

Abstract
A proinflammatory role for glycogen synthase kinase 3β (GSK-3β) has been demonstrated. Here, we addressed its roles on heat-inactivated Staphylococcus aureus-induced microglial inflammation. Heat-inactivated S. aureus induced tumor necrosis factor alpha (TNF-α) and nitric oxide (NO) production, at least in part, via a Toll-like receptor 2-regulated pathway. Neutralization of TNF-α largely blocked heat-inactivated S. aureus-induced NO. Heat-inactivated S. aureus activated GSK-3β, and inhibiting GSK-3β reduced TNF-α production as well as inducible NO synthase (iNOS)/NO biosynthesis. While activation of NF-κB was essential for heat-inactivated S. aureus-induced TNF-α and NO, inhibiting GSK-3β blocked heat-inactivated S. aureus-induced NF-κB p65 nuclear translocation. Additionally, inhibiting GSK-3β enhanced heat-inactivated S. aureus-induced interleukin-10 (IL-10) production (IL-10 is an anti-inflammatory cytokine which inhibits TNF-α production). Neutralization of IL-10 reduced TNF-α downregulation caused by GSK-3β inhibition. These results suggest that GSK-3β regulates heat-inactivated S. aureus-induced TNF-α and NO production in microglia mainly by activating NF-κB and probably by inhibiting IL-10.
Staphylococcus aureus, a gram-positive bacterium, causes a variety of diseases, such as bacteremia, peritonitis, subcutaneous and brain abscess, and life-threatening staphylococcal septic shock (15). The mechanisms that lead to staphylococcal septic shock are multifactorial but involve especially immunogenic and toxic injuries (10, 40). Cell wall components and secreted virulence factors, including enterotoxins and exotoxins, have been shown to be inflammatory and cytotoxic to the host. Pathogen-associated molecular patterns are recognized by the innate immune system through a family of pattern recognition receptors, such as Toll-like receptors (TLRs) (2, 6, 26). Microglia, the resident macrophages in the brain, express TLR2 to recognize S. aureus peptidoglycan and play a critical role in neuroinflammation (7, 35, 37). Induction of neuroinflammation by S. aureus is partially mediated by TLR2- and nuclear factor-κB (NF-κB)-regulated pathways (23, 26, 36, 51).
Infection of S. aureus causes the deregulated production of inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and IL-10, and chemokines, including monocyte chemoattractant protein 1 (MCP-1) and RANTES (regulated on activation, normal T cell expressed and secreted protein) (24, 25, 32, 45). TNF-α, a potent proinflammatory cytokine, causes severe inflammatory responses, including cytokine and chemokine production and inducible nitric oxide (NO) synthase (iNOS)/NO biosynthesis in S. aureus infection (49). The deregulated generation of NO contributes to S. aureus-induced circulatory failure and liver injury (34). IL-10, a potent anti-inflammatory cytokine, inhibits the synthesis of the proinflammatory cytokines (TNF-α, IL-1, IL-6, IL-12, IL-18, and IL-10 itself), chemokines (IL-8, MCP-1, and RANTES), and iNOS/NO (4, 30, 43). IL-10 knockout mice display high mortality and are more susceptible to S. aureus-induced brain abscess (48). Exogenous IL-10 inhibits lethal sepsis, hepatic injury, and TNF-α production induced by staphylococcal enterotoxin B in mice (46, 48).
Inhibiting glycogen synthase kinase 3β (GSK-3β) downregulates TLR-mediated inflammatory responses but increases IL-10 production (41, 53). Since NF-κB is important for inflammatory activation, GSK-3β is also involved in activating NF-κB in response to inflammatory stimuli (17-21, 29, 44, 50, 52). Therefore, GSK-3β inhibitors have been used to confer anti-inflammation against TNF-α administration, endotoxemia, experimental colitis, type II collagen-induced arthritis, ovalbumin-induced asthma, and experimental autoimmune encephalomyelitis (5, 12-14, 18, 20, 31, 41, 50, 52). Notably, current studies also show the effects of GSK-3β inhibition in reducing gram-negative coccobacillus Francisella-induced inflammation (55). GSK-3β inhibitors have also been widely used to reduce microglial inflammation and neurotoxicity (31, 54). In search of strategies against S. aureus-induced microglial inflammation, we investigated the possible effects of GSK-3β inhibition. In the present study, we report that inhibiting GSK-3β blocks NF-κB activation, TNF-α production, and iNOS/NO biosynthesis, but increases IL-10 production in heat-inactivated S. aureus-stimulated microglia.
MATERIALS AND METHODS
Antibodies and reagents.
Neutralizing antibodies specific for TLR2, TNF-α, and IL-10 and isotype-matched antibody control were obtained from RD Systems. Mouse monoclonal antibody specific for β-actin and NF-κB p65 was purchased from Chemicon International, Inc. (Temecula, CA). Monoclonal anti-mouse iNOS was obtained from BD Biosciences (Palo Alto, CA). Alexa Fluor 488- and horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, and donkey anti-goat immunoglobulin G (IgG) antibodies were obtained from Invitrogen Corp. (Carlsbad, CA). Antibodies against phospho-GSK-3β (Ser9), phospho-GSK-3β (Tyr216), GSK-3β, phospho-glycogen synthase (GS) (Ser641), and GS were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Pyrrolidine dithiocarbamate (PDTC), caffeic acid phenethyl ester (CAPE), and BAY11-7821 (BAY) were purchased from Tocris Bioscience (Ellisville, MO). 6-Bromo-indirubin-3′-oxime (BIO) and lithium chloride (LiCl) were obtained from Sigma-Aldrich Co. (St Louis, MO). All drug treatments in cells were assessed for their cytotoxic effects by using a cytotoxicity assay. Doses without cytotoxic effects were used.
Cell culture.
BV-2 immortalized murine microglial cells were obtained from C. C. Huang (Department of Pediatrics, National Cheng Kung University, Tainan, Taiwan). Primary rat microglia-enriched cultures with a purity of >98% were prepared from whole brains of 1-day-old Sprague-Dawley breeder rat pups as previously described (31). The cells were grown in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies, Rockville, MD) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen Life Technologies), 50 U/ml of penicillin, and 50 μg/ml of streptomycin in a humidified atmosphere with 5% CO2 and 95% air.
Bacterial culture and heat inactivation.
Clinical S. aureus isolate S2-1790 is a pediatric isolate from a patient with sternum osteomyelitis-related bacteremia from the National Cheng Kung University Medical College Hospital collection. It shows sensitivity to all antimicrobial agents. To control the standard dosing of bacteria and to avoid the potentially confounding effects by live organisms, heat-inactivated S. aureus was prepared by heating an exponential-phase culture for 1 h at 56°C. Aliquots of heat-inactivated S. aureus were made and stored at −80°C until use.
Cytotoxicity assay.
We used a colorimetric assay (cytotoxicity detection kit; Roche Diagnostics, Lewes, United Kingdom), according to the manufacturer's instructions, to measure lactate dehydrogenase activity, an indicator of cell damage caused by all inhibitors. Aliquots of the incubation medium were transferred to 96-well microplates, and the absorbance was measured using a microplate reader (SpectraMax 340PC; Molecular Devices Corporation, Sunnyvale, CA).
Western blot analysis.
We harvested the cells and lysed them with a buffer containing 1% Triton X-100, 50 mM of Tris (pH 7.5), 10 mM of EDTA, 0.02% NaN3, and a protease inhibitor cocktail (Roche Boehringer Mannheim Diagnostics, Mannheim, Germany). After they had been freeze-thawed once, the cell lysates were centrifuged at 12,000 rpm at 4°C for 10 min. The supernatants were then collected and boiled in sample buffer for 10 min. Following sodium dodecyl sulfate-polyacrylamide gel electrophoresis, proteins were transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA), blocked at 4°C overnight in phosphate-buffered saline (PBS) plus 0.05% Tween 20 containing 5% skim milk, and probed with a 1:1,000 dilution of primary antibodies at 4°C overnight. After they had been washed with PBS plus 0.05% Tween 20, the blots were incubated with a 1:5,000 dilution of horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. The protein bands were visualized using enhanced chemiluminescence (Pierce Biotechnology Inc., Rockford, IL).
Nitrite assay.
We assessed NO production by measuring the accumulated levels of nitrite in the supernatant with Griess reagent as previously described (31). Briefly, 100 μl of the culture supernatant was reacted with 100 μl of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2.5% H3PO4) for 10 min at room temperature. The concentration of nitrite was measured using a microplate reader (SpectraMax 340PC; Molecular Devices) at 540 nm, and the nitrite concentration was calculated using a standard curve of sodium nitrite with an enzyme-linked immunosorbent assay (ELISA) software (Softmax Pro; Molecular Devices).
ELISA.
The concentrations of TNF-α and IL-10 in cell-conditioned culture medium were determined using ELISA kits (RD Systems) according to the manufacturer's instructions.
Immunostaining NF-κB nuclear translocation.
Cells were fixed with 1% formaldehyde in PBS at room temperature for 10 min. After they had been washed twice with PBS, they were stained with anti-mouse NF-κB at a final concentration of 1 μg/ml at room temperature for 1 h and then incubated with a mixture of Alexa Fluor 488-conjugated goat anti-mouse IgG plus 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) at a concentration of 5 μg/ml at room temperature for 1 h. After another washing with PBS, the cells were covered with mounting fluid and visualized under a confocal laser microscope (Leica TCS SPII; Nussloch, Germany). In the untreated group (magnification of ×400), the ratio of nuclear NF-κB-positive cells to total cells was normalized as 100%. The increases are means of the results obtained from two individual experiments.
Statistical analysis.
Student's t test was used to analyze the data (SigmaPlot 8.0 for Windows; Systat Software, Inc., San Jose, CA). Statistical significance was set at P < 0.05.
RESULTS
Heat-inactivated S. aureus induces TLR2-regulated TNF-α and NO production in BV-2 mouse microglia.
Using ELISA and Griess reagent assay, we found that BV-2 mouse microglial cells treated with heat-inactivated S. aureus showed a significant (P < 0.05) increase in TNF-α and NO production, compared to untreated cells, both at the time periods of 12 and 24 h of treatment (Fig. 1A). To further characterize signaling of TLR2, a receptor of gram-positive bacterial peptidoglycan, for heat-inactivated S. aureus, neutralizing IgG specific for TLR2 was used. Results showed that anti-TLR2 neutralizing IgG, but not isotype-matched antibody control, significantly (P < 0.05) blocked the production of heat-inactivated S. aureus-induced TNF-α and NO (Fig. 1B). TNF-α plays a critical role for proinflammatory responses induced by S. aureus infection (36). Using anti-TNF-α neutralizing IgG, we confirmed that heat-inactivated S. aureus induced NO production in a TNF-α-regulated manner (Fig. 1C). These results indicate that heat-inactivated S. aureus induces NO production, at least in part, via TLR2-mediated and TNF-α-regulated pathways.
FIG. 1.

Effects of TLR2 and TNF-α on heat-inactivated S. aureus-induced NO production in microglia. (A) BV-2 mouse microglial cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 105) for 12 and 24 h. After the supernatant had been collected, we determined TNF-α and NO production by using ELISA and Griess reagent, respectively. Sodium nitrite (NaNO2) was used for the standard calculation of nitrite concentration. (B) BV-2 cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 105) for 24 h with cotreatment of isotype-matched antibody control (50 μg/ml) or anti-TLR2 IgG (50 μg/ml). ELISA and Griess reagent were used to determine TNF-α and NO production, respectively. (C) BV-2 cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 105) for 24 h with cotreatment of isotype-matched antibody control (50 μg/ml) or anti-TNF-α IgG (50 μg/ml). Griess reagent was used to determine NO production. The data are means ± the standard deviation (SD) obtained from three individual experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with S. aureus alone or S. aureus plus isotype-matched antibody control.
Heat-inactivated S. aureus activates GSK-3β in BV-2 mouse microglia.
To investigate the activation of GSK-3β in heat-inactivated S. aureus-stimulated BV-2 mouse microglial cells, we used Western blotting to determine the phosphorylation of GSK-3β and its substrate, GS. GSK-3β activity is positively regulated by dephosphorylation of GSK-3β at Ser9 and phosphorylation of GSK-3β at Tyr216 (11, 27, 28, 33). Results showed that heat-inactivated S. aureus did not induce GSK-3β dephosphorylation at Ser9 but significantly induced phosphorylation of GSK-3β (Tyr216) and GS (Ser641) (Fig. 2). Furthermore, GSK-3β inhibitors BIO and LiCl potently blocked these effects (data not shown). These results indicate that heat-inactivated S. aureus induces GSK-3β activation.
FIG. 2.
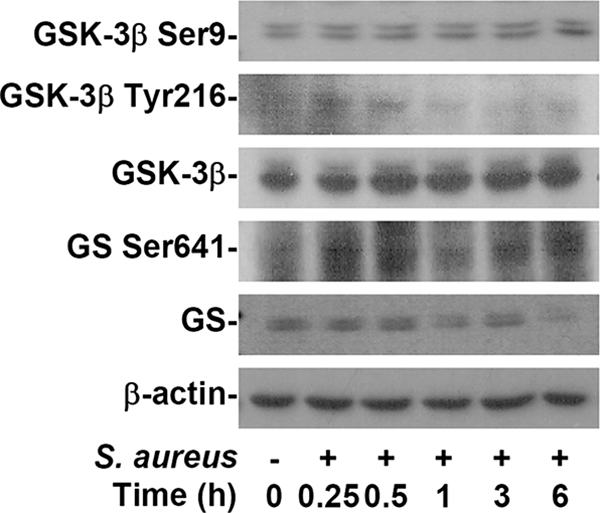
Activation of GSK-3β in heat-inactivated S. aureus-stimulated microglia. BV-2 mouse microglial cells (2.5 × 105) were treated with heat-inactivated S. aureus (2.5 × 106) for the different time periods, as indicated. Western blotting was used to determine phosphorylation of GSK-3β (Ser9 and Tyr216) and GS (Ser641). β-Actin was the internal control. Data shown are representative of the results for three individual experiments.
Inhibiting GSK-3β blocks the production of heat-inactivated S. aureus-induced TNF-α as well as iNOS/NO biosynthesis in microglia.
Inhibiting GSK-3β decreases TLR-mediated inflammation (31, 41). We then tested the effects of GSK-3β inhibition on heat-inactivated S. aureus-induced inflammation. We found, using ELISA, that inhibiting GSK-3β with BIO significantly (P < 0.05) reduced the production of heat-inactivated S. aureus-induced TNF-α (Fig. 3A) and, using Griess reagent, that inhibiting GSK-3β with BIO or LiCl blocked NO production (Fig. 3B) in BV-2 mouse microglial cells as well as in primary rat microglia-enriched cultures. iNOS biosynthesis usually regulates NO production in microglia. To verify that inhibiting GSK-3β causes NO reduction, we further studied iNOS protein expression in heat-inactivated S. aureus-stimulated BV-2 mouse microglial cells. We found, using Western blotting, that BIO and LiCl inhibited iNOS protein expression (Fig. 3B). We also confirmed that GSK-3β regulated NO production in BV-2 cells stimulated with the different diseases of S. aureus infection, including muscle abscess (S2-1065), folliculitis (S2-1509), and bacteremia (S-20) (data not shown). These results provide evidence that inhibiting GSK-3β downregulates TNF-α and iNOS/NO in heat-inactivated S. aureus-stimulated microglia.
FIG. 3.
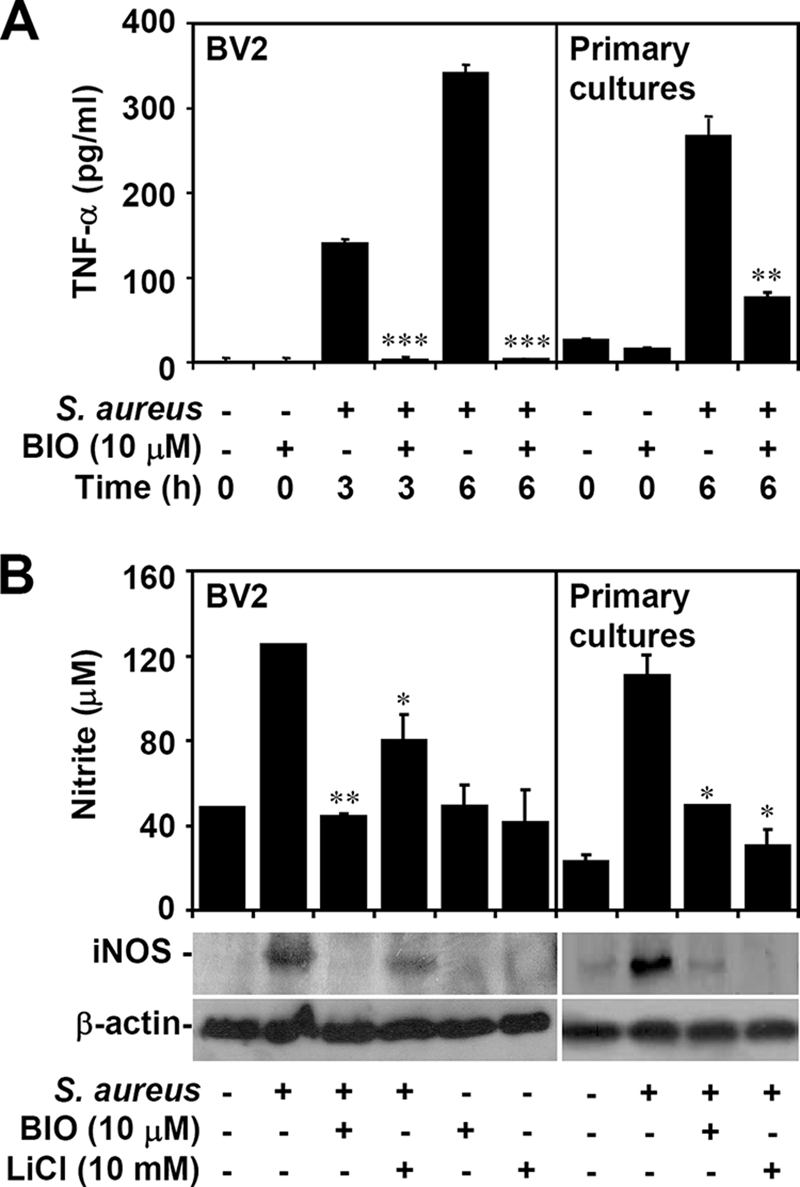
Effects of inhibiting GSK-3β on TNF-α production and iNOS/NO biosynthesis in heat-inactivated S. aureus-stimulated microglia. BV-2 mouse microglial cells or primary rat microglia-enriched cultures (3 × 104) were pretreated with GSK-3β inhibitors BIO (10 μM) or LiCl (10 mM) for 0.5 h and then treated with heat-inactivated S. aureus (3 × 105). For the indicated time periods of treatment, ELISA was used to determine TNF-α production (A). Twenty-four hours posttreatment, we determined NO production and iNOS expression, using Griess reagent and Western blot analysis, respectively (B). β-Actin was the internal control. Data shown are representative of the results for three individual experiments. For ELISA and nitrite assay, the data are means ± SD obtained from three individual cultures. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with S. aureus alone.
Inhibiting GSK-3β blocks TNF-α and NO production by downregulating heat-inactivated S. aureus-activated NF-κB in BV-2 mouse microglia.
GSK-3β may act upstream of NF-κB, an important transcription factor of iNOS and TNF-α (17-21, 29, 44, 50, 52). To confirm the essential role of NF-κB, we evaluated the effects of selective NF-κB inhibitors, including PDTC, CAPE, and BAY. All NF-κB inhibitors significantly (P < 0.05) reduced the production of heat-inactivated S. aureus-induced TNF-α and NO in BV-2 mouse microglial cells (Fig. 4A). To further investigate the effect of GSK-3β on NF-κB, we used immunocytochemistry to determine the nuclear translocation of NF-κB p65. We found that inhibiting GSK-3β blocked heat-inactivated S. aureus-induced NF-κB p65 nuclear translocation (Fig. 4B). These results indicate that inhibiting GSK-3β blocks heat-inactivated S. aureus-induced TNF-α and NO production by inactivating NF-κB.
FIG. 4.
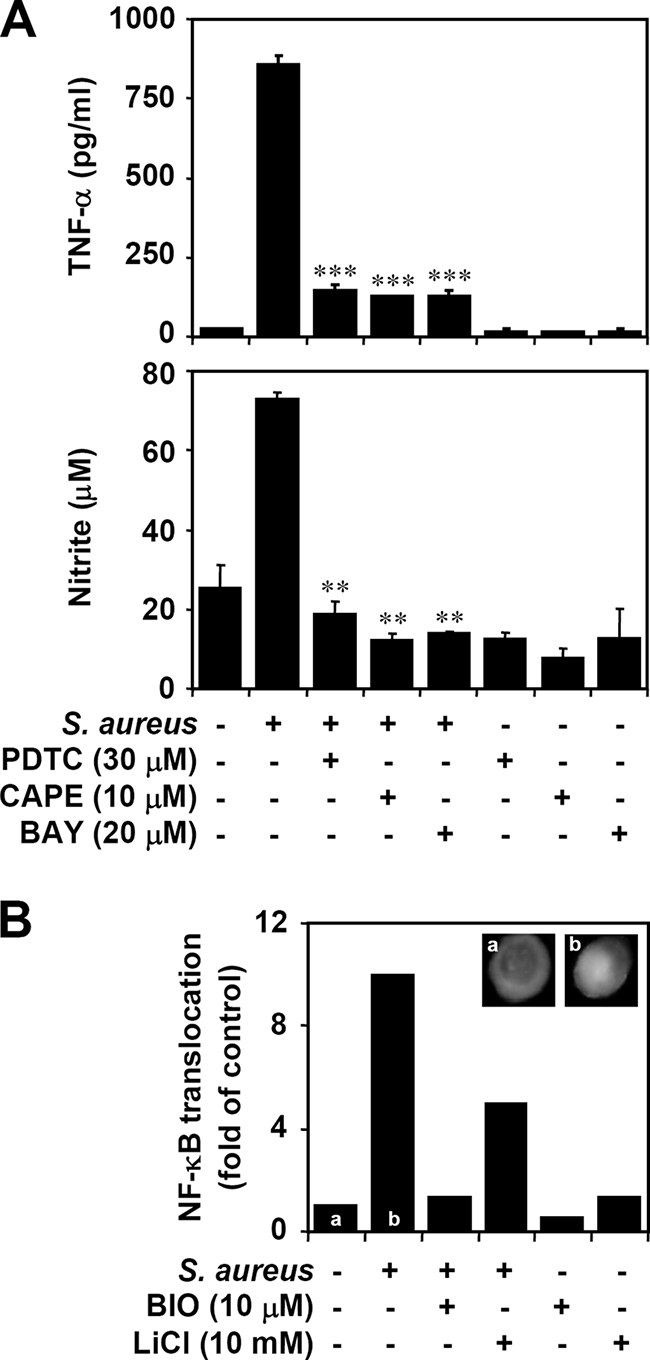
Relationship between NF-κB and GSK-3β on TNF-α and NO production in heat-inactivated S. aureus-stimulated microglia. (A) BV-2 mouse microglia cells (3 × 104) were pretreated with NF-κB inhibitors PDTC (30 μM), CAPE (10 μM), and BAY (20 μM) for 0.5 h and then treated with heat-inactivated S. aureus (3 × 105) for 24 h. ELISA and Griess reagent were used to determine TNF-α and NO production, respectively. The data are means ± SD of the results obtained from three individual experiments. **, P < 0.01; ***, P < 0.001, compared with S. aureus alone. (B) BV-2 cells (3 × 104) were pretreated with GSK-3β inhibitors BIO (10 μM) or LiCl (10 mM) for 0.5 h and then treated with heat-inactivated S. aureus (3 × 105) for 0.5 h. Immunostaining was used to detect the nuclear translocation of NF-κB p65, using primary antibodies followed by Alexa Fluor 488-conjugated secondary antibodies. DAPI was used for nuclear staining. The n-fold increases are means of the results obtained from two individual experiments. a, untreated group; b, heat-inactivated S. aureus-treated group.
Inhibiting GSK-3β increases the production of heat-inactivated S. aureus-induced IL-10 in BV-2 mouse microglia.
TNF-α is critical for the pathogenesis of S. aureus infection, and IL-10 is essential for diminishing S. aureus-induced inflammation (48). Using ELISA, we determined the time kinetics and dose response of heat-inactivated S. aureus on TNF-α and IL-10 production. We found that TNF-α was time-dependently expressed following the early expression of IL-10 (Fig. 5A). To further characterize the effects of IL-10 on the production of heat-inactivated S. aureus-induced TNF-α, neutralizing IgG specific for IL-10 was used. Results showed that anti-IL-10 neutralizing IgG, but not isotype-matched antibody control, significantly (P < 0.05) increased the production of heat-inactivated S. aureus-induced TNF-α (Fig. 5B) as well as NO (data not shown). Early reports showed that inhibiting GSK-3β increases IL-10 in TLR signaling (31, 41). We next investigated the effects of inhibiting GSK-3β on IL-10. Treatment of either BIO or LiCl significantly (P < 0.05) increased the production of heat-inactivated S. aureus-induced IL-10 (Fig. 5C). Our previous study (31) demonstrated that IL-10 is required for inhibiting GSK-3β-regulated anti-inflammation in lipopolysaccharide (LPS)-activated microglia. To evaluate the protective role of upregulated IL-10 by inhibiting GSK-3β, neutralization experiments showed that anti-IL-10 neutralizing IgG, but not isotype-matched antibody control, significantly (P < 0.05) blocked inhibiting GSK-3β-induced downregulation of heat-inactivated S. aureus-induced TNF-α (Fig. 5D) as well as NO (data not shown). These results indicate that inhibiting GSK-3β regulates IL-10 production and IL-10 mediates TNF-α downregulation in heat-inactivated S. aureus-stimulated microglia.
FIG. 5.

Effects of inhibiting GSK-3β on IL-10 production in heat-inactivated S. aureus-stimulated microglia. (A) BV-2 mouse microglial cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 104, 1.5 × 105, and 3 × 105) for the indicated time periods. ELISA was used to determine TNF-α and IL-10 production. (B) BV-2 cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 105) for 6 h with cotreatment of isotype-matched antibody control (50 μg/ml) or anti-IL-10 IgG (50 μg/ml). ELISA was used to determine TNF-α production. (C) BV-2 cells (3 × 104) were pretreated with GSK-3β inhibitors BIO (10 μM) or LiCl (10 mM) for 0.5 h and then treated with heat-inactivated S. aureus (3 × 105) for the indicated time periods. We determined IL-10 production, using ELISA. (D) In the absence or presence of BIO (10 μM) or LiCl (10 mM), BV-2 cells (3 × 104) were treated with heat-inactivated S. aureus (3 × 105) for 6 h with or without cotreatment of isotype-matched antibody control (50 μg/ml) or anti-IL-10 IgG (50 μg/ml). ELISA was used to determine TNF-α production. The data are means ± SD of the results obtained from three individual experiments. *, P < 0.05.
DISCUSSION
Severe microglial inflammation is pathogenic for S. aureus-induced brain abscess (23, 26, 36, 37, 51). S. aureus infection causes TLR2- and NF-κB-regulated microglial inflammation, especially on TNF-α-related inflammatory responses. In search of strategies against S. aureus-induced inflammation in microglia, here we investigated the potential effects of GSK-3β inhibition according to the following rationales. First, GSK-3β has been demonstrated to be a regulator in TLR-regulated NF-κB activation and inflammation (17-21, 29, 44, 50, 52). Second, IL-10 is critical for reducing the progression of S. aureus-induced neuroinflammation (46, 48), and IL-10 can be upregulated by inhibiting GSK-3β in TLR signaling (31, 41, 55). In the present study, we provide evidence that GSK-3β regulates the production of heat-inactivated S. aureus-induced TNF-α and NO by activating NF-κB in microglia. Furthermore, GSK-3β negatively regulates IL-10 production, and IL-10 production may confer protection against heat-inactivated S. aureus-induced microglial inflammation.
Consistent with previous studies (23, 26, 36, 37, 51), we showed that TLR2 is necessary but not sufficient for the production of heat-inactivated S. aureus-induced TNF-α as well as NO in BV-2 mouse microglial cells. In addition to TLR2, other pattern recognition receptors may also be involved in recognition of live or heat-inactivated S. aureus and activation of infected cells. Furthermore, our results showed that proinflammatory TNF-α acts upstream of NO production, thereby implicating TNF-α as the cause of heat-inactivated S. aureus-induced NO. Dysregulation of TNF-α and NO production are contributing factors to S. aureus-induced circulating failure and organ injury (34, 49). Decreasing TNF-α and NO may be useful strategies against S. aureus-induced severe inflammation.
Although GSK-3β is involved in TLR-induced inflammation, the mechanisms of TLR-mediated GSK-3β activation are still unclear. We (31) and others (55) have demonstrated that TLR caused GSK-3β activation. In general, protein phosphatase and Akt are the positive and negative regulators, respectively, of GSK-3β activation by controlling phosphorylation of GSK-3β at Ser9 (11, 27, 28, 33). Furthermore, phosphorylation of GSK-3β at Tyr216 positively regulates its enzymatic activity. Our data show that GSK-3β is transiently activated after heat-inactivated S. aureus stimulation; however, the mechanisms need further investigation.
Inhibiting GSK-3β confers neuronal protection against LPS-induced microglial inflammation in neurodegenerative disorders (9, 31, 42, 54). Additionally, systemic inflammation is also regulated by GSK-3β (5, 12-14, 18, 20, 31, 41, 50, 52). Here, we show that inhibiting GSK-3β blocks the production of heat-inactivated S. aureus-induced TNF-α in BV-2 mouse microglial cells. Consistent with previous studies (5, 12, 13, 17-21, 29, 31, 41, 44, 50, 52), we found that GSK-3β can be regulated by a variety of inflammatory stimuli to cause the production of proinflammatory cytokines, including TNF-α, gamma interferon, IL-1, IL-6, IL-12p40, and COX-2, and chemokines, including RANTES and MCP-1. Furthermore, inhibiting GSK-3β blocked LPS- (31, 54) and heat-inactivated S. aureus-induced iNOS/NO biosynthesis in BV-2 mouse microglial cells. In addition to TLR4, stimulation of the natural ligands of TLR2, including peptidoglycan and lipoteichoic acid, and gram-positive Streptococcus pyogenes also caused GSK-3β-regulated NO production (data not shown). According to our results, iNOS/NO biosynthesis is downregulated due to the diminished expression of TNF-α following GSK-3β inhibition.
GSK-3β is an important regulator for a variety of transcription factors, including activated protein 1, cyclic AMP response element binding protein, heat shock factor 1, nuclear factor of activated T cells, Myc, β-catenin, CCAAT/enhancer binding protein, and NF-κB (3, 11, 27, 28, 33). A mechanistic study showed that NF-κB is inactivated in GSK-3β-deficient mice (29). However, the regulation on NF-κB signaling by GSK-3β remains unclear. Schwabe and Brenner (47) found that NF-κB p65 contains four potential GSK-3β phosphorylation sites within its COOH-terminal domain. This process causes upregulation of NF-κB transactivation in TNF-α stimulation. In the present study, we show that heat-inactivated S. aureus-induced TNF-α and NO production is NF-κB dependent. Furthermore, we demonstrate that inhibiting GSK-3β blocks heat-inactivated S. aureus-induced NF-κB p65 nuclear translocation. Consistent with previous studies, we show that GSK-3β modulates NF-κB activation in LPS-, TNF-α-, type II collagen-, zymosan-, and ovalbumin-induced inflammatory responses (5, 17-21, 44, 50, 52). Thus, we hypothesize that GSK-3β may contribute to NF-κB transactivation and NF-κB-regulated TNF-α and NO production. The molecular mechanisms need further investigation.
Exogenous treatment and transgenic expression of IL-10 have been used to reduce a variety of inflammatory diseases (4, 8, 16, 22, 38). In S. aureus infection, IL-10 plays a key role in reducing severe inflammation (46, 48). In the present study, we confirm that inhibiting IL-10 increases the production of heat-inactivated S. aureus-induced TNF-α. We further provide evidence that inhibiting GSK-3β also enhances the production of heat-inactivated S. aureus-induced IL-10. Nevertheless, the mechanisms need further investigation. Consistent with previous studies (41, 53), we show that inhibiting GSK-3β is able to increase the production of TLR-induced IL-10. Its biological effects by increased IL-10 are still unclear, however. IL-10 acts as an anti-inflammatory cytokine by triggering the expression of the suppressor of cytokine signaling 3 (4, 43). We previously showed that IL-10 was required for the downregulation of TNF-α and RANTES by GSK-3β inhibition in LPS-activated microglia (31). In this study, we also demonstrate that IL-10 is partly essential for inhibiting GSK-3β-conferred protection against S. aureus-induced microglial inflammation. In conclusion, activation of GSK-3β is essential for S. aureus-induced microglial inflammation. Inhibiting GSK-3β raises anti-inflammatory activities against heat-inactivated S. aureus-induced TNF-α and NO production in microglia mainly by inactivating NF-κB and probably by upregulating IL-10.
Acknowledgments
This work was supported by grants NSC 96-2815-C-006-073-B and NSC 96-2320-B-006-018-MY3 from the National Science Council, Taiwan, and the Landmark Project C020 of National Cheng Kung University, Taiwan.
We acknowledge Robert Anderson for critical reading of the manuscript.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 13 July 2009.
REFERENCES
- 1.Reference deleted.
- 2.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124783-801. [DOI] [PubMed] [Google Scholar]
- 3.Ali, A., K. P. Hoeflich, and J. R. Woodgett. 2001. Glycogen synthase kinase-3: properties, functions, and regulation. Chem. Rev. 1012527-2540. [DOI] [PubMed] [Google Scholar]
- 4.Asadullah, K., W. Sterry, and H. D. Volk. 2003. Interleukin-10 therapy—review of a new approach. Pharmacol. Rev. 55241-269. [DOI] [PubMed] [Google Scholar]
- 5.Bao, Z., S. Lim, W. Liao, Y. Lin, C. Thiemermann, B. P. Leung, and W. S. Wong. 2007. Glycogen synthase kinase-3beta inhibition attenuates asthma in mice. Am. J. Respir. Crit. Care Med. 176431-438. [DOI] [PubMed] [Google Scholar]
- 6.Beutler, B., Z. Jiang, P. Georgel, K. Crozat, B. Croker, S. Rutschmann, X. Du, and K. Hoebe. 2006. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 24353-389. [DOI] [PubMed] [Google Scholar]
- 7.Block, M. L., and J. S. Hong. 2007. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 351127-1132. [DOI] [PubMed] [Google Scholar]
- 8.Braat, H., P. Rottiers, D. W. Hommes, N. Huyghebaert, E. Remaut, J. P. Remon, S. J. van Deventer, S. Neirynck, M. P. Peppelenbosch, and L. Steidler. 2006. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn's disease. Clin. Gastroenterol. Hepatol. 4754-759. [DOI] [PubMed] [Google Scholar]
- 9.Chuang, D. M. 2005. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann. N. Y. Acad. Sci. 1053195-204. [DOI] [PubMed] [Google Scholar]
- 10.Cohen, J. 2002. The immunopathogenesis of sepsis. Nature 420885-891. [DOI] [PubMed] [Google Scholar]
- 11.Cohen, P., and S. Frame. 2001. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2769-776. [DOI] [PubMed] [Google Scholar]
- 12.Cuzzocrea, S., R. Di Paola, E. Mazzon, C. Crisafulli, T. Genovese, C. Muia, M. Abdelrahman, E. Esposito, and C. Thiemermann. 2007. Glycogen synthase kinase 3beta inhibition reduces the development of nonseptic shock induced by zymosan in mice. Shock 2797-107. [DOI] [PubMed] [Google Scholar]
- 13.Cuzzocrea, S., E. Mazzon, R. Di Paola, C. Muia, C. Crisafulli, L. Dugo, M. Collin, D. Britti, A. P. Caputi, and C. Thiemermann. 2006. Glycogen synthase kinase-3beta inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin. Immunol. 12057-67. [DOI] [PubMed] [Google Scholar]
- 14.De Sarno, P., R. C. Axtell, C. Raman, K. A. Roth, D. R. Alessi, and R. S. Jope. 2008. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 181338-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinges, M. M., P. M. Orwin, and P. M. Schlievert. 2000. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 1316-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drazan, K. E., L. Wu, D. Bullington, and A. Shaked. 1996. Viral IL-10 gene therapy inhibits TNF-alpha and IL-1 beta, not IL-6, in the newborn endotoxemic mouse. J. Pediatr. Surg. 31411-414. [DOI] [PubMed] [Google Scholar]
- 17.Dugo, L., M. Abdelrahman, O. Murch, E. Mazzon, S. Cuzzocrea, and C. Thiemermann. 2006. Glycogen synthase kinase-3beta inhibitors protect against the organ injury and dysfunction caused by hemorrhage and resuscitation. Shock 25485-491. [DOI] [PubMed] [Google Scholar]
- 18.Dugo, L., M. Collin, D. A. Allen, O. Murch, S. J. Foster, M. M. Yaqoob, and C. Thiemermann. 2006. Insulin reduces the multiple organ injury and dysfunction caused by coadministration of lipopolysaccharide and peptidoglycan independently of blood glucose: role of glycogen synthase kinase-3beta inhibition. Crit. Care Med. 341489-1496. [DOI] [PubMed] [Google Scholar]
- 19.Dugo, L., M. Collin, D. A. Allen, N. S. Patel, I. Bauer, E. Mervaala, M. Louhelainen, S. J. Foster, M. M. Yaqoob, and C. Thiemermann. 2007. Inhibiting glycogen synthase kinase 3beta in sepsis. Novartis Found. Symp. 280128-142. [PubMed] [Google Scholar]
- 20.Dugo, L., M. Collin, D. A. Allen, N. S. Patel, I. Bauer, E. M. Mervaala, M. Louhelainen, S. J. Foster, M. M. Yaqoob, and C. Thiemermann. 2005. GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit. Care Med. 331903-1912. [DOI] [PubMed] [Google Scholar]
- 21.Dugo, L., M. Collin, and C. Thiemermann. 2007. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock 27113-123. [DOI] [PubMed] [Google Scholar]
- 22.Eaton, S., and G. Martin. 2002. Clinical developments for treating ARDS. Expert. Opin. Investig. Drugs 1137-48. [DOI] [PubMed] [Google Scholar]
- 23.Esen, N., F. Y. Tanga, J. A. DeLeo, and T. Kielian. 2004. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the gram-positive bacterium Staphylococcus aureus. J. Neurochem. 88746-758. [DOI] [PubMed] [Google Scholar]
- 24.Faulkner, L., A. Cooper, C. Fantino, D. M. Altmann, and S. Sriskandan. 2005. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J. Immunol. 1756870-6877. [DOI] [PubMed] [Google Scholar]
- 25.Florquin, S., Z. Amraoui, D. Abramowicz, and M. Goldman. 1994. Systemic release and protective role of IL-10 in staphylococcal enterotoxin B-induced shock in mice. J. Immunol. 1532618-2623. [PubMed] [Google Scholar]
- 26.Fournier, B., and D. J. Philpott. 2005. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 18521-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frame, S., and P. Cohen. 2001. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 3591-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grimes, C. A., and R. S. Jope. 2001. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65391-426. [DOI] [PubMed] [Google Scholar]
- 29.Hoeflich, K. P., J. Luo, E. A. Rubie, M. S. Tsao, O. Jin, and J. R. Woodgett. 2000. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 40686-90. [DOI] [PubMed] [Google Scholar]
- 30.Hu, S., C. C. Chao, L. C. Ehrlich, W. S. Sheng, R. L. Sutton, G. L. Rockswold, and P. K. Peterson. 1999. Inhibition of microglial cell RANTES production by IL-10 and TGF-beta. J. Leukoc. Biol. 65815-821. [DOI] [PubMed] [Google Scholar]
- 31.Huang, W. C., Y. S. Lin, C. Y. Wang, C. C. Tsai, H. C. Tseng, C. L. Chen, P. J. Lu, P. S. Chen, L. Qian, J. S. Hong, and C. F. Lin. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology, in press. [DOI] [PMC free article] [PubMed]
- 32.Jedrzkiewicz, S., G. Kataeva, C. M. Hogaboam, S. L. Kunkel, R. M. Strieter, and D. M. McKay. 1999. Superantigen immune stimulation evokes epithelial monocyte chemoattractant protein 1 and RANTES production. Infect. Immun. 676198-6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jope, R. S., and G. V. Johnson. 2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2995-102. [DOI] [PubMed] [Google Scholar]
- 34.Kengatharan, K. M., S. J. De Kimpe, and C. Thiemermann. 1996. Role of nitric oxide in the circulatory failure and organ injury in a rodent model of gram-positive shock. Br. J. Pharmacol. 1191411-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kielian, T., N. Esen, and E. D. Bearden. 2005. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia 49567-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kielian, T., A. Haney, P. M. Mayes, S. Garg, and N. Esen. 2005. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect. Immun. 737428-7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kielian, T., P. Mayes, and M. Kielian. 2002. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J. Neuroimmunol. 13086-99. [DOI] [PubMed] [Google Scholar]
- 38.Leon, L. R., W. Kozak, and M. J. Kluger. 1998. Role of IL-10 in inflammation. Studies using cytokine knockout mice. Ann. N. Y. Acad. Sci. 85669-75. [DOI] [PubMed] [Google Scholar]
- 39.Reference deleted.
- 40.Marshall, J. C. 2001. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit. Care Med. 29S99-S106. [DOI] [PubMed] [Google Scholar]
- 41.Martin, M., K. Rehani, R. S. Jope, and S. M. Michalek. 2005. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6777-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez, A., A. Castro, I. Dorronsoro, and M. Alonso. 2002. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med. Res. Rev. 22373-384. [DOI] [PubMed] [Google Scholar]
- 43.Moore, K. W., R. de Waal Malefyt, R. L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19683-765. [DOI] [PubMed] [Google Scholar]
- 44.Mühl, H., and J. Pfeilschifter. 2006. Controlling the cytokine storm by insulin: glycogen synthase kinase-3 as a target in systemic inflammation. Crit. Care Med. 341567-1569. [DOI] [PubMed] [Google Scholar]
- 45.Nagaki, M., Y. Muto, H. Ohnishi, S. Yasuda, K. Sano, T. Naito, T. Maeda, T. Yamada, and H. Moriwaki. 1994. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 106450-458. [DOI] [PubMed] [Google Scholar]
- 46.Nagaki, M., M. Tanaka, A. Sugiyama, H. Ohnishi, and H. Moriwaki. 1999. Interleukin-10 inhibits hepatic injury and tumor necrosis factor-alpha and interferon-gamma mRNA expression induced by staphylococcal enterotoxin B or lipopolysaccharide in galactosamine-sensitized mice. J. Hepatol. 31815-824. [DOI] [PubMed] [Google Scholar]
- 47.Schwabe, R. F., and D. A. Brenner. 2002. Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 283G204-G211. [DOI] [PubMed] [Google Scholar]
- 48.Stenzel, W., J. Dahm, M. Sanchez-Ruiz, H. Miletic, M. Hermann, C. Courts, H. Schwindt, O. Utermohlen, D. Schluter, and M. Deckert. 2005. Regulation of the inflammatory response to Staphylococcus aureus-induced brain abscess by interleukin-10. J. Neuropathol. Exp. Neurol. 641046-1057. [DOI] [PubMed] [Google Scholar]
- 49.Stenzel, W., S. Soltek, H. Miletic, M. M. Hermann, H. Korner, J. D. Sedgwick, D. Schluter, and M. Deckert. 2005. An essential role for tumor necrosis factor in the formation of experimental murine Staphylococcus aureus-induced brain abscess and clearance. J. Neuropathol. Exp. Neurol. 6427-36. [DOI] [PubMed] [Google Scholar]
- 50.Takada, Y., X. Fang, M. S. Jamaluddin, D. D. Boyd, and B. B. Aggarwal. 2004. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J. Biol. Chem. 27939541-39554. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 1655392-5396. [DOI] [PubMed] [Google Scholar]
- 52.Whittle, B. J., C. Varga, A. Posa, A. Molnar, M. Collin, and C. Thiemermann. 2006. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br. J. Pharmacol. 147575-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodgett, J. R., and P. S. Ohashi. 2005. GSK3: an in-Toll-erant protein kinase? Nat. Immunol. 6751-752. [DOI] [PubMed] [Google Scholar]
- 54.Yuskaitis, C. J., and R. S. Jope. 2009. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 21264-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, P., J. Katz, and S. M. Michalek. 2009. Glycogen synthase kinase-3beta (GSK3beta) inhibition suppresses the inflammatory response to Francisella infection and protects against tularemia in mice. Mol. Immunol. 46677-687. [DOI] [PMC free article] [PubMed] [Google Scholar]