

Abstract
Vibrio vulnificus is an estuarine bacterium capable of causing serious and often fatal wound infections and primary septicemia. We used alkaline phosphatase insertion mutagenesis to identify genes necessary for the virulence of this pathogen. One mutant had an in-frame fusion of ′phoA to the gene encoding RseB, a periplasmic negative regulator of the alternative sigma factor σE. σE controls an extensive regulon involved in responding to cell envelope stresses. Colonies of the rseB mutant were less opaque than wild-type colonies and underwent phase variation between translucent and opaque morphologies. rseB mutants were attenuated for virulence in subcutaneously inoculated iron-dextran-treated mice. To obtain insight into the role of rseB and the extracytoplasmic stress response in V. vulnificus, mutants with defined mutations in rseB and two important members of the extracytoplasmic stress regulon, rpoE and degP, were constructed for analysis of virulence, colony morphology, and stress-associated phenotypes. Deletion of rseB caused reversible phase variation in the colony morphotype that was associated with extracellular polysaccharides. Translucent and transparent morphotype strains were attenuated for virulence. rpoE and degP deletion mutants were sensitive to membrane-perturbing agents and heat but were not significantly attenuated for V. vulnificus virulence in mice. These results reveal complex relationships between regulation of the extracytoplasmic stress response, exopolysaccharides, and the virulence of V. vulnificus.
Vibrio vulnificus is a gram-negative estuarine bacterium responsible for severe opportunistic infections (for a review, see reference 17). Ingestion of raw contaminated seafood can lead to septicemia in susceptible patients, while contact with contaminated seawater or seafood can cause wound infection, which may progress to necrotizing fasciitis and sepsis. Mortality rates for sepsis and wound infection can be as high as 75% and 50%, respectively. Predisposing conditions for septicemia include liver disease, cirrhosis, hemochromatosis, diabetes, and immune compromise, while wound infection can occur in otherwise healthy persons.
Several virulence factors have been identified for V. vulnificus, most notably the antiphagocytic capsule (55, 65), RtxA toxin (26, 28, 32), and iron acquisition systems (31, 64). For a review, see reference 17. However, much is yet to be discovered, particularly the mechanisms of extreme tissue damage and rapid growth in host tissues (17). To examine these traits, we focused on the factors that are localized to the bacterial cell surface or are secreted into the extracellular space, considering that most virulence factors are exported to interact with the host. Alkaline phosphatase (phoA) mutagenesis is a useful tool for identifying genes encoding exported products (33), based on the principle that alkaline phosphatase is active only when it is exported beyond the bacterial cytoplasm. Randomly generated phoA gene fusions, most often generated via TnphoA (33), must be in genes encoding exported proteins to have enzyme activity detected by plating on the chromogenic substrate 5-bromo-4-chloro-3-indolylphosphate (BCIP). In our studies, TnphoA did not work effectively in V. vulnificus for unknown reasons. We therefore created a mini-Tn5 transposon-based ′phoA delivery system, miniTn5phoA (8), that works well in V. vulnificus.
Using this method, we identified a phoA mutant that had an in-frame fusion of ′phoA to the gene encoding RseB, a periplasmic negative regulator of sigma E (σE) activity. The rseB mutant exhibited several interesting phenotypes, including phase variation between translucent and opaque colony morphologies and attenuated virulence. σE is an alternative RNA polymerase sigma factor that controls an extensive regulon involved in responding to cell envelope stresses (48). This response, termed the extracytoplasmic stress response (ESR), is essential for maintaining the envelope integrity of gram-negative bacteria under certain stress conditions (for a review, see reference 48). Because rseB is involved in the ESR, we determined the role of the σE-mediated ESR in V. vulnificus. We also investigated the possible reasons for the translucent morphology of RseB variants by comparing these variants with an acapsular translucent mutant of V. vulnificus. This study uncovered a possible role for RseB in phase variation of extracellular polysaccharide (EPS) expression and is the first study to investigate the role of the ESR in the virulence of V. vulnificus.
MATERIALS AND METHODS
Bacterial strains, media, and chemicals.
Bacterial strains are listed in Table 1. All strains were grown in Luria-Bertani broth containing 0.85% (wt/vol) NaCl (LB-N) or on LB-N plates containing 1.5% (wt/vol) agar. When required, antibiotics were used as follows: rifampin (rifampicin), kanamycin, chloramphenicol, and tetracycline at concentrations of 50 μg/ml, 40 μg/ml, 30 μg/ml, and 12.5 μg/ml, respectively, for Escherichia coli and at concentrations of 50 μg/ml, 300 μg/ml, 3 μg/ml, and 6.25 μg/ml, respectively, for V. vulnificus. VVM (4) and LB-N containing 105 U/liter colistin were used to select against E. coli during conjugations.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| EC100D pir+ | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara-leu)7697 galU galK λ−rpsL nupG pir+(DHFR) | Epicentre |
| MG1655 | F− λ−ilvG rfb-50 rph-1 | 19 |
| S17-1λpir | λpir lysogen; thi pro hsdR hsdM+recA RP4-2 Tc::Mu-Km::Tn7(Tpr Smr) | 54 |
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| V. vulnificus strains | ||
| CMCP6 | Clinical isolate | 25 |
| CMIT232 | MO6/24-O wza::TnphoA | 62 |
| FLA399 | Spontaneous rifampin-resistant derivative of CMCP6 | 3 |
| FLA609 | FLA399 rseB::mini-Tn5Km2phoA | This study |
| FLA609-O | Opaque colony isolate of FLA609 | This study |
| FLA609-Tl | Translucent colony isolate of FLA609 | This study |
| FLA610 | CMCP6 ΔrseB | This study |
| FLA610-O | Opaque colony isolate of FLA610 | This study |
| FLA610-Tl | Translucent colony isolate of FLA610 | This study |
| FLA610-Tp | Transparent colony isolate of FLA610 | This study |
| FLA611-R | Spontaneous rugose isolate of FLA399 | This study |
| FLA1001 | CMCP6 ΔrpoE::aph | This study |
| FLA1002 | CMCP6 ΔdegP::aph | This study |
| FLA1009 | CMCP6 wza::TnphoA via chitin transformation with genomic DNA from CMIT232 | 18 |
| Plasmids | ||
| pCR2.1 | T/A cloning vector, lacZα multiple cloning site, Apr Kmr | Invitrogen |
| pCVD442 | R6K ori-based suicide plasmid, mob RP4, sacB, Apr | 9 |
| pGTR201 | pUTmini-Tn5Tag3phoA (′phoA delivery vector) | This study |
| pGTR1129 | pCVD442 with lacZα from pUC19 with USER Friendly cloning oligonucleotide linker incorporated, cat | 18 |
| pGTR1160 | pRK437 with USER Friendly cloning oligonucleotide linker incorporated | 18 |
| pGTR2005 | rseB cloned into pGTR1160 for complementation of FLA609 and FLA610 | This study |
| pGTR2006 | 500 bp upstream and downstream of rseB USER cloned into pGTR1129 for deletion of rseB | This study |
| pGTR2008 | rpoE cloned into pGTR1160 for expression from the lac promoter | This study |
| pGTR2011 | 500 bp upstream and downstream of rpoE USER cloned into pGTR1129 for deletion of rpoE | This study |
| pGTR2013 | pGTR2011 with aph from pUC4K cloned into SmaI, ΔrpoE::aph | This study |
| pGTR2014 | 500 bp upstream and downstream of degP USER cloned into pGTR1129 for deletion of degP | This study |
| pGTR2015 | pGTR2014 with aph from pUC4K cloned into SmaI, ΔdegP::aph | This study |
| pRK437 | Expression vector, mob RK2, lacZα multiple cloning site, Tcr | 51 |
| pRT291 | IncP1, TnphoA, Kmr Tcr, source of ′phoA | 60 |
| pUC4K | Kmr derivative of pUC4 | 59 |
| pUTmini-Tn5Tag3 | mini-Tn5Tag3 delivery vector; R6K ori, mob RP4, Apr Kmr | 29 |
Strains were stored at −80°C in LB-N with 35% (vol/vol) glycerol. For mouse infection experiments, static overnight starter cultures of bacteria were grown in culture tubes at room temperature. Before infection, starter cultures were diluted 1:20 in prewarmed LB-N and shaken at 37°C until the optical density at 600 nm (OD600) was 0.4 to 0.6 (exponential-phase growth). The bacteria were diluted in phosphate-buffered saline (PBS) containing 0.01% (wt/vol) gelatin (43) to obtain the appropriate inoculum for infection. Further dilution in PBS containing 0.01% (wt/vol) gelatin and plating were used to confirm the number of CFU/ml inoculated.
Unless noted otherwise, components of growth media were obtained from Difco (Franklin Lakes, NJ), chemicals were obtained from Sigma (St. Louis, MO), DNA extraction and purification kits were obtained from Qiagen (Valencia, CA), molecular genetics enzymes were obtained from New England Biolabs (Ipswich, MA), and oligonucleotides were obtained from IDT (Coralville, IA).
Construction of mini-Tn5Km2phoA mutagenesis vector pGTR201.
′phoA was PCR amplified from pRT291 (60) using Taq polymerase and primers engineered to insert NotI sites flanking the product, PhoA3′-NotI-2 and TnPhoA5′-NotI-3 (see Table S1 in the supplemental material). The PCR product was cloned into pCR2.1-TOPO according to the instructions of a TOPO TA cloning kit (Invitrogen) and electroporated into E. coli TOP10 for blue-white screening of inactivation of lacZα using 40 μg/ml of 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal). Plasmid mini-extracts (QIAprep spin miniprep kit; Qiagen) of the resulting clones were digested with NotI to excise the1.3-kb ′phoA fragment, which was purified by gel extraction (QIAquick gel extraction kit; Qiagen) and ligated to NotI-digested pUTmini-Tn5Tag3 (29), a derivative of pUTmini-Tn5Km2 (8). The ligated product was electroporated into E. coli EC100D pir+ for selection on LB-N agar plates containing 40 μg/ml kanamycin. Plasmid mini-extracts of the resulting clones were resolved on agarose gels to confirm the size, and restriction digestion of the plasmids further confirmed the correct orientation of the ′phoA insert. The plasmid was designated pGTR201.
Use of pGTR201 for PhoA mutagenesis.
PhoA mutagenesis plasmid pGTR201 was electroporated into E. coli S17-1λpir for conjugation into V. vulnificus FLA399 by filter mating (56, 57). Transconjugants were selected on LB-N agar plates containing rifampin, kanamycin, 40 μg/ml of BCIP, and 0.2% (wt/vol) glucose. VVM agar (4) was used to confirm that colonies were V. vulnificus. Colonies that appeared to be bluer than wild-type strain FLA399 colonies on BCIP-containing plates were considered PhoA+. Genomic DNA was extracted (DNeasy blood and tissue kit; Qiagen) from PhoA+ strains for sequencing using primer phoA5′rev (see Table S1 in the supplemental material) at the University of Florida Interdisciplinary Center for Biotechnology Research DNA Sequencing Core Laboratory.
Deletion of rseB, rpoE, and degP.
To delete specific genes, approximately 500-bp portions of DNA sequences upstream and downstream of the open reading frames were amplified using the oligonucleotides described in Table S1 in the supplemental material. For example, to delete rseB, the upstream sequences were PCR amplified using primers delrseBup5′USER and delrseBup3′USER, and the downstream sequences were PCR amplified using primers delrseBdn5′USER and delrseBdn3′USER. Similarly, the upstream and downstream sequences of rpoE and degP were amplified. The upstream and downstream sequences were cloned in a single step into the allelic exchange suicide vector pGTR1129 using USER Friendly cloning methods described elsewhere (3, 18). Briefly, the outside USER ends of the amplicons were compatible for ligation into the USER sequences of pGTR1129; i.e., after they were treated with USER enzyme (New England Biolabs) (1), complementary 8-bp overhangs that were unique for the upstream and downstream fragments were created. The inside ends of the upstream and downstream fragments that were to be joined had unique compatible USER ends with a SmaI restriction enzyme site (see Table S1 in the supplemental material). The upstream and downstream PCR products were treated with the USER enzyme. pGTR1129 was treated with XbaI and Nt.BbvCI to create 8-bp 3′ overhangs. The vector and PCR fragments were combined and ligated for 15 min at room temperature using T4 DNA ligase. Every junction to be ligated had a unique USER enzyme-generated 8-bp overhang, facilitating the ligation. The ligation products were electroporated into E. coli EC100D pir+ with selection for chloramphenicol resistance. The resulting plasmids used for deletion of rseB, rpoE, and degP were designated pGTR2006, pGTR2011, and pGTR2014, respectively. To allow selection of the deletion mutation upon recombination into the V. vulnificus genome, an aph kanamycin resistance cassette was inserted into the SmaI site between the upstream and downstream sequences of pGTR2011 and pGTR2014, forming plasmids pGTR2013 and pGTR2015, respectively.
To delete rseB, pGTR2006 was electroporated into E. coli S17-1λpir and conjugated into V. vulnificus wild-type strain CMCP6 with plating on LB-N agar containing colistin and chloramphenicol to select for integration of pGTR2006 into the V. vulnificus genome. A standard two-step, sucrose-mediated allelic exchange method was used to recombine the ΔrseB mutation into the V. vulnificus genome from pGTR2006. After verification of appropriate integration by PCR analysis, the single-crossover strain was grown in the absence of chloramphenicol and plated on LB-N agar containing 10% (wt/vol) sucrose to select for loss of pGTR2006. The sucrose resistance of the resulting excision isolate was confirmed by plating, the loss of pGTR1129 vector sequences was confirmed using chloramphenicol sensitivity and PCR, and appropriate deletion of rseB was confirmed by PCR. The resulting strain was designated FLA610.
The rpoE::aph and degP::aph mutations were recombined in the V. vulnificus genome by using chitin-induced transformation of linear DNA and the method of Meibom et al. for Vibrio cholerae (37), as described by Gulig et al. (18). Briefly, V. vulnificus cultures were grown to exponential phase in LB-N at 37°C and then washed and suspended in seawater diluted to 25 ppt NaCl. Bacteria were incubated with sterile pieces of crab shell without agitation at 30°C for 18 to 24 h in a 12-well tissue culture plate. The growth medium was removed from the plate and replaced with 2 ml of fresh seawater at 25 ppt NaCl, which was followed by addition of 2 μg of linearized DNA from plasmid pGTR2013 or pGTR2015. After 24 h of growth at 30°C, the crab shell pieces were moved to 50-ml conical tubes containing 2 ml of PBS and vortexed vigorously for 30 s. Each crab shell culture was diluted and plated on LB-N agar containing 100 μg/ml kanamycin. Appropriate incorporation of the ΔrpoE::aph and ΔdegP::aph mutations was confirmed by PCR analysis. The resulting strains were designated FLA1001 (ΔrpoE::aph strain) and FLA1002 (ΔdegP::aph strain).
Cloning of rseB and rpoE for expression and/or complementation.
For cloning of genes that were to be expressed, pGTR1160, a USER vector derived from pRK437 (18, 51), was used. The rseB gene was PCR amplified with oligonucleotide primers rseB5′USER and rseB3′USER, and rpoE was PCR amplified with primers SigmaE5′rbs-USER and SigmaE3′rbs-USER (see Table S1 in the supplemental material). The amplicons were treated with the USER enzyme mixture, pGTR1160 was treated with XbaI and Nt.BbvCI, and the fragments were ligated and electroporated into EC100D pir+, yielding plasmid pGTR2005 for rseB and plasmid pGTR2008 for rpoE. In these constructs the cloned genes were expressed from the lac promoter of pGTR1160. The plasmids were electroporated into E. coli S17-1λpir for conjugation into V. vulnificus by filter mating. Transconjugants were selected on LB-N agar plates containing 6.25 μg/ml tetracycline and 105 U/liter colistin.
Rates of phase variation.
To assess the rates of phase variation in V. vulnificus ΔrseB mutants compared to the wild type, wild-type strain CMCP6 and ΔrseB FLA610 variants were grown to exponential phase from static overnight cultures inoculated with a single colony. Aliquots were diluted and plated on LB-N agar (≥6 plates per strain) for observation. Phase variation was expressed as the fraction of colonies that switched to a new phenotype divided by the total number of colonies on the plate.
Infection of mice.
The subcutaneous (s.c.) inoculation, iron-dextran-treated mouse model of Starks et al. (56, 57) was used to assess virulence with 7- to 10-week-old female ICR mice (Harlan Sprague-Dawley, Indianapolis, IN) housed under specific-pathogen-free conditions. Mice were pretreated with iron-dextran (Sigma) by intraperitoneal injection, which was followed by s.c. inoculation of bacteria in the lower back. When the rectal temperature dropped below 33°C (a surrogate for death) or up to 22 h postinoculation, mice were euthanized, and the numbers of CFU in skin lesions and the liver were determined by homogenization and plating. Strains with expression plasmids were plated both nonselectively (LB-N agar) and selectively (LB-N agar with 6.25 μg/ml tetracycline.) Samples were not taken from mice with no visible skin lesions, and the minimum number of detectable CFU/g was used for these mice for statistical analysis. The minimum numbers of detectable CFU/g were 104 CFU/g and 102.5 CFU/g for the skin and liver, respectively.
Sensitivity to membrane stress.
Disk diffusion tests were used to observe inhibition of growth by membrane-perturbing agents. Eight-millimeter disks of Millipore absorbent pads (Millipore, Billerica, MA) were aseptically placed on lawns of 107 CFU plated on LB-N agar. Aliquots of inhibitory agents were added to the disks as follows: 40 μl of 1% (wt/vol) sodium dodecyl sulfate (SDS), 40 μl of 100% (vol/vol) ethanol, and 40 μl of 3% (vol/vol) hydrogen peroxide. The plates were incubated overnight at 37°C, and the diameter of the zone of inhibition surrounding each filter was measured. At least three filters were used for each strain, and at least two separate assays were performed.
Serum sensitivity.
Overnight starter cultures grown without agitation at room temperature were diluted 1:20 in LB-N and grown to an OD600 of 0.4 to 0.6. Culture volumes were adjusted so the concentration was 108 CFU/ml, and cultures were serially diluted to obtain 103 CFU/ml for plating to quantify the input CFU. An aliquot (20 μl) of the 10−1 dilution (107 CFU/ml) was incubated with 180 μl of untreated or heat-inactivated (30 min at 56°C) rat serum for 2 h and then plated to quantify survival. The difference in the log-transformed numbers of CFU in untreated and heat-inactivated serum is reported below. At least two separate assays were performed.
Analysis of EPS.
A modification of the method of Enos-Berlage and McCarter (10) was used for analysis of EPS. An entire petri plate containing LB-N agar was inoculated with cells from a single colony and incubated overnight at 37°C. The lawn of bacteria was scraped from the plate and suspended in 5 ml of PBS with vigorous vortexing for 1 min. A 20-μl aliquot of the suspension was diluted and plated to quantify CFU. The remaining suspension was shaken at 200 rpm on a rotary shaker for 1.5 h at 37°C. The vortexing and shaking process was repeated, and cells and debris were removed by centrifugation at 10,000 × g for 15 min at room temperature. The supernatant containing EPS was moved to a 50-ml conical tube. RNase A (50 μg/ml), DNase I (50 μg/ml), and MgCl2 (10 mM) (final concentrations) were added, and the tube was incubated for 8 h at 37°C. Proteinase K (200 μg/ml) was added, the tube was incubated for 17 h at 37°C, and 750 μl was removed and extracted twice with phenol-chloroform using a phase-lock heavy gel tube (Eppendorf, Westbury, NY). Polysaccharides in the sample were precipitated with 2.5 volumes of 95% ethanol, the sample was centrifuged at 8,000 × g for 30 min at 4°C, and the supernatant was decanted. The pellet was washed with 3 ml of 95% ethanol, air dried, and suspended in 200 μl of water. Samples were stored at −20°C.
Twenty-five microliters of EPS extract was diluted 1:1 with Laemmli sample buffer and was resolved on a Criterion 4 to 20% Tris-HCl gel (Bio-Rad) for 1.5 h at 100 V. Alcian blue staining was performed by using a modification of the method of Møller et al. (39). After SDS-polyacrylamide gel electrophoresis (PAGE), the gels were fixed overnight at room temperature in fresh Alcian blue fixing solution (30% [vol/vol] ethanol and 10% [vol/vol] acetic acid). The gels were stained with 0.2% (wt/vol) Alcian blue dissolved in 40% (vol/vol) ethanol-10% (vol/vol) acetic acid for 1 to 3 h with gentle rocking and were destained slowly with 40% (vol/vol) ethanol-10% (vol/vol) acetic acid. Staining with Stains-all was done using the method of Kelley and Parker (23). Gels were scanned using an Epson 1670 scanner.
qRT-PCR.
Quantitative reverse transcription-PCR (qRT-PCR) was performed exactly as described by Brown and Gulig (3). Briefly, RNA from mid-log-phase cultures (OD600, 0.4 to 0.6) grown in LB-N was treated with DNase I and reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc.). iQ SYBR green Supermix (Bio-Rad) was used for detection with the iQ real-time PCR detection system (Bio-Rad) in triplicate. Relative expression was determined by calculating 2−ΔΔCt using the 16S rRNA gene as an internal control. Primers used for amplification of 16S rRNA, degP, and rpoE are described in Table S1 in the supplemental material.
Statistical analysis.
The Student t test was used to examine the significance of differences between pairs of means. Groups of more than two means were analyzed by using analysis of variance, followed by the Bonferroni posttest to identify significant differences between the groups. χ2 tests were used in mouse experiments to determine if the numbers of mice with detectable CFU were significantly different for mutant and wild-type infections. Statistical analyses were done using Microsoft Excel or GraphPad Prism5 software. Values were considered significantly different if the P value was <0.05. Quantitative experiments were repeated at least once.
RESULTS
Identification of an rseB::mini-Tn5Km2phoA mutation that attenuates the virulence of V. vulnificus.
We used our mini-Tn5Km2phoA suicide donor plasmid, pGTR201, to mutagenize a spontaneous rifampin-resistant derivative of V. vulnificus CMCP6, FLA399, as described in Materials and Methods. Colonies that were blue on LB-N agar plates containing 0.2% (wt/vol) glucose and 40 μg/ml BCIP were assumed to have fusions of the PhoA sequence of the mini-transposon with exported proteins and were examined further. One mini-Tn5Km2phoA mutant, FLA609, appeared to be less opaque than the wild type on LB-N agar plates. This result suggested that it could have been a capsular polysaccharide (CPS) mutant, as acapsular mutants of V. vulnificus appear to be translucent (66). Single-colony passaging of FLA609 yielded colonies with different translucent and opaque morphologies (Fig. 1). Passaging of translucent colonies always yielded a mixture of translucent and opaque colonies. The percentage of opaque variants obtained from a translucent colony ranged from 20% to over 90%. It appeared that FLA609 underwent high-frequency phase variation from translucent to opaque forms. The reverse switch, from opaque to translucent forms, was less frequent, in the range of 0.01%.
FIG. 1.

Colony morphologies of the rseB::mini-Tn5Km2phoA FLA609 strain. (A) A single translucent colony of FLA609 was grown in LB-N without agitation overnight, diluted, and grown to log phase with shaking at 37°C, and an aliquot was diluted and plated on LB-N agar. More than 30% of the colonies switched to a more opaque morphology during the growth period. Black arrow, translucent colony; white arrow, opaque colony. (B) Subculturing of a single opaque (O) or translucent (T) colony of FLA609 compared to the wild type (W). (C) Close-up of the translucent sector in panel B, showing opaque variants (white specks) scattered throughout the mainly translucent culture.
The initial test for virulence of FLA609 involved s.c. infection of three iron-dextran-treated mice with approximately 1,000 CFU of the translucent variant (FLA609-Tl), which is three times the minimum lethal dose of the wild-type parent. Considerable switching in colony morphology occurred while the culture was grown for inoculation, as shown by the 10% opaque colonies on plates of the inoculum used for titration. One of the three inoculated mice failed to develop a skin lesion. Bacteria were recovered from only one skin sample from the two mice that developed skin lesions. None of the liver samples yielded detectable CFU. For the mouse whose skin lesion yielded bacteria, a mixture of translucent and opaque bacteria was observed on the plates, and the concentration was 107.8 CFU/g skin. Typical values for wild-type V. vulnificus FLA399 in this mouse model are 108 CFU/g skin and 104.7 CFU/g liver. Therefore, FLA609-Tl was attenuated for virulence. Considerable switching to the opaque morphology occurred during infection of mice, suggesting that there was selective pressure against the translucent form in mice.
Because of the attenuated virulence of FLA609-Tl and the variant colony morphologies, we determined the DNA sequence of the genomic locus into which the mini-Tn5Km2phoA had inserted. The nucleotide sequence of FLA609 showed an in-frame phoA fusion to VV1_1561 encoding a protein designated a “negative regulator of σE activity” (Fig. 2). Analysis of the amino acid sequence using PSORT (12) predicted with 94% probability that the protein was localized to the periplasm. BLAST searches showed that the VV1_1561 protein sequence had high levels of similarity to the sequences of the “negative regulator of σE” from other Vibrio species (72% identical to the Vibrio splendidus sequence and 68% identical to the V. cholerae sequence). Analysis of the genomic region surrounding VV1_1561 in V. vulnificus revealed a possible operon that included VV1_1559, designated “similar to σ24”; VV1_1560, another “negative regulator of σE activity”; and VV1_1562, a “positive regulator of σE activity” (Fig. 2). The organization of this genomic region was identical to that of the well-studied E. coli rpoE operon, and VV1_1561 was 45% identical to the E. coli protein RseB, a periplasmic negative regulator of σE activity. Thus, we designated VV1_1561 rseB and assumed that the other genes were rpoE, rseA, and rseC, as annotated for E. coli.
FIG. 2.

V. vulnificus rpoE rseABC locus. Similar to the well-studied E. coli rpoE rseABC locus, the V. vulnificus genome contains homologues of rpoE (VV1_1559), rseA (VV1_1560), rseB (VV1_1561), and rseC (VV1_1562) that appear to form an operon. The location of the ′phoA insertion in VV1_1561 is indicated by the arrowhead. The putative signal sequence of RseB is indicated by a black box. The V. vulnificus CMCP6 genome can be accessed at www.ncbi.nlm.nih.gov/nuccore/NC_004459 and www.ncbi.nlm.nih.gov/nuccore/NC_004460.
In subsequent infections with larger numbers of mice, the translucent rseB::mini-Tn5Km2phoA variant (FLA609-Tl) was not significantly attenuated for local skin infection but was attenuated for systemic liver infection compared to the wild type, and there was a nearly 100-fold decrease in the number of CFU/g liver compared to the results for wild-type strain FLA399 (P = 0.01) (Fig. 3A). Bacteria harvested from mice infected with FLA609-Tl were always enriched for opaque colonies. In fact, translucent bacteria were almost never recovered from liver samples; apparently, the bacteria that switched to the opaque phenotype were the bacteria most capable of causing systemic infection. The opaque variant of FLA609 (FLA609-O) showed wild-type levels of skin infection but variable levels of systemic infection. When the data for three infections with FLA610-O were combined for analysis (14 mice), the mean value obtained was 8.1 ± 0.25 log CFU/g skin, while the mean value for the liver was 3.8 ± 1.8 log CFU/g, which was greater variation than normally seen with wild-type infections (the standard deviations were approximately 50% and 20%, respectively, of the means) (Fig. 3B). Moreover, skin CFU were recovered from 100% of mice inoculated with FLA609-O, while liver CFU were recovered from only 50% of these mice (P = 0.001 for a comparison with the wild type, as determined by a χ2 test). For the 50% of the mice that yielded liver CFU the mean level of recovery was nearly typical for the wild type (Fig. 3B). Therefore, with an inoculum containing 1,000 CFU, FLA609-O exhibited a stochastic infection profile, either full virulence in about one half of the mice or severe attenuation in the other half.
FIG. 3.

Virulence of translucent and opaque variants of the rseB::mini-Tn5Km2phoA FLA609 strain in s.c. inoculated iron-dextran-treated mice. (A) Iron-dextran-treated mice were inoculated with 1,000 CFU of translucent variant FLA609-Tl, opaque variant FLA609-O, or wild-type (WT) strain FLA399. The fractions below the bars indicate the frequencies of detectable skin or liver infection. *, P = 0.04 for a comparison with the wild type for CFU/g liver (Student t test); †, P = 0.038 for a comparison with the wild type for the frequency of detectable liver infection (χ2 test). (B) Scatter plot showing the results for three separate mouse infections with FLA609-O combined to illustrate the trends for skin and liver infection. Means and standard deviations are indicated.
Deletion of rseB from wild-type strain CMCP6.
Because an RseB-PhoA fusion protein could have resulted in unintended phenotypes, we deleted the rseB open reading frame from V. vulnificus wild-type strain CMCP6 as described in Materials and Methods, creating FLA610. Deletion of rseB from CMCP6 reproduced the phase-variable phenotype observed for the rseB::mini-Tn5Km2phoA mutant with opaque (FLA610-O) and translucent (FLA610-Tl) variants, in addition to a form that was more translucent than FLA610-Tl, which we designated transparent strain FLA610-Tp (data not shown).
The virulence of ΔrseB FLA610 variants in mice was examined (Fig. 4). When an inoculum containing 1,000 CFU was used, FLA610-Tp was avirulent in mice; none of five mice developed skin lesions, and the mice remained healthy throughout the 20-h course of infection. FLA610-Tl was recovered from only two of five mice, and the mean levels were 105.3 CFU/g skin and 103.7 CFU/g liver. As observed for the translucent rseB::mini-Tn5Km2phoA mutant, a mixture of translucent and opaque bacteria was recovered from tissue samples of mice infected with FLA610-Tl (20 to 50% opaque bacteria). FLA610-O was recovered from only two of five mice, and the mean levels were 105 CFU/g skin and 103.2 CFU/g liver. As observed for the opaque rseB::mini-Tn5Km2phoA variant, subsequent infections showed that FLA610-O had variable levels of virulence; it was fully virulent in 100% of mice in some infections, but its virulence was significantly attenuated in other infections with inocula containing approximately 1,000 CFU (Fig. 4B). The mean level for a total of 20 mice inoculated with FLA610-O (four separate infections) was 106.4 CFU/g skin, which was nearly 100-fold lower than the level in a typical wild-type infection. However, for 12 of these mice the levels were greater than 107 CFU/g skin, while for 7 of them the levels were approximately 104 CFU/g or less (minimum detectable level). For 7 of 20 mice inoculated with ΔrseB mutant FLA610-O there was no detectable level in the liver (P = 0.0014 for a comparison with the wild type, as determined by a χ2 test). The mean level in the liver for 20 infected mice was 104.5 CFU/g, a value which is somewhat inflated because the minimum detectable level, 102.5 CFU/g, was used for mice in which no detectable CFU were found. Furthermore, for liver samples with detectable CFU, the mean was 105.6 CFU/g, which is in the range of the wild-type values. Therefore, as was the case for the opaque rseB::mini-Tn5Km2phoA mutant, the virulence of FLA610-O was enigmatic; in some mice this strain was fully virulent, while in others it was severely attenuated.
FIG. 4.

Virulence of ΔrseB variants in s.c. inoculated iron-dextran-treated mice. (A) Mice were inoculated with 1,000 CFU of transparent variant FLA610-Tp (ΔrseB-Tp), translucent variant FLA610-Tl (ΔrseB-Tl), and opaque variant FLA610-O (ΔrseB-O) or these strains containing plasmid pGTR2005 encoding rseB (Tp-compl, Tl-comp, and O-comp). Five mice were used for each experiment. For complementation, the results of plating on nonselective media are shown (there was no statistical difference between the plate counts on selective and nonselective media). The numbers below the bars indicate the numbers of mice with detectable bacteria in skin or liver samples from five inoculated mice. The Bonferroni t test was used to compare each mutant to its corresponding complemented strain (*, P = 0.01; **, P = 0.008). (B) Scatter plot showing the results for four mouse infections (five mice each) with FLA610-O.
To further examine the extent of attenuation of the ΔrseB transparent variant (FLA610-Tp), mice were inoculated with 105 CFU. We considered the possibility that the transparent form was acapsular, so we also infected mice with 105 CFU of an acapsular strain, the CMCP6 wza::TnphoA mutant FLA1009 (18). Wild-type strain CMCP6 was inoculated using a dose of 103 CFU as a control. Inoculation of all three strains resulted in >107.5 CFU/g skin (Fig. 5). However, the numbers of skin CFU of unencapsulated mutant FLA1009 were significantly higher than the numbers of skin CFU of wild-type strain CMCP6 (P = 3 × 104 ) that had been inoculated using a 100-fold-lower dose, and the numbers of skin CFU of FLA1009 were significantly higher than the numbers of skin CFU of FLA610-Tp that was inoculated using the same dose (P = 3 × 104). In contrast, for both FLA610-Tp and FLA1009, liver CFU were detected in only one mouse each, compared with uniform liver infection by CMCP6 (P ≤ 0.02). Therefore, ΔrseB transparent variant FLA610-Tp was at least as attenuated as the acapsular mutant FLA1009; an inoculum that was more than 100 times the wild-type inoculum failed to result in consistent liver infection in spite of high levels of skin infection.
FIG. 5.

Highly attenuated virulence of ΔrseB transparent variant FLA610-Tp. Iron-dextran-treated mice were inoculated s.c. with 105 CFU of FLA610-Tp (ΔrseB-Tp) or nonencapsulated FLA1009 (CPS-) or 103 CFU of wild-type strain CMCP6 (WT). A χ2 test was used to compare the frequency of detectable liver infection with the results for the wild type (†, P = 0.01; ††, P = 0.02). An analysis of variance with the Bonferroni t test was used to compare the values for mutants with the results for the wild type (*, P ≤ 2 × 10−5; **, P ≤ 0.006). ***, P = 3 × 10−4 for a comparison of the number of skin CFU with the results for FLA610-Tp.
Complementation in trans with cloned wild-type rseB did not restore the wild-type colony morphology to ΔrseB variants; phase variation continued to be observed. The virulence of the complemented ΔrseB variants was tested using mice and an inoculum containing 1,000 CFU (Fig. 4A). The complemented transparent variant was recovered from the skin lesion of only one of five mice, and the concentration was 105.2 CFU/g. All five mice infected with the complemented transparent variant maintained a healthy appearance and had normal temperatures (mean, 37.2 ± 0.6°C) throughout the 22-h course of infection. Thus, the virulence of the ΔrseB-Tp mutant was not restored to wild-type virulence by complementation. The complemented translucent variant was recovered from four of five skin and liver samples, and the levels were 106.1 CFU/g skin and 102.4 CFU/g liver. The mean numbers of skin and liver CFU obtained were not statistically significantly different from those obtained for the uncomplemented translucent mutant. The complemented opaque variant was recovered from 100% of mouse skin and liver samples, and the levels were 108 CFU/g skin and 105.3 CFU/g liver (P = 0.012 for skin CFU and P = 0.008 for liver CFU for comparisons with the uncomplemented mutant). In summary, analysis of the virulence of ΔrseB mutants revealed that an opaque variant had variable attenuation that could be complemented. However, the attenuation of the translucent and transparent ΔrseB variants, which was apparently the result of induced phase variation, could not be complemented because complementation did not reverse the phase switch. We therefore focused on the nature of the colony morphology variation of the rseB mutants.
Characterization of RseB phase variants. (i) CPS and carbohydrate expression.
To determine if the colony variants of the ΔrseB FLA610 strain were the result of variations in capsule production, the EPSs of these variants were compared with those of wild-type strain CMCP6, the acapsular wza::TnphoA FLA1009 strain, and a spontaneous rugose mutant of FLA399, FLA611-R. These strains were plated on LB-N agar containing the carbohydrate-binding dye Congo red (41). The wild-type, ΔrseB-O, and rugose strains were bright red on Congo red agar (data not sown). The acapsular mutant was slightly less red than the wild type. The ΔrseB-Tl and ΔrseB-Tp strains bound the least Congo red and were light pink, and the ΔrseB-Tp strain was slightly lighter than the ΔrseB-Tl strain. The ΔrseB-Tp and ΔrseB-Tl strains resembled each other more than they resembled the acapsular mutant. Therefore, the ΔrseB-Tp and ΔrseB-Tl strains were not simply acapsular variants.
The abilities of the strains to bind the fluorescent stain calcofluor, a fluorochrome that binds to cellulose, chitin, and other carbohydrates (20), were examined by growing them on calcofluor-containing agar plates and observing fluorescence under UV light. The wild-type, ΔrseB-O, and rugose mutant strains had the highest levels of fluorescence, followed by the acapsular mutant, and the translucent and transparent ΔrseB variants did not fluoresce (Fig. 6). Therefore, like plating on Congo red-containing agar, plating on calcofluor-containing agar indicated that the nonopaque FLA610 ΔrseB variants were more similar to each other than to an acapsular mutant and produced the lowest levels of extracellular carbohydrates.
FIG. 6.

Analysis of EPS by calcofluor binding. Strains were grown on LB-N agar containing the fluorescent carbohydrate-binding agent calcofluor overnight at 37°C and then at room temperature for 24 h. The photographs were taken under UV light. (A) WT, wild-type strain CMCP6; Tp, ΔrseB-Tp mutant FLA610-Tp; Tl, ΔrseB-Tl mutant FLA610-T; O, ΔrseB-O mutant FLA610-O; C, wza::TnphoA mutant FLA1009; R, FLA611-R. (B) WT, wild-type strain CMCP6; P, ΔdegP mutant FLA1002; E, ΔrpoE mutant FLA1001.
EPSs were then extracted from each of these strains, resolved by SDS-PAGE, and stained with Stains-all or Alcian blue. Wild-type strain CMCP6 produced a broad dark smear when it was stained with Alcian blue or with Stains-all (Fig. 7). EPS extracts of the FLA610 ΔrseB-Tp strain and the acapsular mutant did not stain to as great an extent. The ΔrseB-Tl strain exhibited slightly darker staining with Alcian blue and Stains-all than the ΔrseB-Tp strain or the acapsular mutant, but it did not stain as intensely as the wild type. EPS from the rugose strain, FLA611-R, bound Alcian blue and Stains-all more strongly than EPS from any other strain in this group, consistent with the suggestion that rugose variants overexpress EPS or may have a different EPS composition than the wild type (16). These results suggested that the transparent and translucent ΔrseB variants may have decreased opacity due to loss of or decreased amounts of EPS.
FIG. 7.

SDS-PAGE analysis of EPS extracts. Purified EPS extracts from the FLA610 ΔrseB transparent (lane Tp), translucent (lane Tl), and opaque (lane O) variants, acapsular strain FLA1009 (lane CPS), rugose strain FLA611-R (lane R), ΔrpoE strain FLA1001 (lane E), ΔdegP strain FLA1002 (lane P), and wild-type strain CMCP6 (lane WT) cells were resolved on SDS-PAGE gels and stained with Alcian blue (A) or Stains-all (B) to observe exopolysaccharides. The volume of the ΔdegP sample loaded was 1/10 the volume of the other samples.
Chatzidaki-Livanis et al. (5) reported that genetic rearrangements in a V. vulnificus CPS locus detectable by PCR caused a loss of or decreased capsule expression. To examine if genetic rearrangements in the capsule region were responsible for the translucent phenotypes of ΔrseB mutants, PCR was performed as described by Chatzidaki-Livanis et al. (5) using oligonucleotide primers CMCP6wza5′ and CMCP6wzc3′ (see Table S1 in the supplemental material) that were designed to target the group 1 CPS operon of V. vulnificus CMCP6. Genomic DNA templates from ΔrseB mutants FLA610-Tp, FLA610-Tl, and FLA610-O generated the same 4.8-kb PCR amplicon as wild-type strain CMCP6 templates, indicating that no gross genetic rearrangements had occurred in the group 1 CPS genes of the variants (data not shown).
(ii) Resistance to stress and serum complement.
σE that is negatively regulated by RseB is essential for resistance to environmental stresses in many bacteria (61). We hypothesized that ΔrseB mutants would show altered responses to external stresses compared to the wild type due to dysregulation of the σE-mediated extracytoplasmic stress response. Disk diffusion assays were performed to assess the sensitivity of ΔrseB mutants to membrane-perturbing agents. There were no statistically significant differences between the ΔrseB variants and the wild type in sensitivity to 100% (vol/vol) ethanol, 3% (vol/vol) hydrogen peroxide, or 1% (wt/vol) SDS (Table 2). The ΔrseB variants and the acapsular mutant did not exhibit altered sensitivity to incubation at 42°C compared to the wild type (not shown). Conversely, the rugose mutant showed a marked decrease in viability when it was incubated at 42°C. Capsule mutants of V. vulnificus are more sensitive to complement than wild-type bacteria (53). We assayed the sensitivities of the colony variants and mutants to 90% (vol/vol) normal rat serum. A serum-sensitive E. coli strain, MG1655, was used as a positive control for complement-mediated killing. While wild-type strain CMCP6, FLA610-Tl, and FLA610-O survived as well in intact serum as in heat-inactivated serum, the capsule mutant FLA1009 and transparent FLA610-Tp strain were complement sensitive (Table 3). Taken together, these results suggest that RseB is not essential for resistance to envelope stress but that the apparent loss of EPS in the transparent variant rendered it complement sensitive.
TABLE 2.
Sensitivity of mutants to envelope stressa
| Strain | Genotype | Zone of inhibition (% of wild type) with:
|
||
|---|---|---|---|---|
| Ethanol | H2O2 | SDS | ||
| FLA610-Tp | ΔrseB-Tp | 115 ± 9 | 106 ± 7 | 100 ± 9 |
| FLA610-Tl | ΔrseB-Tl | 116 ± 6 | 100 ± 6 | 98 ± 7 |
| FLA610-O | ΔrseB-O | 113 ± 16 | 99 ± 9 | 95 ± 11 |
| FLA1001 | ΔrpoE | 115 ± 6b | 129 ± 6c | 125 ± 3c |
| FLA1002 | ΔdegP | 106 ± 3 | 132 ± 4c | 127 ± 4c |
Inhibition by 100% ethanol, 3% hydrogen peroxide, or 1% SDS was determined by measuring zones of inhibition, using ≥6 filters per experiment. Each experiment was repeated at least once, or the data are from combined experiments. The values are percentages of the inhibition by the wild type.
P = 0.006, as determined by an unpaired t test comparing mutant zones of inhibition to wild-type zones of inhibition.
P ≤ 9 × 10−7, as determined by an unpaired t test comparing mutant zones of inhibition to wild-type zones of inhibition.
TABLE 3.
Sensitivity of ΔrseB mutants to serum complementa
| Strain | Genotype | Δlog CFU |
|---|---|---|
| CMCP6 | Wild type | −0.7 |
| FLA610-Tp | ΔrseB-Tp | −5.0 |
| FLA610-Tl | ΔrseB-Tl | −1.3 |
| FLA610-O | ΔrseB-O | −1.0 |
| FLA1009 | wza::TnphoA | −4.6 |
| E. coli MG1655 | Wild type | −6.5 |
Wild-type strain CMCP6, ΔrseB transparent, translucent, and opaque variants (FLA610-Tp, FLA610-Tl, and FLA610-O), acapsular strain FLA1009, and complement-sensitive E. coli MC1655 were incubated in untreated serum or heat-inactivated rat serum for 2 h. The values are the differences between the number of log-transformed CFU for untreated serum and the number of log-transformed CFU for heat-treated serum. Similar results were obtained in an independent experiment.
(iii) Expression of rpoE.
RseB is a negative regulator of σE in E. coli (7, 38). To test if V. vulnificus rseB had a similar function, transcription of rpoE in ΔrseB mutants was measured and compared to that in wild-type strain CMCP6 by using qRT-PCR performed with mRNA isolated from exponential-phase cells grown in LB-N. RseB negatively regulates σE in a posttranslational manner in concert with RseA by tethering σE to the inner membrane (49). However, changes in the amount σE can be measured at the transcriptional level because rpoE autoregulates its own transcription (46). Thus, in the absence of RseB, there should be elevated levels of free σE and increased transcription of rpoE. The levels of rpoE were 13- to 24-fold greater in the FLA610-Tp, FLA610-Tl, and FLA610-O strains than in the wild type (P ≤ 0.0001 for ΔCt values for each mutant compared to the wild type) (Table 4). As a further test for negative regulation of σE in rseB mutants, we examined expression of a putative σE-regulated gene, VV1_0603, a homolog of degP (see below). Expression of degP is positively regulated by σE in E. coli and other bacteria (49). The expression of VV1_0603 was 28- to 100-fold greater in the ΔrseB variants than in the wild type (Table 4). Collectively, these gene expression results confirmed that V. vulnificus RseB negatively regulates σE.
TABLE 4.
Analysis of expression of rpoE and degP in ΔrseB mutants by qRT-PCR
| Strain | Genotype | Change (fold)a
|
|
|---|---|---|---|
| rpoE | degP | ||
| FLA610-Tp | ΔrseB-Tp | 24 ± 4b | 70 ± 30c |
| FLA610-Tl | ΔrseB-Tl | 20 ± 2b | 100 ± 25c |
| FLA610-O | ΔrseB-O | 13 ± 3b | 28 ± 6c |
The values indicate the changes in raw cycle threshold values (2−ΔΔCt) compared to the wild-type value, normalized to 16S rRNA as an endogenous control.
P ≤ 0.0001, as determined by Student's t test.
P ≤ 0.01, as determined by Student's t test.
Effects of overexpression of rpoE in wild-type V. vulnificus.
Because we confirmed that V. vulnificus ΔrseB mutants had increased transcription of rpoE, we hypothesized that the rseB phenotypes were due to overexpression of σE. Furthermore, we expected that overexpression of σE in wild-type V. vulnificus would recapitulate ΔrseB phenotypes. pGTR2008, which expresses V. vulnificus rpoE (VV1_1559) from a lac promoter, was introduced into CMCP6 by conjugation. CMCP6(pGTR2008) appeared to be identical to wild-type strain CMCP6 on LB-N agar plates. No differences in carbohydrate binding were observed when the organisms were plated on calcofluor or Congo red agar plates (not shown). These results suggested that the rseB mutant phenotypes might be independent of σE or that the level of σE expression in CMCP6(pGTR2008) was different from that in the ΔrseB mutants. To examine the virulence of CMCP6(pGTR2008) in mice, 300 CFU of wild-type strain CMCP6 and 300 CFU of rpoE-overexpressing strain CMCP6(pGTR2008) were inoculated into iron-dextran-treated mice (Fig. 8A). Plating of the CMCP6(pGTR2008) inoculum nonselectively on LB-N agar showed that the inoculum contained 340 bacteria, whereas selective plating on LB-N agar with tetracycline showed that only one-half of the inoculum was Tetr (i.e., contained the plasmid). Nonetheless, the rpoE-overexpressing strain showed decreased systemic virulence compared to the wild type, and the number of CFU/g liver was nearly 1,000-fold lower than the number for the wild type (P = 0.03). Similarly, the temperatures of mice infected with the rpoE-overexpressing strain (37.1 ± 2.4°C) were higher than those of mice infected with wild-type bacteria (30.3 ± 4.2°C) (P = 0.01). The numbers of skin CFU for mice inoculated with rpoE-overexpressing bacteria and wild-type bacteria were similar (log 7.2 ± 0.7 CFU/g and log 7.7 ± 1.4 CFU/g, respectively). In other experiments, there were decreased numbers of CFU in skin lesions as well as in the liver. Thus, as observed with the ΔrseB mutants, overexpression of rpoE in wild-type strain CMCP6 attenuated virulence.
FIG. 8.

Effects of overexpression of σE and deletion of rpoE and degP on virulence of CMCP6. (A) Iron-dextran-treated mice were inoculated s.c. with 300 CFU of wild-type CMCP6 (WT) or CMCP6 overexpressing rpoE from plasmid pGTR2008 (rpoE++). Plate counts on selective medium are shown for the rpoE-overexpressing strain. The results for the nonselective plates were not significantly different. Student's t test was used to compare the number of CFU/g liver or temperature for the rpoE-overexpressing strain to the results for the wild type (*, P = 0.03; **, P = 0.01). (B) Iron-dextran-treated mice were inoculated with 1,000 CFU of wild-type strain CMCP6, ΔrpoE mutant FLA1001, or ΔdegP mutant FLA1002. The only significant difference was the difference in the temperatures for FLA1002 and CMCP6 (P = 0.004); however, most mice were still considered moribund, as determined by a temperature less than 33°C. The fractions below the bars are the frequencies of detectable skin and liver infection.
Role of the σE-mediated ESR in virulence of V. vulnificus. (i) Characterization of V. vulnificus ΔrpoE mutant.
σE is important for virulence and stress resistance in several bacterial pathogens, including V. cholerae (27), Salmonella enterica (22), and Pseudomonas aeruginosa (2, 15). For a review, see reference 48. To probe the role of the σE-mediated ESR in V. vulnificus, rpoE was deleted from wild-type strain CMCP6 by allelic exchange via chitin-induced transformation, yielding FLA1001. On LB-N agar ΔrpoE mutants had a whitish appearance that suggested that they might be more heavily encapsulated than wild-type strain CMCP6 (data not shown).
Several reports have indicated that rpoE mutants of gram-negative bacteria are more susceptible to environmental stresses (for a review, see reference 48). The sensitivity of FLA1001 to envelope stresses was compared to that of wild-type strain CMCP6 using disk diffusion assays (Table 2). The increases in the sensitivity of FLA1001 to stresses were 15% for 100% (vol/vol) ethanol (P = 0.006), 29% for 3% (vol/vol) hydrogen peroxide (P = 1 × 10−7), and 25% for 1% (wt/vol) SDS (P = 3 × 10−7). The ΔrpoE mutant showed a slight increase in heat sensitivity compared to the wild type (Fig. 9). Conversely, there was no difference between wild-type strain CMCP6 and the ΔrpoE FLA1001 mutant in plating efficiency when the strains were incubated for 18 h at 4°C and then overnight at room temperature (data not shown).
FIG. 9.
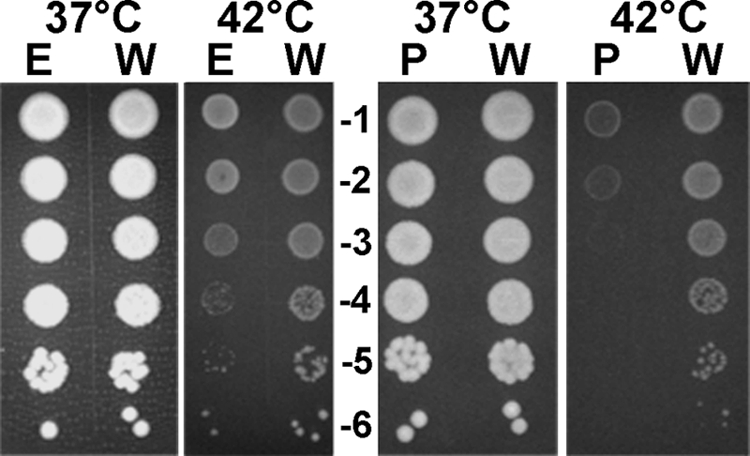
Sensitivities of ΔrpoE mutant FLA1001 and ΔdegP mutant FLA1002 to heat. Serial 10-fold dilutions (numbers in the center) of exponential-phase bacteria were spotted on plates and incubated at the temperatures indicated for 18 h. E, ΔrpoE mutant; W, wild-type strain CMCP6; P, ΔdegP mutant. Each experiment was repeated at least once.
When the inoculum contained 1,000 CFU, FLA1001 exhibited full virulence compared to the wild type. The mean levels of recovery for FLA1001 were 108.2 CFU/g skin and 106 CFU/g liver (Fig. 8B). With a smaller inoculum (300 CFU), FLA1001 showed decreased levels of skin (106.1 CFU/g) and liver (103.2 CFU/g) infection compared to the wild type (means, 107.7 CFU/g skin and 105.1 CFU/g liver) (not shown). Because the ΔrpoE mutant was attenuated for virulence only when the inoculum was small, we concluded that σE does not have a major role in the virulence of V. vulnificus in the s.c. inoculation iron-dextran-treated mouse model.
(ii) Identification and characterization of V. vulnificus degP.
An important mediator of the ESR is the periplasmic protease DegP, also known as HtrA and DO (6, 11, 30, 58). DegP binds to and degrades misfolded proteins and acts as a chaperone to direct proper folding of some envelope proteins (52, 58). We therefore investigated the role of DegP as a possible effector of the ESR in V. vulnificus. To find the V. vulnificus homolog of DegP, BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi) searches were performed using DegP homologues from V. cholerae (protease DO), Vibrio fischeri (DegP), Vibrio parahaemolyticus (protease DO), and E. coli (protease DO). VV1_0603, annotated as protease DO, had the highest level of homology (56 to 84%). There was also significant similarity between the vibrio degP homologues and the gene immediately downstream of VV1_0603, VV1_0604, annotated as degS. In all of the Vibrio species examined, degP is followed by degS. This result indicated that VV1_0603 could be a degP homolog. Interestingly, the genomic region surrounding degP in the Vibrio species was almost identical to the region surrounding another degP homolog, degQ, in E. coli. Therefore, there was a chance that V. vulnificus DO might be DegQ and not DegP. This was an important distinction because E. coli degP is regulated by σE but degQ is regulated only by σ70 (42).
Using the E. coli σE promoter consensus sequence −10 T/CGGTCAAAA, −35 GGAACTTTT (44) as a guide, the following putative σE-responsive promoter sequence was located upstream of VV1_0603: −10 CTGTCTATT, −35 TGAACTTTT. More importantly, expression of VV1_0603 was significantly increased in ΔrseB mutants compared to the wild type as determined by qRT-PCR (Table 4), suggesting that VV1_0603 was regulated by σE. VV1_0603 had two C-terminal PDZ domains (an essential feature of DegP proteins [42]) at amino acids 257 to 346 and 368 to 444, based on data from the NCBI conserved domain database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=cdd). None of the V. vulnificus genes annotated as “serine endoprotease” or “periplasmic protease” shared this feature. Therefore, VV1_0603 most likely encodes a DegP orthologue in V. vulnificus, and we designated this gene degP.
VV1_0603 was deleted from wild-type strain CMCP6 by allelic exchange and chitin transformation, as described in Materials and Methods, yielding FLA1002. The ΔdegP mutant formed slightly smaller colonies than the wild type on LB-N agar plates and was whitish, similar to rpoE mutants (data not shown). degP mutants of several other bacteria were reported to be less resistant to oxidative and other envelope stresses (42). To see if this was true for V. vulnificus, the sensitivities of FLA1002 and wild-type strain CMCP6 to 3% (vol/vol) hydrogen peroxide, 100% (vol/vol) ethanol, and 1% (wt/vol) SDS were tested by using disk diffusion assays (Table 2). FLA1002 was 32% more sensitive to hydrogen peroxide (P = 1 × 10−6) and 27% more sensitive to SDS (P = 1 × 10−7) than CMCP6. FLA1002 exhibited no statistically significant increase in sensitivity to 100% (vol/vol) ethanol, and FLA1002 exhibited a marked decrease in viability after incubation at 42°C for 18 h (Fig. 9). These results indicated that DegP is essential for the envelope stress resistance and, in particular, the heat resistance of V. vulnificus.
The increased opacity of the ΔdegP and ΔrpoE mutants suggested that they might overexpress CPS. As a test for increased production of exopolysaccharides, EPS extracts of the ΔdegP and ΔrpoE mutants were resolved by SDS-PAGE and stained with Alcian blue and Stains-all as described above for the ΔrseB mutants. Although there was only a small increase in the level of exopolysaccharides in the ΔrpoE sample compared to the wild type, there was a substantial increase in the level of exopolysaccharides in the ΔdegP mutant, as 10-fold-fewer ΔdegP cells were able to produce wild-type levels of staining (Fig. 7).
DegP has been identified as a virulence factor for several bacterial pathogens, including S. enterica serovar Typhimurium, Legionella pneumophila, and Streptococcus pyogenes (48). To test the virulence of the V. vulnificus ΔdegP mutant, mice were infected with 1,000 CFU of the ΔdegP FLA1002 mutant or wild-type strain CMCP6 (Fig. 8B). The mean skin (108.3 CFU/g) and liver (104.8 CFU/g) levels for FLA1002 were similar to the wild-type levels (107.9 CFU/g skin and 105.4 CFU/g liver). While the temperatures of mice infected with FLA1002 were significantly higher than the temperatures of mice infected with the wild type (means, 33.0°C and 30.9°C, respectively; P = 0.004), all but one mouse had a temperature below 33°C (range, 32.5°C to 33.8°C), indicating that the mice were moribund. Thus, FLA1002 was not attenuated at this inoculum when the s.c. route of infection was used, and DegP appears to be dispensable for virulence in this animal model of disease.
DISCUSSION
Identification of a V. vulnificus rseB::mini-Tn5Km2phoA mutation that caused altered colony morphology and attenuated virulence.
V. vulnificus rseB::mini-Tn5Km2phoA mutant FLA609 initially appeared to be less opaque than the wild type on LB-N agar and underwent high-frequency phase variation to a more opaque morphology (Fig. 1). The translucent variant was attenuated for virulence in s.c. inoculated iron-dextran-treated mice (Fig. 3) and switched to a more opaque form in vivo. In contrast, the opaque variant was not consistently attenuated for virulence and did not appear to undergo phase variation in the mouse host. It is possible that the opaque variant underwent phase variation to a translucent morphology during infection, but such variants would have been selected against. Deletion of rseB from wild-type strain CMCP6 essentially recapitulated these phenotypes with additional appearance of a transparent variant, FLA610-Tp, which was highly attenuated for virulence (Fig. 5). The only reports of an rseB homolog having a role in virulence involve mucB of P. aeruginosa. Alginate production in P. aeruginosa is controlled by AlgU/σE (34). Mutations in MucB, the periplasmic negative regulator of σE, lead to the alginate-overproducing, mucoid phenotype associated with the establishment of lethal disease in cystic fibrosis patients (13, 14, 35, 36). This scenario is opposite that for V. vulnificus, in which the rseB mutation caused decreased expression of EPSs and decreased virulence. To our knowledge, this is the first report of an rseB mutation causing attenuated virulence.
EPS and the altered colony morphology of V. vulnificus ΔrseB mutants.
V. vulnificus rseB::mini-Tn5Km2phoA and ΔrseB mutants had a translucent colony phenotype that showed high frequencies of phase variation to a more opaque form (Fig. 1 and data not shown). Colony opacity has been correlated with CPS expression in V. vulnificus (62, 63, 65). However, Chatzidaki-Livanis et al. (5) noted that a genetic event that led to a phase-variable translucent phenotype (designated Tr1) did not correspond to genetic rearrangements in the CPS biosynthesis operon. Rosche et al. (45) reported that V. vulnificus can exhibit a morphology that is intermediate between the translucent and opaque forms, which is designated intermediate or Int and is characterized by decreased expression of CPS. While the ΔrseB-Tl variant examined in the present study had a level of opacity that was between that of the ΔrseB-O variant and that of the ΔrseB-Tp variant, we do not believe that the ΔrseB-Tl variant is the Int morphotype of Rosche et al. (45) because the frequencies of phase variation that Rosche et al. reported were very different from our frequencies of phase variation (data not shown). More importantly, while the translucent ΔrseB variant FLA610-Tl was not as translucent as the acapsular mutant FLA1009 on LB-N agar, FLA610-Tl was less red than FLA1009 on Congo red agar and exhibited less fluorescence than FLA1009 on calcofluor agar (Fig. 6). These results indicated that the translucent ΔrseB variant expressed smaller amounts of some exopolysaccharides than the acapsular mutant FLA1009 expressed.
There is increasing evidence that V. vulnificus possesses EPSs in addition to CPS. A mutation in ntrC (encoding a transcriptional regulator) or in gmhD (encoding a sugar epimerase regulated by NtrC and by RpoN) caused a significant decrease in the EPS level but did not cause a decrease in colony opacity (24). This result suggested that V. vulnificus makes several kinds of EPSs. Interestingly, RpoN, which regulates expression of the sugar epimerase GmhD, is a member of the core σE regulon (44). It was recently reported that increased cyclic diguanylate levels in V. vulnificus caused expression of an EPS that was distinct from CPS (40). Translucent strains of V. vulnificus with mutations in the wzy CPS polymerase gene, the rmlC sugar epimerase gene, or the wzc tyrosine autokinase gene could be restored to an opaque morphology via expression of a diguanylate cyclase gene, dcpA, in trans. Moreover, the non-CPS EPS affected by ntrC and gmhD mutations (24) and cyclic diguanylate changes caused by dcpA (40) appeared to be composed of acidic carbohydrate moieties as they stained with Stains-all and with Alcian blue. It is possible, therefore, that the altered colony morphology of some of the V. vulnificus ΔrseB variants described in the present study could be due to decreased expression of EPS determinants that are distinct from CPS. This could explain the fact that ΔrseB-Tp and ΔrseB-Tl variants appeared to be different from the acapsular mutant on the media containing carbohydrate-binding calcofluor (Fig. 6) or Congo red. The differences in binding could also be the result of differences in the amount of CPS expressed between the ΔrseB and acapsular mutants. It is necessary to analyze the chemical compositions of the exopolysaccharides of the various strains to determine if the ΔrseB mutants are deficient in CPS or another EPS.
Comparison of the ΔrseB-Tp mutant and an acapsular V. vulnificus mutant.
The ΔrseB-Tp mutant was less opaque than the ΔrseB-Tl mutant and the translucent rseB::mini-Tn5Km2phoA strain. In fact, the ΔrseB-Tp mutant most closely resembled an acapsular mutant of V. vulnificus CMCP6 in terms of staining of EPS with Alcian blue and Stains-all (Fig. 7), increased serum sensitivity (Table 3), and highly attenuated virulence (Fig. 5). Also, similar to the acapsular mutant, the transparent ΔrseB variant did not appear to switch to a more opaque form in vivo. The major phenotypic differences observed between the ΔrseB-Tp mutant and the acapsular mutant were differences in morphology on LB-N agar and in binding of calcofluor (Fig. 6) and Congo red. While there was no evidence of genetic rearrangements in the CPS locus in the ΔrseB-Tp mutant compared to the wild type, it is possible that the ΔrseB-Tp mutant is nearly or completely acapsular.
Possible reasons for the attenuated virulence of ΔrseB mutants.
There are two main possibilities to explain the attenuated virulence of the V. vulnificus ΔrseB mutant. Because the translucent and transparent variants showed decreased fitness in vivo and switched to a more opaque form, it is plausible that attenuation was due to a decrease in the level of exopolysaccharides that may have been directly or indirectly due to the rseB mutation. The second possibility is that the attenuation was due solely to the deleterious effects of overexpression of σE. CMCP6 expressing rpoE from pGTR2008 did not exhibit decreased opacity but was attenuated for virulence when an inoculum containing 300 CFU was used (Fig. 8A). When the opaque ΔrseB mutant was examined for virulence in 20 mice (four separate infections with five mice each) using an inoculum containing 1,000 CFU, 35% of the skin and liver samples did not yield detectable CFU (Fig. 3B). Thus, even without being translucent, the ΔrseB-O variant that overexpressed rpoE showed decreased virulence in mice.
Regulation of σE in V. vulnificus.
V. vulnificus ΔrseB variants showed 13- to 24-fold increases in rpoE expression compared to the wild type (Table 4). E. coli RseB was reported to be the minor negative regulator of σE; deletion of rseB resulted in a twofold increase in transcription from σE-dependent promoters, whereas deletion of rseA led to a ninefold increase (7, 38). Conversely, inactivation of either MucA (RseA) or MucB (RseB) in P. aeruginosa can result in comparably large increases in AlgU activity (47, 50), supporting the hypothesis that both proteins have major regulatory roles. Studies to compare the relative roles of rseB and rseA in regulation of σE levels in V. vulnificus are needed. Nevertheless, the high degree of regulation of σE by rseB in V. vulnificus suggests that rseB is an important mediator of the ESR in this pathogen.
σE and DegP are important for stress resistance but not for virulence in V. vulnificus.
Deletion of rpoE or degP from V. vulnificus CMCP6 resulted in significantly increased fluorescence with calcofluor compared to the wild-type fluorescence (Fig. 6), suggesting that EPS expression is altered. Both mutants also exhibited increased sensitivity to the membrane-perturbing agents ethanol, peroxide, and SDS (Table 2) and to heat (Fig. 9). Interestingly, a ΔrpoE mutant of the closely related species V. cholerae showed different patterns of stress sensitivity (27). The V. cholerae ΔrpoE mutant was hypersensitive to 3% ethanol but was similar to the wild type in resistance to other envelope stresses, including growth at 43°C, peroxide, and antimicrobial peptides. Similarly, the growth of an S. enterica serovar Typhimurium rpoE mutant at high temperatures was not significantly affected, but this mutant was more sensitive than the wild type to peroxide, paraquat, and antimicrobial peptides (22). As in V. vulnificus, in E. coli rpoE is required for growth at a high temperature (21). Taken together, the data clearly show that while the general function of σE is regulation of the extracellular stress response, its specific functions are tailored differently to the needs and niches of different bacteria.
Despite the overall increase in envelope stress sensitivity in the ΔrpoE mutant, this mutant was not attenuated compared to the wild type when an inoculum containing 1,000 CFU was used (Fig. 8B). The ΔdegP mutant showed only a marginal decrease in virulence compared to the wild type when an inoculum containing 1,000 CFU was used (Fig. 8B), and this effect was seen as a higher temperature that still represented a lethal endpoint for four of the five infected mice. Therefore, it appears that σE and DegP have little importance in the virulence of V. vulnificus in iron-dextran-treated mice when the s.c. route is used, despite their importance in resisting external stresses in vitro. It is possible that σE and DegP are essential for survival in the environment but not for causing disease or that they have a role in infection that could not be assessed by our animal model. As previously noted, AlgU (σE) of P. aeruginosa is important for respiratory infection but not for systemic infection (48). Likewise, HtrA (DegP) of S. enterica serovar Typhimurium is essential for systemic disease but not for enteric disease (48). A ΔrpoE mutant of V. cholerae was significantly attenuated for virulence in orally inoculated suckling mice, demonstrating the importance of σE for intestinal colonization and survival (27). Therefore, a role for σE or DegP in virulence of V. vulnificus may be discovered using a different animal model or route of infection, e.g., oral inoculation, in which V. vulnificus must survive gut acidity and colonize the intestines. Because members of the σE regulon are virulence factors in many bacterial pathogens (48), identifying the full V. vulnificus σE regulon may reveal novel virulence factors for this pathogen.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 056056.
We thank Patrick Thiaville, Jennifer Joseph, Julio Martin, and Aaron Mittel for their assistance.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 29 June 2009.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Bitinaite, J., M. Rubino, K. H. Varma, I. Schildkraut, R. Vaisvila, and R. Vaiskunaite. 2007. USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res. 351992-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boucher, J. C., H. Yu, M. H. Mudd, and V. Deretic. 1997. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 653838-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, R. N., and P. A. Gulig. 2008. Regulation of fatty acid metabolism by FadR is essential for Vibrio vulnificus to cause infection of mice. J. Bacteriol. 1907633-7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerda-Cuellar, M., J. Jofre, and A. R. Blanch. 2000. A selective medium and a specific probe for detection of Vibrio vulnificus. Appl. Environ. Microbiol. 66855-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatzidaki-Livanis, M., M. K. Jones, and A. C. Wright. 2006. Genetic variation in the Vibrio vulnificus group 1 capsular polysaccharide operon. J. Bacteriol. 1881987-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dartigalongue, C., D. Missiakas, and S. Raina. 2001. Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 27620866-20875. [DOI] [PubMed] [Google Scholar]
- 7.De Las Peñas, A., L. Connolly, and C. A. Gross. 1997. The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Mol. Microbiol. 24373-385. [DOI] [PubMed] [Google Scholar]
- 8.de Lorenzo, V., M. Herrero, U. Jakubzik, and K. N. Timmis. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 1726568-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 594310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enos-Berlage, J. L., and L. L. McCarter. 2000. Relation of capsular polysaccharide production and colonial cell organization to colony morphology in Vibrio parahaemolyticus. J. Bacteriol. 1825513-5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erickson, J. W., and C. A. Gross. 1989. Identification of the σE subunit of Escherichia coli RNA polymerase: a second alternate sigma factor involved in high-temperature gene expression. Genes Dev. 31462-1471. [DOI] [PubMed] [Google Scholar]
- 12.Gardy, J. L., C. Spencer, K. Wang, M. Ester, G. E. Tusnady, I. Simon, S. Hua, K. deFays, C. Lambert, K. Nakai, and F. S. Brinkman. 2003. PSORT-B: improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 313613-3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilligan, P. H. 1991. Microbiology of airway disease in patients with cystic fibrosis. Clin. Microbiol. Rev. 435-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg, J. B., W. L. Gorman, J. L. Flynn, and D. E. Ohman. 1993. A mutation in algN permits trans activation of alginate production by algT in Pseudomonas species. J. Bacteriol. 1751303-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Govan, J. R., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grau, B. L., M. C. Henk, and G. S. Pettis. 2005. High-frequency phase variation of Vibrio vulnificus 1003: isolation and characterization of a rugose phenotypic variant. J. Bacteriol. 1872519-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulig, P. A., K. L. Bourdage, and A. M. Starks. 2005. Molecular pathogenesis of Vibrio vulnificus. J. Microbiol. 43118-131. [PubMed] [Google Scholar]
- 18.Gulig, P. A., M. S. Tucker, P. C. Thiaville, J. L. Joseph, and R. N. Brown. 2009. USER friendly cloning coupled with chitin-based natural transformation enables rapid mutagenesis of Vibrio vulnificus. Appl. Environ. Microbiol. 754936-4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guyer, M. S., R. R. Reed, J. A. Steitz, and K. B. Low. 1981. Identification of a sex-factor-affinity site in Escherichia coli as gamma delta. Cold Spring Harbor Symp. Quant. Biol. 45135-140. [DOI] [PubMed] [Google Scholar]
- 20.Harrington, B. J., and K. B. Raper. 1968. Use of a fluorescent brightener to demonstrate cellulose in the cellular slime molds. Appl. Microbiol. 16106-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiratsu, K., M. Amemura, H. Nashimoto, H. Shinagawa, and K. Makino. 1995. The rpoE gene of Escherichia coli, which encodes σE, is essential for bacterial growth at high temperature. J. Bacteriol. 1772918-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphreys, S., A. Stevenson, A. Bacon, A. B. Weinhardt, and M. Roberts. 1999. The alternative sigma factor, σE, is critically important for the virulence of Salmonella typhimurium. Infect. Immun. 671560-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley, J. T., and C. D. Parker. 1981. Identification and preliminary characterization of Vibrio cholerae outer membrane proteins. J. Bacteriol. 1451018-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, H. S., M. A. Lee, S. J. Chun, S. J. Park, and K. H. Lee. 2007. Role of NtrC in biofilm formation via controlling expression of the gene encoding an ADP-glycero-manno-heptose-6-epimerase in the pathogenic bacterium, Vibrio vulnificus. Mol. Microbiol. 63559-574. [DOI] [PubMed] [Google Scholar]
- 25.Kim, Y. R., S. E. Lee, C. M. Kim, S. Y. Kim, E. K. Shin, D. H. Shin, S. S. Chung, H. E. Choy, A. Progulske-Fox, J. D. Hillman, M. Handfield, and J. H. Rhee. 2003. Characterization and pathogenic significance of Vibrio vulnificus antigens preferentially expressed in septicemic patients. Infect. Immun. 715461-5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, Y. R., S. E. Lee, H. Kook, J. A. Yeom, H. S. Na, S. Y. Kim, S. S. Chung, H. E. Choy, and J. H. Rhee. 2008. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell. Microbiol. 10848-862. [DOI] [PubMed] [Google Scholar]
- 27.Kovacikova, G., and K. Skorupski. 2002. The alternative sigma factor σE plays an important role in intestinal survival and virulence in Vibrio cholerae. Infect. Immun. 705355-5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, J. H., M. W. Kim, B. S. Kim, S. M. Kim, B. C. Lee, T. S. Kim, and S. H. Choi. 2007. Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J. Microbiol. 45146-152. [PubMed] [Google Scholar]
- 29.Lehoux, D. E., F. Sanchagrin, and R. C. Levesque. 1999. Defined oligonucleotide tag pools and PCR screening in signature-tagged mutagenesis of essential genes from bacteria. BioTechniques 26473-480. [DOI] [PubMed] [Google Scholar]
- 30.Lipinska, B., S. Sharma, and C. Georgopoulos. 1988. Sequence analysis and regulation of the htrA gene of Escherichia coli: a σ32-independent mechanism of heat-inducible transcription. Nucleic Acids Res. 1610053-10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litwin, C. M., T. W. Rayback, and J. Skinner. 1996. Role of catechol siderophore synthesis in Vibrio vulnificus virulence. Infect. Immun. 642834-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, M., A. F. Alice, H. Naka, and J. H. Crosa. 2007. The HlyU protein is a positive regulator of rtxA1, a gene responsible for cytotoxicity and virulence in the human pathogen Vibrio vulnificus. Infect. Immun. 753282-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manoil, C., and J. Beckwith. 1985. TnphoA: a transposon probe for protein export signals. Proc. Natl. Acad. Sci. USA 828129-8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin, D. W., B. W. Holloway, and V. Deretic. 1993. Characterization of a locus determining the mucoid status of Pseudomonas aeruginosa: AlgU shows sequence similarities with a Bacillus sigma factor. J. Bacteriol. 1751153-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin, D. W., M. J. Schurr, M. H. Mudd, and V. Deretic. 1993. Differentiation of Pseudomonas aeruginosa into the alginate-producing form: inactivation of mucB causes conversion to mucoidy. Mol. Microbiol. 9497-506. [DOI] [PubMed] [Google Scholar]
- 36.Martin, D. W., M. J. Schurr, M. H. Mudd, J. R. Govan, B. W. Holloway, and V. Deretic. 1993. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 908377-8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu, and G. K. Schoolnik. 2005. Chitin induces natural competence in Vibrio cholerae. Science 3101824-1827. [DOI] [PubMed] [Google Scholar]
- 38.Missiakas, D., M. P. Mayer, M. Lemaire, C. Georgopoulos, and S. Raina. 1997. Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol. Microbiol. 24355-371. [DOI] [PubMed] [Google Scholar]
- 39.Møller, H. J., D. Heinegård, and J. H. Poulsen. 1993. Combined alcian blue and silver staining of subnanogram quantities of proteoglycans and glycosaminoglycans in sodium dodecyl sulfate-polyacrylamide gels. Anal. Biochem. 209169-175. [DOI] [PubMed] [Google Scholar]
- 40.Nakhamchik, A., C. Wilde, and D. A. Rowe-Magnus. 2008. Cyclic-di-GMP regulates extracellular polysaccharide production, biofilm formation, and rugose colony development by Vibrio vulnificus. Appl. Environ. Microbiol. 744199-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novelli, A. 1953. New method of staining of bacterial capsules in films and sections. Experientia 934-35. [DOI] [PubMed] [Google Scholar]
- 42.Pallen, M. J., and B. W. Wren. 1997. The HtrA family of serine proteases. Mol. Microbiol. 26209-221. [DOI] [PubMed] [Google Scholar]
- 43.Provence, D. L., and R. Curtiss III. 1994. Gene transfer in gram-negative bacteria, p. 317-347. In P. Gerhardt, R. G. E. Murray, W. A. Wood, and N. R. Krieg (ed.), Methods for general and molecular bacteriology. American Society for Microbiology, Washington, DC.
- 44.Rhodius, V. A., W. C. Suh, G. Nonaka, J. West, and C. A. Gross. 2006. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol. 4e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosche, T. M., B. Smith, and J. D. Oliver. 2006. Evidence for an intermediate colony morphology of Vibrio vulnificus. Appl. Environ. Microbiol. 724356-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rouviere, P. E., A. De Las Peñas, J. Mecsas, C. Z. Lu, K. E. Rudd, and C. A. Gross. 1995. rpoE, the gene encoding the second heat-shock sigma factor, σE, in Escherichia coli. EMBO J. 141032-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowen, D. W., and V. Deretic. 2000. Membrane-to-cytosol redistribution of ECF sigma factor AlgU and conversion to mucoidy in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Mol. Microbiol. 36314-327. [DOI] [PubMed] [Google Scholar]
- 48.Rowley, G., M. Spector, J. Kormanec, and M. Roberts. 2006. Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat. Rev. Microbiol. 4383-394. [DOI] [PubMed] [Google Scholar]
- 49.Ruiz, N., and T. J. Silhavy. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8122-126. [DOI] [PubMed] [Google Scholar]
- 50.Schurr, M. J., H. Yu, J. M. Martinez-Salazar, J. C. Boucher, and V. Deretic. 1996. Control of AlgU, a member of the σE-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 1784997-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott, H. N., P. D. Laible, and D. K. Hanson. 2003. Sequences of versatile broad-host-range vectors of the RK2 family. Plasmid 5074-79. [DOI] [PubMed] [Google Scholar]
- 52.Seol, J. H., S. K. Woo, E. M. Jung, S. J. Yoo, C. S. Lee, K. J. Kim, K. Tanaka, A. Ichihara, D. B. Ha, and C. H. Chung. 1991. Protease Do is essential for survival of Escherichia coli at high temperatures: its identity with the htrA gene product. Biochem. Biophys. Res. Commun. 176730-736. [DOI] [PubMed] [Google Scholar]
- 53.Shinoda, S., M. Kobayashi, H. Yamada, S. Yoshida, M. Ogawa, and Y. Mizuguchi. 1987. Inhibitory effect of capsular antigen of Vibrio vulnificus on bactericidal activity of human serum. Microbiol. Immunol. 31393-401. [DOI] [PubMed] [Google Scholar]
- 54.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1784-791. [Google Scholar]
- 55.Simpson, L. M., V. K. White, S. F. Zane, and J. D. Oliver. 1987. Correlation between virulence and colony morphology in Vibrio vulnificus. Infect. Immun. 55269-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Starks, A. M., K. L. Bourdage, P. C. Thiaville, and P. A. Gulig. 2006. Use of a marker plasmid to examine growth and death of Vibrio vulnificus in infected mice. Mol. Microbiol. 61310-323. [DOI] [PubMed] [Google Scholar]
- 57.Starks, A. M., T. R. Schoeb, M. L. Tamplin, S. Parveen, T. J. Doyle, P. E. Bomeisl, G. M. Escudero, and P. A. Gulig. 2000. Pathogenesis of infection by clinical and environmental strains of Vibrio vulnificus in iron-dextran-treated mice. Infect. Immun. 685785-5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strauch, K. L., and J. Beckwith. 1988. An Escherichia coli mutation preventing degradation of abnormal periplasmic proteins. Proc. Natl. Acad. Sci. USA 851576-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor, L. A., and R. E. Rose. 1988. A correction in the nucleotide sequence of the Tn903 kanamycin resistance determinant in pUC4K. Nucleic Acids Res. 16358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor, R. K., C. Manoil, and J. J. Mekalanos. 1989. Broad-host-range vectors for delivery of TnphoA: use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J. Bacteriol. 1711870-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warne, S. R., J. M. Varley, G. J. Boulnois, and M. G. Norton. 1990. Identification and characterization of a gene that controls colony morphology and auto-aggregation in Escherichia coli K12. J. Gen. Microbiol. 136455-462. [DOI] [PubMed] [Google Scholar]
- 62.Wright, A. C., J. L. Powell, J. B. Kaper, and J. G. Morris, Jr. 2001. Identification of a group 1-like capsular polysaccharide operon for Vibrio vulnificus. Infect. Immun. 696893-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wright, A. C., J. L. Powell, M. K. Tanner, L. A. Ensor, A. B. Karpas, J. G. Morris, Jr., and M. B. Sztein. 1999. Differential expression of Vibrio vulnificus capsular polysaccharide. Infect. Immun. 672250-2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wright, A. C., L. M. Simpson, and J. D. Oliver. 1981. Role of iron in the pathogenesis of Vibrio vulnificus infections. Infect. Immun. 34503-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wright, A. C., L. M. Simpson, J. D. Oliver, and J. G. Morris, Jr. 1990. Phenotypic evaluation of acapsular transposon mutants of Vibrio vulnificus. Infect. Immun. 581769-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoshida, S., M. Ogawa, and Y. Mizuguchi. 1985. Relation of capsular materials and colony opacity to virulence of Vibrio vulnificus. Infect. Immun. 47446-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.