pBC-DHFR-41E2comp with EcoRV and AvrII digests and replacing it with an identical piece containing the HA tag. In order to insert an HA tag on the N terminus of the protein, primers 41E2-5′HAF (5′-GCGGCCGCTACCCATACGACGTCCCAGACTACGCGGGAGGCAGCTCGAACTGCG-3′) and 41E2_seqR were used to amplify a 1.9-kb product, which was digested with NotI and AvrII and subcloned into pBC-DHFR-41E2. To create TgTSA1 expressed from the α-tubulin promoter with a C-terminal HA tag, primers 41E2NsiF (5′-ATGCATTGTGTGGAGGTTCTGGAGCTG-3′) and 41E2PacR (5′-TTAATTAAGCCAGGCCCACCATTTTTCTGC-3′) were used to amplify the open reading frame from pBC-DHFR-41E2HA. NsiI and PacI sites were engineered on the primers for subcloning into pT/230 (35), which expresses the open reading frame from an α-tubulin promoter. Twenty-five micrograms of the linearized constructs were electroporated with 1 × 107 T. gondii 41E2 mutants, and stable transformants were selected as resistant to 1 μM pyrimethamine.
Mouse infections.
Eight-week-old C57BL/6 female mice (NCI, Frederick, MD) were intraperitoneally infected with 1 × 104 and 1 × 105 tachyzoites. Parasite suspensions used for infections were applied to HFF monolayers for plaque counts to ensure approximately equal numbers of viable parasites were injected. Mice were sacrificed at 22 days, and brains were collected, macerated, fixed with 3% formaldehyde, and then permeabilized and blocked in 3% bovine serum albumin (BSA)-0.2% Triton X-100. Brain cysts were stained with fluorescein-labeled Dolichos biflorus agglutinin (Vector Laboratories). The cysts were enumerated in three 5-μl samples, using fluorescence microscopy. The chronic infection experiment was performed twice using four mice per strain and at two different doses. Significance of the data was determined using Student's t test.
Immunofluorescence assay.
Confluent host cells on coverslips were infected with recently lysed parasites for 24 h. For extracellular parasites, recently lysed parasites in DMEM were applied to coverslips and allowed to attach for 30 min. The monolayer or extracellular parasites were fixed with 3% formaldehyde and then either permeabilized and blocked in 3% BSA-0.2% Triton X-100 or just blocked in 3% BSA. Mouse anti-HA (Covance Innovative Antibodies, Princeton, NJ) was used as the primary antibody for HA-tagged proteins and costained with either SAG1, IMC1, or GRA2. Secondary antibodies were Alexa Fluor 633 goat anti-mouse or anti-rabbit and Alexa Fluor 488 goat anti-mouse or anti-rabbit (Molecular Probes). Coverslips were mounted onto slides, using VectaShield mounting medium containing DAPI (Vector Laboratories). Samples were examined using a motorized Zeiss Axiplan II microscope equipped with a rear-mounted excitation filter wheel, a triple-pass (DAPI/fluorescein isothiocyanate/Texas Red) emission cube, differential interference contrast optics, and a Hamamatsu ORCA-AG charge-coupled-device camera. Serial image stacks (0.2-μm Z-increment) were collected with a 100× PlanApo oil immersion lens (1.4 numerical aperture) and deconvolved and pseudocolored using OpenLabs 4.0 software (Improvision, Waltham, MA).
Western immunoblotting.
Freshly lysed tachyzoites were collected by centrifugation (425 × g for 10 min). Bradyzoites were harvested after 5 days under bradyzoite growth conditions by syringing with a 30-gauge needle to release parasites and centrifuged to collect parasites. An equal number of tachyzoites and bradyzoites was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto Immobilon-P membrane (Millipore, Billerica, MA). The membrane was incubated with a primary antibody (anti-HA, 1:5,000; anti-BAG1, 1:2,500; anti-β-tubulin, 1:5,000) for 1 hour followed by a 1-hour incubation with a secondary antibody (donkey anti-mouse or donkey anti-rabbit immunoglobulin G [IgG]) conjugated to horseradish peroxidase (1:5,000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Detection was performed with the Amersham ECL Western blotting system for chemiluminescence according to manufacturer's protocol (GE Healthcare, Buckinghamshire, United Kingdom).
Growth of 41E2 in bone marrow-derived dendritic cells.
Dendritic cells were isolated from murine bone marrow following 7 days of maturation in the presence of interleukin-4 and granulocyte-monocyte colony-stimulating factor as previously described (15). On the seventh day of maturation, dendritic cells were seeded on coverslips in the presence or absence of 100 ng/ml LPS. After 6 hours, dendritic cells were infected with 1 × 105 tachyzoites. Coverslips were processed for immunofluorescence as described above 24 h after infection, using SAG1 to visualize the parasites in order to count the number of parasites per vacuole. Student's t test was used to determine significance of the data.
Peptide production and purification.
A region of TgTSA1 was amplified with primers 41E2pepF (5′-CCATGGGCGCGAAGCGCTTGGCACAACTTCAG-3′) and 41E2pepR (5′-CTCGAGCTGTTCGAGTTCGGTCCC-3′), which added an NcoI site on the 5′ end and an XhoI site on the 3′ end. The PCR product was cloned into pCR 2.1 (Invitrogen) according to the manufacturer's protocols and verified by sequencing with M13F and M13R vector primers. The plasmid was then digested with NcoI and XhoI to obtain a 273-bp fragment that was subcloned into pET-28a(+) (Novagen, Madison, WI) that was linearized with NcoI and XhoI. The construct was transformed into a Rosetta strain to allow for induction. The peptide was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h at 37°C and the soluble fraction, a 14-kDa protein with a C-terminal six-His tag, was purified using the His·Bind Quick 900 cartridges (Novagen) according to the manufacturer's protocol.
Immunoblotting with chronic serum.
The TgTSA1 peptide was separated on a denaturing 20% acrylamide gel and transferred onto Immobilon-P (Millipore). Ponceau S staining (0.1% [wt/vol] Ponceau S in 5% acetic acid) was done to confirm that transfer was complete. Serum was collected from mice infected with T. gondii for 22 days and used as the primary antibody. Serum from an uninfected mouse was also used as a primary antibody. The secondary antibody was donkey anti-mouse IgG conjugated to horseradish peroxidase (1:5,000; Jackson ImmunoResearch Laboratories, Inc.). Chemiluminescent detection was done with the Amersham ECL Western blotting system (GE Healthcare).
RESULTS
Identification of insertional mutants that exhibit defective replication in activated macrophages.
Thirty-nine mutants impaired in their ability to establish a chronic infection in mice (11) underwent a secondary screen to identify those with replication defects in activated macrophages. Parasites were allowed to infect naïve macrophages for 3 hours, and then the infected macrophages were activated with IFN-γ and LPS for 24 h. After incubation, cells were fixed, stained with a polyclonal antibody to T. gondii, and analyzed by immunofluorescence. Wild-type parasites were predominantly phase dense, bow shaped, and within tight-fitting vacuoles of two or four parasites (Fig. 1A and B). In contrast, four mutants (41E2, 37B2, 40E4, 57G3) consistently failed to replicate beyond one parasite per vacuole after 28 h of culture and appeared amorphous/degraded (typical vacuoles shown in Fig. 1B). Vacuoles containing parasite mutants did not colocalize with LAMP1, suggesting that the mutants were not in parasitophorous vacuoles that had fused with lysosomes (data not shown). To ensure that the replication defect of the mutants was associated with macrophage activation, the growth rate of the four mutants was examined in naïve macrophages. All four mutants replicated similarly to wild-type parasites in naïve macrophages, with the mean parasite number per vacuole as follows: wild type, 7.4 ± 0.14; 41E2, 8.68 ± 0.54; 57G3, 7.2 ± 0; 40E4, 7.2 ± 0.28; 37B2, 7.0 ± 0.21.
FIG. 1.

Screen of parasite mutants in macrophages activated post-parasite invasion. (A) Twenty-four hours after macrophage activation, parasites were analyzed microscopically by phase-contrast and immunofluorescence microscopy. Shown are the percentages of vacuoles that had two or four parasites per vacuole (replication), one parasite per vacuole (stasis), or a single amorphous parasite (degraded/amorphous). The majority of wild-type (WT) parasites had two to four parasites per vacuole 25 h after activation. In contrast, the majority of mutant parasites had a single parasite per vacuole that appeared either intact or amorphous. A minimum of 100 vacuoles were counted per experiment, and the experiment is representative of at least three experiments. (B) Typical images of wild-type (WT) parasites (two and four parasites per vacuole are shown) compared to the amorphous vacuoles and parasites of the mutants. Phase-contrast images are shown in the left column, and immunofluorescence images are shown in the right column. Parasites are indicated with a white arrow.
Characterization of the gene interrupted in the 41E2 mutant.
The insertion site in mutant 41E2 was determined to be at location 2,191,215 of ME49 chromosome XI. This site is 61 bp after a predicted translational start site of T. gondii annotation TGME49_111670 (www.toxodb.org; identified as 583.m05443 in reference 12), generating essentially a deletion of this 440-amino-acid protein (Fig. 2A). Mapping of the transcript using reverse transcription-PCR and 5′ and 3′ rapid amplification of cDNA ends (Ambion) confirmed that TGME49_111670 has no introns and has a 711-bp 5′ UTR and a 1,050-bp 3′ UTR. The protein product of TGME49_111670 is predicted to have one transmembrane domain between amino acids 60 and 80 (Fig. 2A), creating an intracellular 59-amino-acid N terminus (www.ch.embnet.org/software/TMPRED_form.html). TGME49_111670 does not contain any other known functional domains, including a signal peptide (www.toxodb.org), and it does not share significant homology with any proteins of known function. Neospora canium does have a TGME49_111670 ortholog that is annotated as a hypothetical (www.toxodb.org).
FIG. 2.

TgTSA1 is a transmembrane domain-containing surface antigen. (A) Diagram of the TgTSA1 protein. In the 41E2 mutant, the insertion site of the mutagenesis plasmid was 20 amino acids in this 440-amino-acid protein. A single transmembrane domain (TM) is predicted at amino acid 59. A 129-amino-acid polypeptide near the C terminus was purified from E. coli to investigate the interactions of this protein with the immune system (Fig. 7). (B) Parasites expressing a C-terminal HA-tagged version of TgTSA1 (green) were costained for SAG1 (red). Intracellular parasites are shown in the first three rows of panels. HFFs are the host cells for row 1, naïve macrophages (Mφ) are in row 2, and activated macrophages (Act. Mφ) are in row 3. Extracellular parasites expressing a C-terminal (rows 4 and 5) or N-terminal (rows 6 and 7) HA-tagged version of TgTSA1 were stained following permeabilization (rows 4 and 6) or without permeabilization (rows 5 and 7).
TGME49_111670 is a transmembrane domain-containing surface antigen with an intracellular N terminus.
To localize the TGME49_111670 protein, PRUΔHPT was engineered to express TGME49_111670 from the α-tubulin promoter with a C-terminal HA epitope tag. Immunofluorescence assays on tachyzoite-infected HFFs using antibody against the HA to visualize the tagged protein revealed the protein colocalizes with SAG1, a tachyzoite surface antigen (Fig. 2B, row 1). Given that the mutant has diminished replication in activated macrophages, we examined the localization of the protein in naïve and activated macrophages. The protein was also seen on the parasite surface in naïve and activated macrophages (Fig. 2B, rows 2 and 3). To confirm the surface localization of TGME49_111670, immunofluorescence was performed on extracellular parasites expressing an HA-tagged TGME49_111670 protein expressed from the endogenous promoter with or without membrane permeabilization. TGME49_111670 with a C-terminal HA tag was detectable with and without permeabilization, similar to SAG1 (Fig. 2B, rows 4 and 5). No staining was seen using an antibody to a dense granule protein 2 in the absence of permeabilization (data not shown). These data show that TGME49_111670 is on the parasite surface and that the C terminus of the protein is external. To confirm this topology, immunofluorescence of PRUΔHPT expressing TGME49_111670 with an N-terminal HA tag was performed. Without membrane permeabilization, the N-terminal HA TGME49_111670 had a lower signal with identical exposure times to the C-terminal HA (Fig. 2B, compare rows 5 and 7). Light staining of the N-terminal HA protein without permeabilization is likely due to the fixation conditions as a signal was also detected in the absence of permeabilization with anti-IMC1 antibody (data not shown; 37). With permeabilization, the N-terminal HA TGME49_111670 protein had fluorescence intensity similar to that of the C-terminal HA (Fig. 2B, rows 4 and 6). These data are consistent with TGME49_111670 being a surface protein in T. gondii with a single transmembrane domain that orients the N terminus inside the parasite and the C terminus outside. To our knowledge, TGME49_111670 is the first characterized surface protein of T. gondii that contains a transmembrane domain. Therefore, we have named TGME49_111670 T. gondii transmembrane surface antigen 1 (TgTSA1).
TgTSA1 is developmentally regulated.
Most members of the SAG, SRS, and SUSA families of GPI-anchored surface antigens are developmentally regulated between tachyzoites and bradyzoites. To investigate whether transmembrane domains containing surface proteins were also developmentally controlled, we examined expression of TgTSA1 with a C-terminal HA tag under the control of its native promoter. Western immunoblotting using an antibody against the HA tag showed that TgTSA1 was expressed in tachyzoites but not in bradyzoites (Fig. 3). The size of this tachyzoite protein is close to the predicted size of 47 kDa. The blot was striped and reprobed with anti-T. gondii β-tubulin to show that each lane contained approximately equal amounts of parasites. The blot was reprobed again with antibodies against the bradyzoite-specific heat shock protein Bag1 to show the level of bradyzoite induction (Fig. 3). These data show that, similar to the GPI-anchored surface antigens of T. gondii, the transmembrane domain-containing TgTSA1 surface protein is developmentally regulated between tachyzoites and bradyzoites.
FIG. 3.
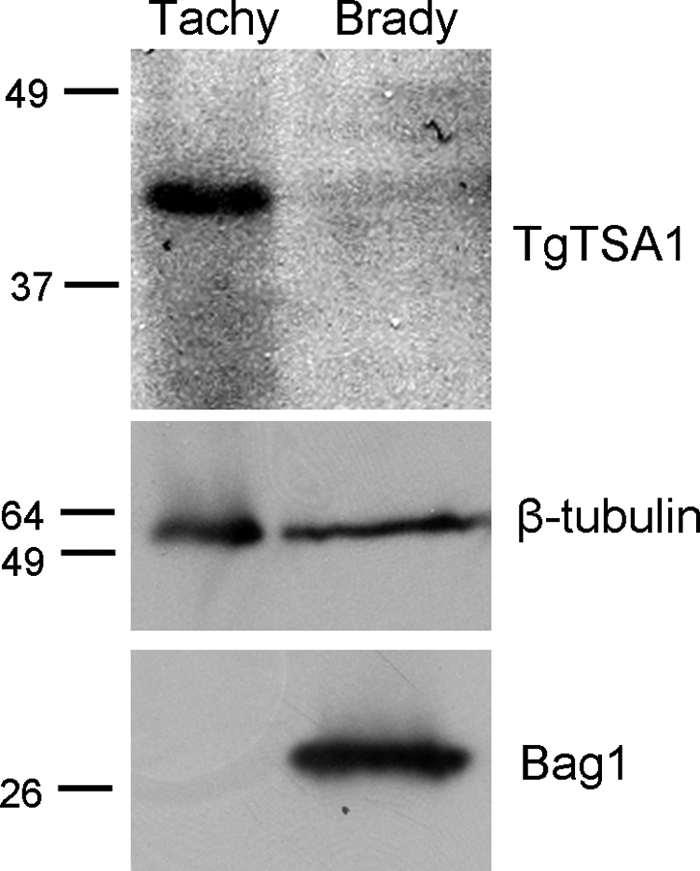
TgTSA1 is downregulated in bradyzoites. Equal numbers of Comp2-HA tachyzoites (Tachy) and bradyzoites (Brady) were analyzed by Western immunoblotting for expression of TgTSA1, using HA to detect the tagged version of the protein. β-Tubulin is shown as a loading control, and Bag1 expression indicates the level of bradyzoite induction.
Replication of the 41E2 mutant is defective in activated dendritic cells.
The 41E2 mutant was identified due to its diminished ability to replicate in activated macrophages. We wanted to determine if this defect was present in dendritic cells, which also play an important role in the immune interactions with T. gondii. Similar to naïve macrophages, the mutant was able to grow to wild-type levels in naïve dendritic cells; the mean number of parasites per vacuole was 3 ± 0.31 for the parent and 3 ± 0.6 for the 41E2 mutant. However, the 41E2 mutant displayed a significant decrease in its growth rate at 36 h postinfection in activated dendritic cells (Fig. 4A) (P < 0.005 for one parasite per vacuole; P < 0.025 for two parasites per vacuoles). Comparable results were seen at 24 h postinfection (data not shown). Thus, similar to macrophages, the replication rate of the 41E2 mutant in activated dendritic cells is reduced.
FIG. 4.
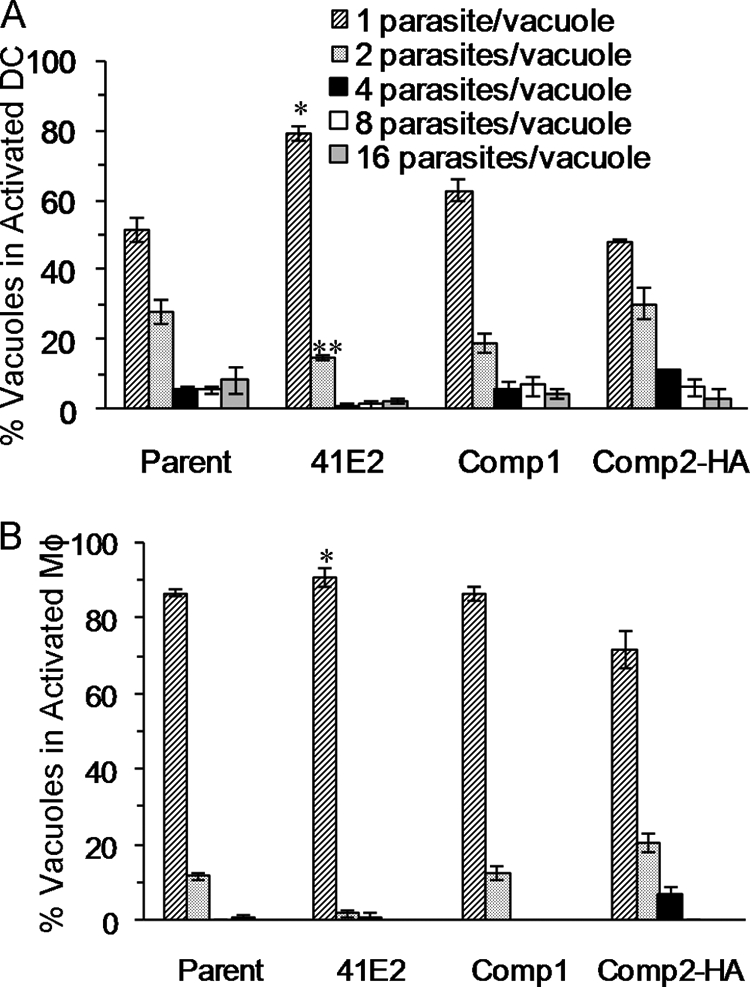
Genetic complementation of 41E2 restores replication in activated immune cells. (A) Dendritic cells (DC) were infected with parasites 6 h after induction with LPS. Infections were terminated at 36 h postinfection, and the number of parasites/vacuole was determined using immunofluorescence. (B) Macrophages (Mφ) were induced with LPS and IFN-γ 3 hours after infection. The number of parasites/vacuole was determined by immunofluorescence 36 h after infection. For panels A and B, data from a representative experiment are displayed as the average percentage of total vacuoles ± standard error determined from triplicate counts. P value is determined as a comparison to the parent and is denoted as follows: *, <0.005; **, <0.025. While the relative growth rates among strains are consistently seen in replicate experiments, the amount of parasite growth fluctuates due to the various degrees of induction of the host cells between experiments.
Complementation of 41E2 with TgTSA1 restores replication in activated immune cells.
A plasmid containing a region of genomic DNA spanning the TgTSA1 coding region, promoter, 5′ UTR, and 3′ UTR was stably transfected into the 41E2 mutant for complementation studies. Complementation with TgTSA1 containing a C-terminal HA tag (Comp2-HA) was also investigated to ensure that the localization described above was functional. In activated dendritic cells, growth of 41E2 complemented with TgTSA1, either with or without the HA tag, was not significantly different from that of the parent strain. These results reveal that reintroduction of TgTSA1 into 41E2 corrects the growth defect (Fig. 4A). The growth of the complement strains was assessed in activated macrophages to resolve whether the growth defect seen in the mutant is due to the interruption in TgTSA1. The mutant has a significant growth defect in activated macrophages 24 and 36 h postinfection, but the defect is more pronounced at 36 h (data not shown; Fig. 4B) (P < 0.005 for two parasites per vacuoles). At 36 h, complement strains are able to grow at rates similar to that of the parent strain (Fig. 4B).
TgTSA1 confers increased resistance to NO without decreasing NO production.
To identify the mechanism responsible for the replication defect of the 41E2 mutant in activated macrophages, we employed aminoguanidine, a commonly used inhibitor of NOS, to disrupt NO production in the host cells. We infected bone marrow-derived macrophages with either parental or 41E2 mutant parasites for 3 hours and then either (i) treated with aminoguanidine but did not activate, (ii) treated with aminoguanidine and activated with LPS and IFN-γ, or (iii) activated with LPS and IFN-γ only. The growth of parent and 41E2 parasites in activated macrophages treated with aminoguanidine was not significantly different than that of the parent in naïve macrophages treated with aminoguanidine (Fig. 5A). These results indicate that the reduction in replication of both the parent and 41E2 mutant in activated macrophages is dependent on NO. The similarity of the replication rates between the parent and 41E2 mutant in activated macrophages treated with aminoguanidine highlights that TgTSA1 provides additional protection against NO.
FIG. 5.
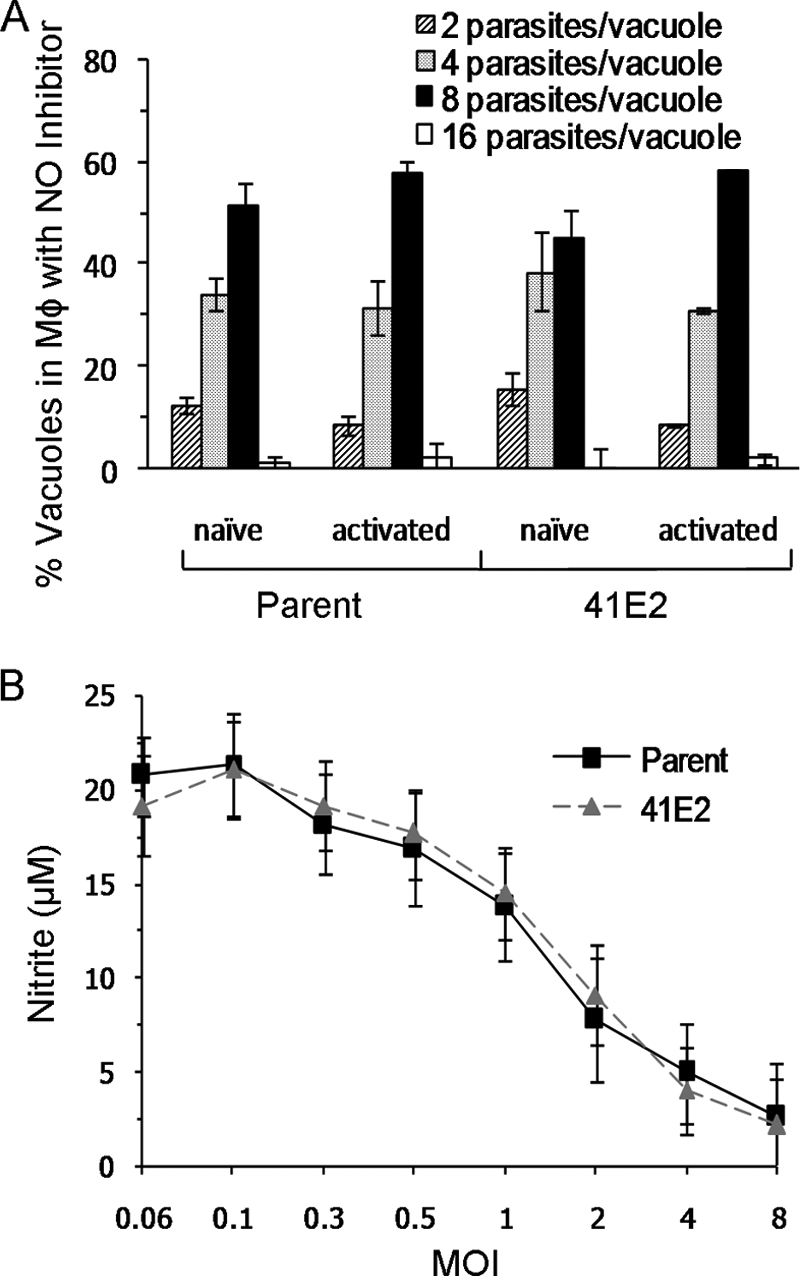
TgTSA1 increases resistance to NO without changing NO production. (A) Infected macrophages (Mφ) were activated in the presence of 1 mM aminoguanidine. The number of parasites/vacuole was determined by immunofluorescence 33 h after infection. Data from a representative experiment are displayed as the average percentage of total vacuoles ± standard error determined from triplicate counts. (B) Macrophages (Mφ) were induced with LPS and IFN-γ 3 hours after infection. Twenty-four hours postinfection, media from the infected wells were tested for the presence of nitrite. Samples were tested in triplicate. Data from a representative experiment are displayed as the average level of nitrite (μM) detected ± standard error. MOI, multiplicity of infection.
To understand how TgTSA1 confers increased resistance to NO, we determined the amount of NO produced by activated macrophages infected with either parent or 41E2 parasites. T. gondii can suppress the ability of activated macrophages to produce NO (21, 34). The amount of NO present can be determined by the Griess reaction, which measures the downstream product nitrite. The amount of nitrite present in cultures of bone marrow-derived macrophages infected with the parent and the amount in those infected with the 41E2 mutant were not significantly different (Fig. 5B). Thus, it is unlikely that TgTSA1 inhibits iNOS induction and that the mechanism of the additional protection against NO conferred by TgTSA1 is not dependent on the amount of NO produced.
Complementation of 41E2 restores cyst production in mice.
The original defect described for the 41E2 mutant was a reduction in the number of brain cysts in chronically infected mice (11). To investigate whether the complementation of the replication defect in activated immune cells would also restore the cyst production in mice, we inoculated mice with the parent strain, the 41E2 mutant, or the two complement strains (Comp1 and Comp2-HA). Twenty-two days after infection with either 104 or 105 parasites, the number of cysts per brain was determined. As expected, the mutant displayed a statistically significant decrease in its ability to produce cysts compared to the parental strain, with both infectious doses (Fig. 6). Similar to the complementation seen in the activated immune cells, brain cyst levels were restored in the complement strains to levels seen in the parent strain (Fig. 6), highlighting the importance of the TgTSA1 protein for the establishment of a chronic infection in mice.
FIG. 6.

Cyst production defect of 41E2 complemented with TgTSA1. Chronic infections were performed with parasite doses of 104 (black bars) and 105 (white bars). Mice were sacrificed at 22 days postinfection, and brains were analyzed for cyst burden. Cyst burden is reported as the average number of cysts per brain ± standard error. P value is determined as a comparison to parent and is denoted as follows: *, < 0.01. Each bar represents the result of eight mice, except the 105 dose for Comp2-HA is from seven mice due to one mouse succumbing to acute infection.
TgTSA1 is recognized by the host immune system.
Proteins on the surface of T. gondii have been shown to interact with the host immune system; therefore, we wanted to determine if TgTSA1 was an antigen recognized during a natural infection. Knowing that the C-terminal three-quarters of the TgTSA1 protein was on the outside of the parasite membrane, we chose an immunogenic 129-amino-acid polypeptide near the C-terminal end for these studies (diagrammed in Fig. 2). This TgTSA1 polypeptide was purified from E. coli (Fig. 7, lane labeled Ponceau S) and used for Western immunoblots with serum from mice infected with T. gondii PRU for 22 days. Serum samples from two independent infections of mice were reactive with the polypeptide (Fig. 7, lanes labeled infected mouse 1 and 2). When serum from an uninfected mouse or no chronic infection serum was used, reactivity to the TgTSA1 polypeptide was not seen (Fig. 7, lane labeled uninfected mouse; data not shown). Chronic infection serum was not reactive with purified lysate from E. coli not expressing the TgTSA1 peptide (data not shown). The reactivity of T. gondii chronic infection serum indicates that TgTSA1 interacts with the immune system of the host.
FIG. 7.
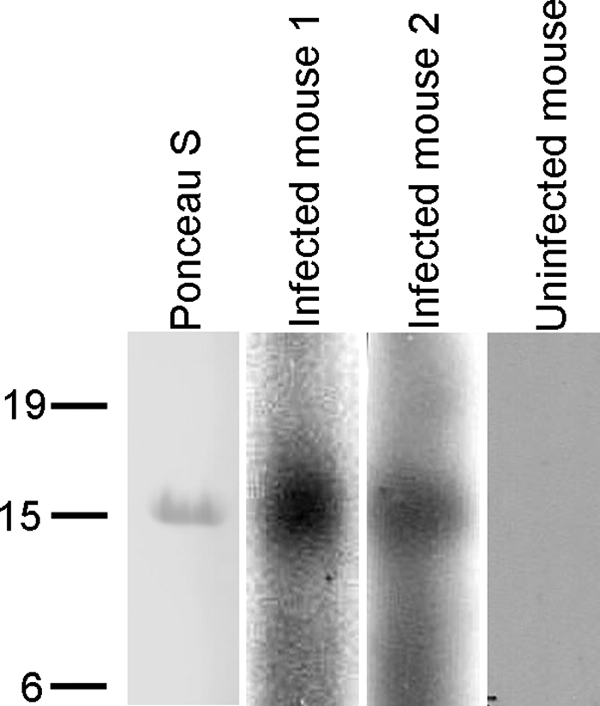
TgTSA1 is detected by mouse chronic infection serum samples. A 14-kDa polypeptide of TgTSA1 was visualized by Ponceau S staining following transfer. Serum samples collected from mice infected with T. gondii from two independent experiments detected the polypeptide (infected mouse 1 and infected mouse 2). Serum from an uninfected mouse did not react with the polypeptide (uninfected mouse).
DISCUSSION
TgTSA1 is annotated in the T. gondii database as a hypothetical protein that does not share homology with any other proteins of known function or contain any functional domains. Our previous work highlighted that TgTSA1 may be important for parasite virulence (11). This current study confirms that TgTSA1 is indeed important for the establishment of a chronic infection in mice. To characterize this virulence defect further, we show that TgTSA1 is important for replication in activated macrophage and dendritic cells. We also show that TgTSA1 is a tachyzoite-specific, transmembrane domain-containing surface protein. The discontinuous staining of the outer membrane seen with the HA antibody may indicate that the protein is found within lipid rafts on the parasite surface. Due to its surface location and its role in protecting parasites from immune cell activation, it is not surprising that TgTSA1 interacts with the immune system and is detected by chronic infection sera.
As an obligate intracellular parasite, T. gondii must invade host cells upon initiation of an infection. The parasite then uses these cells to replicate and disseminate throughout the host during acute infection, with the intended goal of spreading to muscle and brain tissue to form a chronic infection. Some of the cells T. gondii uses as a vehicle during infection are also immune system cells responsible for controlling and terminating the infection. Macrophages and dendritic cells are involved in cell-mediated immunity through the production of cytokines. Additionally, once activated, these cells are able to limit the growth of the parasite (1, 2, 21). To circumvent the unfriendly environment within macrophages and dendritic cells, T. gondii is able to manipulate signaling cascades involved in the activation of these infected cells (6, 24). The production of NO is one of the signaling cascades suppressed in cells infected with T. gondii. The use of aminoguanidine to inhibit NO synthase in activated macrophages shows that TgTSA1 confers increased resistance to NO (Fig. 5A). It is possible that the TgTSA1 interacts with host cell receptors to curb the production of NO or that the absence of TgTSA1 in the parasite membrane damages the membrane integrity, making it more susceptible to NO. The 41E2 mutant was shown to suppress NO in a previous screen (26) and this study (Fig. 5B), highlighting that TgTSA1 mediates resistance to NO rather than inhibits its production. Surface proteins help maintain membrane integrity and facilitate initial contacts between the host and the parasite. Alterations of membrane components could make the parasite more susceptible to host elements and decrease the fitness of the parasite. This diminished fitness would more likely be evident in restrictive environments, such as activated immune cells.
NO production by the host has been shown to be important to control a T. gondii chronic infection. iNOS knockout mice infected with T. gondii are able to survive acute infection and succumb to infection 3 to 4 weeks after inoculation. Mortality is associated with uncontrolled tachyzoite replication in the brain and necrotizing encephalitis. These results indicate that while the killing of tachyzoites by NO is not necessary for control of acute infection, NO is important for controlling tachyzoite replication in the brain during early chronic infection (33). Microglial cells, which are responsible for immune defense in the central nervous system, that are activated with LPS and IFN-γ control T. gondii replication in an NO-dependent mechanism (5). Rozenfeld et al. (31) showed that T. gondii-infected microglial cells have a significant decrease in iNOS expression. Neighboring microglial cells activated by IFN-γ did not display a reduction in NO production, indicating that the parasite protein responsible for this activity is not secreted (31). The 41E2 mutant is impaired in its ability to establish a chronic infection but is able to kill mice during acute infection at the same rate as the parent strain (Fig. 6 and data not shown). It is quite possible that the mutant is able to disseminate normally during acute infection, but encounters difficulties with activated microglial cells, which results in a reduction of brain cysts. The reduction in cysts seen in the brain could be due to an inability to prevent NO-mediated killing of the parasites by microglial cells prior to conversion to bradyzoites. Further studies will examine the growth of the mutant in activated microglial cells to further characterize the infection defect of this mutant.
The surface of T. gondii is decorated with an abundance of proteins, many belonging to the SAG, SRS, and SUSA families. When SAG1 is blocked by antibodies, parasites have a diminished ability to attach to and invade host cells (25). This result indicates that SAG1 is involved in these vital processes, and that other surface antigens may play redundant roles. The surface localization of TgTSA1 makes it a possible player in interactions with the host. Results of immunofluorescence studies indicate that the C terminus of the TgTSA1 is extracellular. This orientation would expose the majority of the protein, 370 amino acids, to the dynamic environment of the host during an infection. Through interactions with parasitophorous vacuole membrane (PVM) proteins, TgTSA1 could manipulate contact with host cell proteins, such as immunity-related GTPase (IRG) family members. IRGs traffic to the PVM to disrupt parasite membranes (23, 40). Virulent strains of T. gondii are not susceptible to IRG-mediated membrane disruption, likely due to polymorphisms between strains that do not allow some IRG proteins to bind (40). Although TgTSA1 displays a low degree of polymorphism (www.toxodb.org), TgTSA1 may facilitate some interaction with IRG proteins. Efforts are currently underway to crystallize the extracellular C terminus of TgTSA1 (W. Anderson, personal communication) in hopes that structural data will help elucidate a role for this protein.
The sheer number and immunodominance of the GPI-anchored proteins covering the surface of the parasite have made it difficult to identify surface transmembrane proteins. In an attempt to identify integral membrane proteins that are not GPI anchored, Gilk et al. (13) used a photoactivatable compound, 5-[125I]iodonaphthalene-1-azide to tag membrane-embedded proteins. Using this method, they were able to detect and identify two characterized proteins of the inner membrane complex (GAP45 and GAP50) and a novel protein associated with the parasite periphery (PhIL1). Analysis of the secondary structure of PhIL1 revealed no transmembrane domain or lipid anchor addition signals. Other potential transmembrane proteins were detected but not identified or characterized (13). TgTSA1 is the first T. gondii transmembrane domain-containing surface protein to be characterized.
The GPI-anchored surface proteins are further classified by their stage specificity. For example, SAG1 and SRS1 are tachyzoite specific, while SRS9 and SAG4A are expressed only in bradyzoites (19). Many of the surface proteins are recognized by the immune system, reinforcing the idea that the proteins interact with the host immune system. The high expression of tachyzoite-specific SAG1 may assist the host in recognizing and clearing tachyzoites (19). Misexpression of SRS9 in tachyzoites resulted in a greater number of parasites being cleared during acute infection (17). Changing of the parasite surface proteins throughout the life cycle may allow immune evasion and infection persistence. It is interesting that similar to most GPI-anchored surface antigens, the transmembrane domain-containing surface protein TgTSA1 is regulated between the tachyzoite and bradyzoite stages. Characterization of the many hypothetical proteins, such as TgTSA1, within the T. gondii genome may better define vital processes in diverse life cycle stages of the parasite and address the gaps in our knowledge of T. gondii biology.
Acknowledgments
We sincerely thank Taylor Dagenais and Steve Giles for assistance with dendritic cells, Jay Bangs for the use of his microscope, and the following individuals for antibodies: David Sibley (β-tubulin and GRA2), Gary Ward (IMC1), John Boothroyd (SAG1), and Louis Weiss (Bag1).
This research was supported by NIH Awards AI054603 (L.J.K.), AI072817 (A.M.P.), and AI072028 (D.G.M.).
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 6 July 2009.
REFERENCES
- 1.Alexander, J., and C. A. Hunter. 1998. Immunoregulation during toxoplasmosis. Chem. Immunol. 7081-102. [DOI] [PubMed] [Google Scholar]
- 2.Aline, F., D. Bout, and I. Dimier-Poisson. 2002. Dendritic cells as effector cells: gamma interferon activation of murine dendritic cells triggers oxygen-dependent inhibition of Toxoplasma gondii replication. Infect. Immun. 702368-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bliss, S. K., Y. Zang, and E. Y. Denkers. 1999. Murine neutrophil stimulation by Toxoplasma gondii antigen drives high level production of IFN-γ independent IL-12. J. Immunol. 1632081-2088. [PubMed] [Google Scholar]
- 4.Boothroyd, J. C., A. Hehl, L. J. Knoll, and I. D. Manger. 1998. The surface of Toxoplasma: more or less. Int. J. Parasitol. 283-9. [DOI] [PubMed] [Google Scholar]
- 5.Chao, C. C., W. R. Anderson, S. Hu, G. Gekker, A. Martella, and P. K. Peterson. 1993. Activated microglia inhibit multiplication of Toxoplasma gondii via a nitric oxide mechanism. Clin. Immunol. Immunopathol. 67178-183. [DOI] [PubMed] [Google Scholar]
- 6.Denkers, E. Y., and B. A. Butcher. 2005. Sabotage and exploitation in macrophages parasitized by intracellular protozoans. Trends Parasitol. 2135-41. [DOI] [PubMed] [Google Scholar]
- 7.Denkers, E. Y., and R. T. Gazzinelli. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11569-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donald, R. G., and D. S. Roos. 1993. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. USA 9011703-11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubey, J. P. 1994. Toxoplasmosis. J. Am. Vet. Med. Assoc. 2051593-1598. [PubMed] [Google Scholar]
- 10.Dubey, J. P. 1998. Advances in the life cycle of Toxoplasma. Int. J. Parasitol. 281019-1024. [DOI] [PubMed] [Google Scholar]
- 11.Frankel, M. B., D. G. Mordue, and L. J. Knoll. 2007. Discovery of parasite virulence genes reveals a unique regulator of chromosome condensation 1 ortholog critical for efficient nuclear trafficking. Proc. Natl. Acad. Sci. USA 10410181-10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gazzinelli, R. T., S. Hieny, T. Wynn, S. Wolf, and A. Sher. 1993. IL-12 is required for the T-cell independent induction of IFN-γ by an intracellular parasite and induces resistance in T-cell deficient hosts. Proc. Natl. Acad. Sci. USA 906115-6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilk, S. D., Y. Raviv, K. Hu, J. M. Murray, C. J. Beckers, and G. E. Ward. 2006. Identification of PhIL1, a novel cytoskeletal protein of the Toxoplasma gondii pellicle, through photosensitized labeling with 5-[125I]iodonaphthalene-1-azide. Eukaryot. Cell 51622-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimwood, J., and J. E. Smith. 1996. Toxoplasma gondii: the role of parasite surface and secreted proteins in host cell invasion. Int. J. Parasitol. 26169-173. [DOI] [PubMed] [Google Scholar]
- 15.Inaba, K., W. J. Swiggard, R. M. Steinman, N. Romani, and G. Schuler. 2003. Isolation of dendritic cells. p. 3.7.1-3.7.15. In J. E. Coligan, A. M. Krusibeck, D. H. Margulies, E. M. Shevach, and W. Strobe (ed.), Current protocols in immunology. John Wiley & Sons, Indianapolis, IN.
- 16.Jung, C., C. Y.-F. Lee, and M. E. Grigg. 2004. The SRS superfamily of Toxoplasma surface proteins. Int. J. Parasitol. 34285-296. [DOI] [PubMed] [Google Scholar]
- 17.Kim, S.-K., A. Karasov, and J. C. Boothroyd. 2007. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect. Immun. 751626-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert, H., N. Hitziger, I. Dellacasa, M. Svensson, and A. Barragan. 2006. Induction of dendritic cell migration upon Toxoplasma gondii infection potentates parasite dissemination. Cell. Microbiol. 81611-1623. [DOI] [PubMed] [Google Scholar]
- 19.Lekutis, C., D. J. P. Ferguson, M. E. Grigg, M. Camps, and J. C. Boothroyd. 2001. Surface antigens of Toxoplasma gondii: variations on a theme. Int. J. Parasitol. 311285-1292. [DOI] [PubMed] [Google Scholar]
- 20.Liu, C.-H., Y. Fan, A. Dias, L. Esper, R. A. Corn, A. Bafica, F. S. Machado, and J. Aliberti. 2006. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 17731-35. [DOI] [PubMed] [Google Scholar]
- 21.Lüder, C. G., M. Algner, C. Lang, N. Bleicher, and U. Gross. 2003. Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int. J. Parasitol. 33833-844. [DOI] [PubMed] [Google Scholar]
- 22.Luft, B. J., Y. Naot, F. G. Araujo, E. B. Stinson, and J. S. Remington. 1983. Primary and reactivated Toxoplasma infection in patients with cardiac transplants. Ann. Intern. Med. 9927-31. [DOI] [PubMed] [Google Scholar]
- 23.Martens, S., I. Parvanova, J. Zerrahn, G. Griffiths, G. Shell, G. Reichmann, and J. C. Howard. 2005. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 1e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKee, A. S., F. Dzierszinski, M. Boes, D. S. Roos, and E. J. Pearce. 2004. Functional inactivation of immature dendritic cells by the intracellular parasite Toxoplasma gondii. J. Immunol. 1732632-2640. [DOI] [PubMed] [Google Scholar]
- 25.Mineo, J. R., and L. H. Kasper. 1994. Attachment of Toxoplasma gondii to host cells involves major surface protein, SAG-1 (P30). Exp. Parasitol. 7911-20. [DOI] [PubMed] [Google Scholar]
- 26.Mordue, D. G., C. F. Scott-Weathers, C. M. Tobin, and L. J. Knoll. 2007. A patatin-like protein protects Toxoplasma gondii from degradation in activated macrophages. Mol. Microbiol. 63482-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mordue, D. G., and L. D. Sibley. 2003. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 741015-1025. [DOI] [PubMed] [Google Scholar]
- 28.Pollard, A. M., K. N. Onatolu, L. Hiller, K. Haldar, and L. J. Knoll. 2008. Highly polymorphic family of glycosylphosphatidylinositol-anchored surface antigens with evidence of developmental regulation in Toxoplasma gondii. Infect. Immun. 76103-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter, S. B., and M. Sande. 1992. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N. Engl. J. Med. 3271643-1648. [DOI] [PubMed] [Google Scholar]
- 30.Remington, J. S., R. McLeond, and G. Desmonts. 1990. Toxoplasmosis, p. 89-195. In J. S. Remington and J. O. Klein (ed.), Infectious diseases of the fetus and newborn. WB Saunders Co., Philadelphia, PA.
- 31.Rozenfeld, C., R. Martinez, S. Seabra, C. Sant'anna, J. G. Goncalves, M. Bozza, V. Moura-Neto, and W. De Souza. 2005. Toxoplasma gondii prevents neuron degeneration by interferon-γ-activated microglia in a mechanism involving inhibition of inducible nitric oxide synthase and transforming growth factor-β1 production by infected microglia. Am. J. Pathol. 1671021-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharton-Kersten, T. M., T. A. Wynn, E. Y. Denkers, S. Bala, L. Showe, E. Grunvald, S. Hieny, R. T. Gazzinelli, and A. Sher. 1996. In the absence of endogenous IFN-γ mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 1574045-4054. [PubMed] [Google Scholar]
- 33.Scharton-Kersten, T. M., G. Yap, J. Magram, and A. Sher. 1997. Inducible nitric oxide is essential for host control of persistent but not acute infection of intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 1851261-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seabra, S. H., W. de Sousa, and R. A. Damatta. 2002. Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp. Parasitol. 10062-70. [DOI] [PubMed] [Google Scholar]
- 35.Soldati, D., and J. C. Boothroyd. 1995. A selector of transcription initiation in the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol. 1518-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki, Y., M. A. Orellana, R. D. Schreiber, and J. S. Remington. 1988. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science 240516-518. [DOI] [PubMed] [Google Scholar]
- 37.Wichroski, M. J., J. A. Melton, C. G. Donahue, R. K. Tweten, and G. E. Ward. 2002. Clostridium septicum alpha-toxin is active against the parasitic protozoan Toxoplasma gondii and targets members of the SAG family of glycosylphosphatidylinositol-anchored surface proteins. Infect. Immun. 704353-4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong, S. Y., and J. S. Remington. 1993. Biology of Toxoplasma gondii. AIDS 7299-316. [DOI] [PubMed] [Google Scholar]
- 39.Yap, G. S., and A. Sher. 1999. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation, and effector function. Immunobiology 201240-247. [DOI] [PubMed] [Google Scholar]
- 40.Zhao, Y., D. J. P. Ferguson, D. C. Wilson, J. C. Howard, L. D. Sibley, and G. S. Yap. 2009. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J. Immunol. 1823775-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]