

Abstract
Infection by herpesviruses causes a dramatic disturbance of PML oncogenic domains (PODs) that has been suggested to be essential for viral lytic replication. Several proteins from Kaposi's sarcoma-associated herpesvirus (KSHV) have been tested as putative POD-disrupting factors with negative results. Here, we show that LANA2, a viral protein that is absolutely required for the viability and proliferation of KSHV-infected primary effusion lymphoma (PEL) cells, increases the levels of SUMO2-ubiquitin-modified PML and induces the disruption of PODs by a proteasome-mediated mechanism. In addition, we demonstrate that this disruption is largely dependent on both the integrity of a SUMO interaction motif in LANA2 and the lysine 160 from PML. Moreover, silencing of LANA2 expression in PEL cells by RNA interference led to an increase in the PML levels. Finally, we demonstrate that LANA2 relieves PML-mediated transcriptional repression of survivin, a protein that directly contributes to malignant progression of PEL. This represents the first example of inactivation of these important antiviral structures by KSHV.
PML oncogenic domains (PODs), also known as nuclear dots, PML nuclear bodies (NBs), or ND10 domains, are spherical nuclear substructures of multiple cellular proteins that are involved in gene transcription, genomic stability, cell cycle regulation, and apoptosis (6). PML is the major component of PODs and is responsible for the proper localization of all other POD-associated proteins (22). PML acts as a tumor suppressor protein, which is particularly relevant in the lympho-hematopoietic compartment (40, 50), and is involved in the control of viral infections (19). Consistent with the antiviral activity of PML, many nuclear-replicating viruses encode polypeptides that cause the disruption of PODs early during infection. The herpes simplex virus (HSV) ICP0 (10), cytomegalovirus (CMV) IE1 (28), adenovirus E4-ORF3 (9), and human T-cell leukemia virus type 1 Tax (13) proteins alter the localization of at least one POD-associated protein.
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, is a member of the gammaherpesvirus subfamily and is the causal agent of several human malignancies including Kaposi's sarcoma, primary effusion lymphoma (PEL), and multicentric Castleman's disease. Several KSHV proteins have been tested for their ability to disrupt PODs, with negative results. The early lytic cycle KSHV protein K-bZIP (also called K8) is localized in PODs (27) but does not disrupt these structures (25). Other KSHV lytic proteins tested such as ORF50, K2, K8.1, K10, K11, ORF59, and ORF65, as well as latent protein ORF73, did not colocalize with PML (26, 27, 47), raising the hypothesis that KSHV might be unique among herpesviruses in that it might not target PODs for destruction.
The KSHV latent protein LANA2, also called viral interferon regulatory factor 3, is exclusively expressed in KSHV-infected B cells, inhibits apoptosis induced by p53 (42) and the double-stranded RNA-dependent protein kinase R (17), and inhibits NF-κB activation (43), interferon regulatory factor 7-mediated interferon signal transduction (24), and virus-mediated transcriptional activity of the IFNA promoter (32). In addition, LANA2 is required for the survival of KSHV-infected PEL cells (52). Remarkably, although LANA2 is present both in the nucleus and the cytoplasm of PEL cells (31, 36), nuclear LANA2 has a speckled expression pattern, similar to that described for PML (12, 42). The aim of this study was to explore the functional interactions between LANA2 and PODs. Here, we show that LANA2 expression leads to a proteasome-dependent disruption of PODs and interferes with the PML-mediated transcriptional repression of survivin, a protein that contributes to malignant progression of KSHV-infected PEL cells. The LANA2-mediated POD disruption is a result of LANA2-mediated increase in the SUMO2-ubiquitin-modified PML protein levels. Moreover, we demonstrate that this POD disruption is largely dependent on the integrity of a SUMO interaction motif (SIM) in LANA2 and the lysine 160 from PML. Finally, we show that LANA2 is directly implicated in the control of PML in PEL cells.
MATERIALS AND METHODS
Cell lines and transfections.
MCF-7 and HEK-293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Gibco), 5 mmol/liter l-glutamine (Invitrogen), and penicillin-streptomycin (Invitrogen). Suspension cultures of KSHV-positive BC-3 cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, 5 mmol/liter l-glutamine, and penicillin-streptomycin. Transfection of MCF-7 and HEK-293 cells was done using FuGene (Roche) following the manufacturer's instructions. For electroporation, BC-3 cells (107 cells) were washed in RPMI 1640 medium without fetal calf serum, resuspended in 250 μl of the same medium, and placed with 20 μg of plasmid DNA in 0.4-cm gap electroporation cuvettes. Cells were transfected using an electroporator (Bio-Rad Laboratories) at 250 V and 960 μF.
Plasmids and reagents.
Plasmids pCDNA-LANA2 and EGFP-LANA2 (where EGFP is enhanced green fluorescent protein) have been previously described (38). HA-PMLIV (where HA is hemagglutinin) plasmid was kindly provided by Jin-Hyun Ahn (Sungkyunkwan University School of Medicine, Suwon, South Korea). pCDNA3-His-PML4 plasmid was provided by Kun-Sang Chang (MD Anderson Cancer Center, Houston, Texas). Plasmids encoding PML mutants were generated by site-directed mutagenesis on pCDNA3-His-PML4 or HA-PMLIV. Plasmid pCDNA-LANA2-474AAAS477 was generated by site-directed mutagenesis on pCDNA-LANA2. Oligonucleotides used in the site-directed mutagenesis are listed in Table 1. For proteasome inhibition, cells were incubated for 16 h in the presence of 20 μM carbobenzoxy-l-leucyl-l-leucl-l-leucinal (MG132; Sigma).
TABLE 1.
Oligonucleotides for site-directed mutagenesis
| Oligonucleotide | Sequence |
|---|---|
| K65RPMLF | 5′-GCAGGCGGAAGCCAGGTGCCCGAAGCTGCTGCC-3′ |
| K65RPMLR | 5′-GGCAGCAGCTTCGGGCACCTGGCTTCCGCCTGC-3′ |
| K160RPMLF | 5′-GGCACACCAGTGGTTCCTCAGGCACGAGGCCCGG-3′ |
| K160RPMLR | 5′-CCGGGCCTCGTGCCTGAGGAACCACTGGTGTGCC-3′ |
| K490RPMLF | 5′-CCCCAGGAAGGTCATCAGGATGGAGTCTGAGG-3′ |
| K490RPMLR | 5′-CCTCAGACTCCATCCTGATGACCTTCCTGGGG-3′ |
| LANA2AAASF | 5′-GCGCTCGTTTGCTTCGGGTGCTGCGGCATCTTCTCTCCGATCTGGC-3′ |
| LANA2AAASR | 5′-GCCAGATCGGAGAGAAGATGCCGCAGCACCCGAAGCAAACGAGCGC-3′ |
GST pull-down.
Glutathione S-transferase (GST) pull-down experiments were performed by incubating in vitro translated and [35S]methionine-labeled LANA2 or the LANA2-474AAAS477 mutant with GST or GST-SUMO1 bound onto glutathione-Sepharose 4B overnight at 4°C in binding buffer containing 50 mM Tris-HCl (pH 7.8), 150 mM NaCl, 0.5 mM EDTA, 0.1% (vol/vol) Triton X-100, 0.1% (vol/vol) Nonidet P-40, 5 mM MgCl2, 10% (vol/vol) glycerol, 50 μM ZnCl2, and protease inhibitor cocktail. The resin was washed four times with 1 ml of binding buffer, and bound proteins were eluted with sodium dodecyl sulfate (SDS) sample buffer and heated at 95°C for 5 min. Proteins were separated on 8% SDS-polyacrylamide gel electrophoresis (PAGE) gels and detected by fluorography.
Western blot analysis and antibodies.
Cells were washed in phosphate-buffered saline (PBS), scraped in SDS-gel loading buffer, and boiled for 5 min. Proteins of total extracts were separated by 8% SDS-PAGE and transferred to nitrocellulose membrane. The following anti-PML antibodies were used: rabbit H-238 from Santa Cruz and rabbit AB1370 from Chemicon. Rabbit anti-Sp100 was from Chemicon International. Mouse anti-actin antibody was from MP Biomedicals. Mouse anti-LANA2 antibody was from Novus Biologicals. Rabbit anti-SUMO2 antibody was from Zymed laboratories. Mouse anti-ubiquitin antibody was from Santa Cruz. Rat anti-HA antibody was from Roche. Anti-survivin antibody and secondary fluorescein isothiocyanate-conjugated anti-mouse antibody were from Sigma. Cy3-conjugated anti-rabbit and anti-rat antibodies were from Chemicon International.
Immunofluorescence staining.
Cells were grown in 24-well plates on coverslips and transfected with the indicated plasmids, and at 36 h after transfection, cells were washed once with PBS and fixed with 2% paraformaldehyde. Then, cells were rinsed twice in PBS and permeabilized in PBS containing 0.1% Tween-20, incubated with primary antibodies, washed in PBS, and incubated with the appropriate secondary antibodies. Chromosomal DNA was stained with DAPI (4′,6′-diamidino-2-phenylindole). Stained cells were mounted on glass slides and examined in a Nikon Eclipse TE2000-U microscope or a confocal Bio-Rad microscope, and images were processed in Adobe Photoshop, version 7.0.
Quantitative reverse transcription-PCR.
RNA was extracted from NIH 3T3 cells and NIH 3T3 cells stably expressing LANA2 using an RNeasy minikit (Qiagen, Hilden, Germany), and reverse transcription-PCR was performed using a reverse transcription system kit (Promega). Real-time quantitative PCR was performed using an ABI 7700 instrument and TaqMan system (Applied Biosystems). TaqMan probes for detection of murine PML and glyceraldehyde-3-phosphate dehydrogenase were purchased from Applied Biosystems.
In vitro expression of proteins.
In vitro transcription/translation of proteins was performed in the presence of [35S]methionine using 1 μg of plasmid DNA and a rabbit reticulocyte-coupled transcription/translation system according to the instructions provided by the manufacturer (Promega).
Purification of His-tagged PML conjugates.
Cells were lysed in 4 ml of 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, and 0.01 M Tris-HCl, pH 8.0, plus 5 mM imidazole and 10 mM β-mercaptoethanol per 75-cm3 flask. Then, lysates were mixed with 50 μl of Ni2+-nitrilotriacetic acid-agarose beads prewashed with lysis buffer and incubated for 2 h at room temperature. The beads were successively washed with the following: 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, and 0.01 M Tris-HCl, pH 8.0, plus 10 mM β-mercaptoethanol; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 6.3, and 10 mM β-mercaptoethanol (buffer A) plus 0.2% Triton X-100; buffer A; and then buffer A with 0.1% Triton X-100. After the last wash with buffer A, the beads were eluted with 200 mM imidazole in 5% SDS, 0.15 M Tris-HCl, pH 6.7, 30% glycerol, and 0.72 M β-mercaptoethanol. The eluates were subjected to SDS-PAGE (8%), and Western blotting was performed as indicated above.
RESULTS
LANA2 causes significant POD alterations.
To examine the effects of LANA2 on the components of PODs, MCF-7 cells were transfected with a plasmid expressing LANA2 (pCDNA-LANA2), LANA2 protein fused to GFP (GFP-LANA2), or GFP alone as a control, and at 36 h after transfection, immunofluorescence analysis of endogenous PML protein was carried out. As expected, PML with the typical nuclear speckled pattern was observed in the control cells, independently of GFP expression (Fig. 1). In contrast, fewer, larger, and more irregular PML dots were observed in cells transfected with GFP-LANA2 (Fig. 1) or LANA2 (see Fig. S1A in the supplemental material). Quantification of the PML dots in more than 200 cells confirmed that more than 80% of the LANA2-expressing cells have considerably fewer PML dots (less than 10 PML dots per cell) than control GFP-expressing cells (Fig. 1). The distribution of other components of the NBs, such as Sp100, in dots is dependent on PML (56). In order to determine whether the expression of this POD-associated protein is also affected by LANA2, MCF-7 cells transfected as described above were stained with anti-Sp100 antibody. As expected, cells transfected with GFP showed nuclear Sp100 dots that were almost undetectable in more than 80% of cells expressing GFP-LANA2 or LANA2 (see Fig. S1B in the supplemental material). Together, these results demonstrate that LANA2 disturbs the nuclear dot distribution of endogenous PML and Sp100 proteins.
FIG. 1.

LANA2 induces the displacement of PML from PODs. MCF-7 cells were transfected as indicated and stained with anti-PML antibody. Arrows indicate cells that are positively expressing LANA2. The number of PML dots per cell as well as the percentage of cells with fewer than 10 PML dots is presented, counting at least 200 transfected cells. ***, P < 0.0005, compared with cells not expressing LANA2 (Student's test).
The disruption of PODs by LANA2 is mediated by a proteasome-dependent mechanism.
To determine whether LANA2 induced PML protein loss by reducing the PML RNA levels, we analyzed the PML mRNA from NIH 3T3 cells stably expressing LANA2 or an empty vector (pCDNA) (37). The results showed that PML mRNA levels remained constant after expression of LANA2 (data not shown). To test the possibility that POD disruption occurred via a proteasome-dependent process, 24 h after transfection of MCF-7 cells with pCDNA-LANA2 or the GFP-LANA2 expression plasmid, cells were treated with the proteasome inhibitor MG132 for 16 h, and endogenous PML was then analyzed by immunofluorescence staining. MG132 treatment abrogated LANA2-induced displacement of endogenous PML from PODs (Fig. 2A; see also Fig. S2A in the supplemental material). In addition, there was no obvious alteration in Sp100 staining in the majority of the cells treated with MG132 (see Fig. S2B in the supplemental material). We were not able to detect any endogenous PML protein in MCF-7 cells using the anti-PML antibodies we tested by Western blotting. For this reason, PML protein stability was determined after transfection of MCF-7 cells with a plasmid encoding HA-PMLIV together with pCDNA or pCDNA-LANA2. At 24 h of transfection, MG132 was added to half of the cells for an additional 16 h, when treatment with cycloheximide was performed in both MG132-treated and untreated cells. HA-PML protein levels were clearly reduced in cells expressing LANA2 at any time after cycloheximide treatment (Fig. 2B, left panel, compare lanes 1 to 4, 2 to 5, and 3 to 6), and this reduction was abrogated in the presence of MG132 (Fig. 2B, right panel). These results demonstrate that LANA2-induced disruption of PODs is mediated by a proteasome-dependent PML degradation mechanism.
FIG. 2.

Disruption of PODs and degradation of PML by LANA2 are mediated by the proteasome. (A) Disruption of PODs by LANA2 is mediated by the proteasome. MCF-7 cells were transfected and treated with the proteasome inhibitor MG132 as indicated. Endogenous PML was detected by immunofluorescence using anti-PML antibody. The weaker speckled pattern detected in cells expressing GFP-LANA2 is recovered after MG132 treatment. Arrows indicate cells that are positively expressing LANA2. The percentage of transfected cells with fewer than 10 PML dots is presented, counting at least 200 transfected cells. (B) LANA2 expression induces the degradation of HA-PML by a proteasome-dependent mechanism. Cells cotransfected with HA-PMLIV and pCDNA or pCDNA-LANA2 were treated with cycloheximide (CHX) in the presence or absence of the proteasome inhibitor MG312 and harvested at the indicated time points. Cell extracts were then analyzed by Western blotting with the indicated antibodies.
PML residue lysine 160 is required for the disruption of PODs induced by LANA2.
Sumoylation of the human PML is necessary for the formation of PODs (5). Although PML contains three major sites of SUMO modification, only the sumoylation status of PML Lys 160 regulates the nuclear trafficking of PODs (30, 44), the sensitivity to arsenic exposure (30), and the degradation of PML after HSV type 1 (HSV-1) ICP0 protein expression (8). To determine whether sumoylation of PML lysine residues is required for LANA2-mediated POD disruption, MCF-7 cells were cotransfected with plasmids encoding PML mutants in different lysine residues known to covalently interact with SUMO together with an empty vector or the pCDNA-LANA2 plasmid. The integrity of the nuclear dots detected at 36 h after transfection with PML wild type (WT) or each PML mutant was then analyzed by immunofluorescence staining. As expected, PML WT formed multiple punctate foci in the nucleus of transfected cells (Fig. 3). Quantification of the PML WT dots in more than 150 cells confirmed that 74% of the cells cotransfected with pCDNA showed more than 50 PML foci per cell while only 35% of the cells coexpressing LANA2 showed similar staining. In addition, around 19% of the cells expressing LANA2 did not show PML WT dots, and more than 46% showed reorganized PML distribution with fewer, larger, and less regular dots located mainly at the periphery of the cell nucleus (Fig. 3). As previously described, a PML mutant in which three arginines replace the three lysine residues that covalently bind to SUMO formed only a few strong dots (44), and this distribution was not affected by LANA2 expression (cells showed around 8 dots per cell independently of the expression of LANA2) (Fig. 3). Furthermore, cells transfected with PML containing the double mutation K160/490R or LK65/160R revealed similar staining both in cells that expressed LANA2 and in those that did not (a mean of 10 and 6 dots per cell, respectively). However, staining and counting of the dots detected in at least 200 cells transfected with the PMLK65/490R double mutant revealed that nuclear dots were absent or clearly reduced in more than 73% of cells expressing LANA2 (cells expressing LANA2 displayed a mean of four PML dots per cell in comparison with nine dots per cell observed in pCDNA-transfected cells) (Fig. 3). These results indicate that the lysine 160 of PML is required for the disruption of PODs induced by LANA2.
FIG. 3.

PML residue lysine 160 is required for the disruption of PODs induced by LANA2. MCF-7 cells were transiently transfected with pCDNA or pCDNA-LANA2 plasmid together with plasmids that encoded PML WT or the indicated PML mutants and stained for LANA2 (green) and PML (red). PML-3KR, PML with arginine residues replacing the three lysine residues that covalently bind to SUMO.
LANA2 increases the levels of SUMO2-ubiquitin-modified PML.
PML function is regulated by covalent modifications such as the attachment of SUMO (7), which appears to be crucial for the PML-mediated recruitment of other proteins to PODs (22, 56). SUMO1 modification can be implicated in the stabilization of proteins (14) and disruption of the PML NBs by some viral proteins (such as HSV-1 ICP0, CMV IE1, and Epstein-Barr virus BZFL1 proteins) correlates with the abrogation of PML sumoylation (1, 35). This suggested that posttranslational modification of PML with SUMO1 may stabilize these structures (35). However, recent studies have established that SUMO modification can also serve as a proteolytic targeting signal that is recognized by ubiquitin ligases for SUMO conjugates (11, 39, 46, 49, 53) that bind to polysumoylated proteins and prevent their accumulation. In this sense, degradation of PML upon arsenic treatment involves the formation of poly-SUMO2/3-PML chains that are then recognized and ubiquitinated by RNF4 (29, 48, 51). Based on these data, we decided to analyze the effect of LANA2 expression on the conjugation of SUMO2 and ubiquitin to PML. HEK-293 cells were cotransfected with pCDNA-His-PML4 together with a plasmid encoding LANA2 or an empty vector, in the presence or absence of SUMO2. At 60 h after transfection, His-tagged PML was purified using nickel-bound beads under denaturing conditions, and PML modifications were determined by Western blot analysis using antibodies to PML, SUMO2, or ubiquitin. As expected, transfection of SUMO2 in combination with PML resulted in a clear upregulation of the SUMO2-modified forms of His-tagged PML (Fig. 4A, compare lanes 1 and 3 of first and second panels) that resulted in an increase in the ubiquitin-conjugated PML (Fig. 4A, compare lanes 1 and 3 of the third panel). Under these experimental conditions, expression of LANA2 induced a clear reduction in the PML protein (Fig. 4A, compare lanes 1 and 2 in the first panel) to almost undetectable levels in the cells cotransfected with SUMO2 (Fig. 4A, compare lanes 3 and 4 in the first panel). Western blot analysis of the His-purified proteins using antibodies to SUMO2 or ubiquitin revealed that the expression of LANA2 resulted in a significant upregulation in both SUMO2-modified PML (Fig. 4A, compare lanes 1 and 2 in the second panel) and ubiquitin-modified PML (Fig. 4A, compare lanes 1 and 2 in the third panel). When cells were cotransfected with both LANA2 and SUMO2, a slight decrease in the levels of SUMO2-modified PML (Fig. 4A, compare lanes 3 and 4 in the second panel) and ubiquitin-modified PML (Fig. 4A, compare lanes 3 and 4 in the third panel) was observed, suggesting that the upregulation of SUMO2-ubiquitin conjugates of PML by LANA2 accelerates its degradation. To further verify whether LANA2 increases the modification of PML by SUMO2 and ubiquitin, HEK-293 cells were cotransfected with pCDNA-His-PML4 and increasing amounts of the LANA2 expression plasmid. Analysis of total PML and SUMO2- or ubiquitin-modified PML after 36 h of transfection and nickel affinity purification was carried out. Expression of LANA2 resulted in the appearance of high-molecular-mass forms of PML (Fig. 4B, first panel) that resemble the SUMO2-modified forms of PML detected after SUMO2 overexpression (Fig. 4A, black arrows in first panel), in a dose-response fashion. Under these experimental conditions, LANA2 also caused a slight reduction in the major PML band (Fig. 4B, first panel; the band [white arrow] presumably represents unmodified PML). Western blot analysis of the His-purified proteins using antibodies to SUMO2 or ubiquitin demonstrated that LANA2 caused a marked upregulation of both SUMO2- and ubiquitin-modified PML in a dose-response fashion (Fig. 4B, second and third panels). All together, these results suggest a model whereby PML degradation is caused by the increase in the SUMO-dependent ubiquitylation of PML induced by LANA2.
FIG. 4.

Upregulation of SUMO2 and ubiquitin modification of PML by LANA2. (A) MCF-7 cells were transfected with the indicated plasmids, and at 60 h after transfection His-tagged PML protein was purified from the cell extracts by nickel affinity chromatography before analysis. Purified extracts were then analyzed by Western blotting (WB) using antibodies to SUMO2, PML, or ubiquitin (Ub). Input extracts were incubated with anti-LANA2 or anti-actin antibodies. A shorter exposition of the gel is shown in the right-hand panel. (B) MCF-7 cells were transfected with the indicated plasmids, and 36 h after transfection His-tagged PML protein was purified from the cell extracts by nickel affinity chromatography before analysis. Purified extracts were analyzed by Western blotting using antibodies to SUMO2, PML, or ubiquitin. Input extracts were incubated with anti-LANA2 or anti-actin antibodies.
POD disruption depends on the integrity of a LANA2 SIM.
Since modification of PML with SUMO has been demonstrated to be important in the regulation of POD assembly and since lysine 160 in PML is required for the degradation of PML by LANA2, we reasoned that the interaction of LANA2 with SUMO pathways could be required for the disruption of PODs by LANA2. SIMs are expected to play a crucial role in regulating sumoylated proteins. The best-studied SIM domain contains three hydrophobic residues (typically valine, leucine, or isoleucine) in a sequence of four amino acids [(V/L/I)(V/L/I) X(V/L/I) or (V/L/I)X(V/L/I)(V/L/I)] (20, 45). LANA2 protein is sumoylated in several lysine residues (unpublished observations) and also presents a domain that resembles a SIM domain at position 474 to 477 composed of four large nonpolar (LVIV) amino acids. To test whether LANA2 interacts with SUMO1 through its proposed SIM, we generated a LANA2-474AAAS477 mutant by replacing the four large nonpolar amino acids (LVIV) with small nonpolar amino acids (AAAS), as previously described for other SIM-containing proteins. LANA2 WT and the LANA2-474AAAS447 mutant were then synthesized using rabbit reticulocyte lysates and incubated with GST-SUMO1 or GST beads. In vitro translated LANA2 efficiently bound to the GST-SUMO1 resin but did not adhere to the GST resin (Fig. 5A). LANA2-474AAAS477 did not bind to either GST or GST-SUMO1. These results confirmed the functionality of the SIM in LANA2. We then investigated whether the SIM domain of LANA2 is required for the disruption of PODs by LANA2. Staining of endogenous PML or Sp100 at 36 h after transfection of the LANA2-474AAAS477 mutant revealed a distribution of dots very similar to the one observed when the LANA2 mutant was not expressed, indicating that the SIM LANA2 mutant was essentially inactive in reducing the numbers of both PML and Sp100 dots per nuclei (Fig. 5B). These results strongly indicate an essential role for the SIM in LANA2-mediated disruption of the PODs.
FIG. 5.

SIM domain of LANA2 is required for POD disruption. (A) LANA2 WT but not the LANA2 mutant in the SIM domain (LANA2-474AAA477) binds SUMO1 in vitro. 35S-labeled LANA2 or the LANA2-474AAAS477 mutant generated by in vitro transcription/translation was incubated with resins containing GST or GST-SUMO1. The resins were subsequently washed extensively, and the eluates were resolved by 8% SDS-PAGE and visualized by fluorography. Input samples represent 10% of the total amount of labeled protein incubated with the matrices. (B) Expression of the mutant of LANA2 in the SIM domain does not disrupt PODs. MCF-7 cells transfected with LANA2 WT or the LANA2 mutant in the SIM domain (LANA2-474AAAS477) were analyzed by immunofluorescence with the indicated antibodies. Arrows indicate cells that are positively expressing LANA2. The percentage of transfected cells with fewer than 10 PML dots is presented, counting at least 200 transfected cells.
LANA2 expression inhibits the transcriptional repression of the survivin promoter mediated by PML.
PML4 downregulates survivin protein via repression of its promoter (54). Since LANA2 expression led to PML degradation, the effect of LANA2 on the regulation of survivin was analyzed. MCF-7 cells were transfected with pCDNA, LANA2, or the SIM LANA2 mutant plasmids, and cells were left untreated or were treated with MG132. At 48 h after transfection, cell extracts were subjected to Western blotting using anti-survivin antibody. Expression of LANA2 induced an increase in the survivin levels, which was not detected in cells transfected with the SIM LANA2 mutant (Fig. 6). Survivin levels were maintained at high levels after treatment with MG132, independently of the expression of LANA2 WT or LANA2 mutant, due to the inhibition of survivin regulation by the proteasome (55). Moreover, we observed an inhibition of the repression of the survivin promoter mediated by PML after expression of LANA2 WT but not the SIM LANA2 mutant (see Fig. S3 in the supplemental material).
FIG. 6.
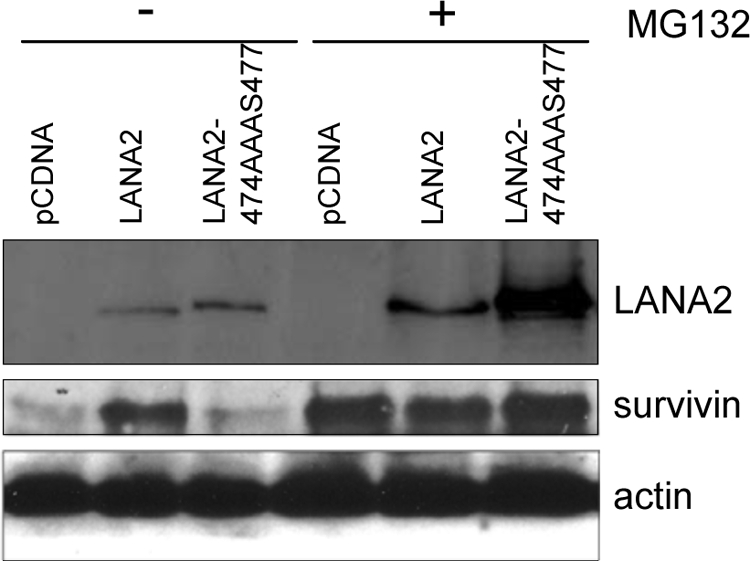
Survivin levels are upregulated in cells expressing LANA2 WT but not the LANA2-474AAAS477 mutant. MCF-7 cells were transfected with the indicated plasmids and treated (+) or not (−) with MG132. At 48 h after transfection, protein extracts were analyzed by Western blotting with the indicated antibodies.
Inhibition of LANA2 expression induces an increase in the PML protein levels.
Anti-LANA2 short hairpin RNA (shRNA) expression vectors were generated in the pSuper background (sh-LANA2) and were shown to effectively block LANA2 expression when cotransfected with the GFP-LANA2 expression vector into HeLa cells (see Fig. S4 in the supplemental material). Since knockdown of LANA2 expression reduced the viability of PEL cells (52), only a short-time and transient LANA2 interference experiment in PEL cells could be done. KSHV-infected BC-3 PEL cells were transfected with either sh-LANA2 vector or with a vector expressing randomized shRNA (sh-control), and 72 h after transfection, the expression of LANA2 and PML was analyzed by Western blotting. Relative levels of LANA2 and PML were then quantified by densitometry analysis of blots corresponding to three independent experiments. As shown in Fig. 7, transfection of sh-LANA2 into BC-3 cells resulted in a statistically significant reduction of LANA2 expression, together with a small but statistically significant increase in the PML levels compared with cells transfected with the sh-control. These results suggest that LANA2 is directly implicated in the control of PML protein in the KSHV-infected PEL cells.
FIG. 7.
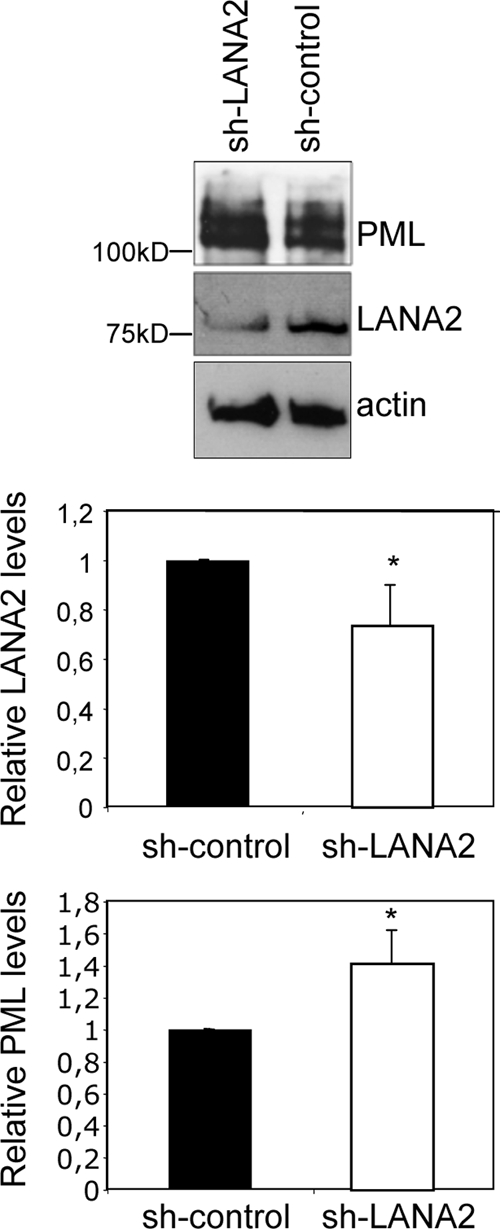
Silencing of LANA2 in KSHV-infected PEL cells induces an increase in PML levels. BC-3 PEL cells were transfected with the anti-LANA2 shRNA expression vector (sh-LANA2) or a randomized shRNA vector (sh-control), and 72 h after transfection, the expression levels of LANA2 and PML were analyzed by Western blotting. The relative abundance of LANA2 and PML was then quantitated by densitometric analysis of the blots obtained in three different experiments. All values were normalized to the actin loading control and are represented as a ratio of the control. *, P < 0.05, compared with sh-control-transfected cells (Student's test).
DISCUSSION
PML is a ring finger interferon-inducible protein typically concentrated within discrete speckled nuclear structures called PODs (21). In addition to PML, PODs contains several other proteins, such as Sp100, Sp140, ISG20, CBP, pRb, Daxx, BLM, and p53 (34). PML acts as a tumor suppressor, antagonizing initiation, promotion, and progression of tumors of various histological origins and, particularly, of the lympho-hematopoietic compartment (50). PML is also implicated in the regulation of infection by a variety of RNA viruses (41), adenoviruses (15), and human CMV (2). Apart from being disorganized in a number of human diseases, PODs have been shown to be highly sensitive to environmental stimuli such as response to stress or interferon as well as to viral infection (33). In this sense, many studies have shown that different viral proteins induce POD disruption (18). Members of all alpha-, beta- and gammaherpesvirus subfamilies contain proteins that counteract this host defense mechanism, suggesting that this function may be universally important for efficient herpesvirus replication. However, no information about POD-modifying proteins is available for the gammaherpesvirus KSHV. Several groups have analyzed the putative disruption of PODs by different KSHV proteins, with negative results. However, to our knowledge the regulation of PODs by the latent protein LANA2 has not been previously examined. Our results show that LANA2 expression induces the displacement of PML or Sp100 from PODs and the degradation of PML. In addition, we demonstrate that the disruption of PODs induced by LANA2 is dependent on proteasome activity since it can be prevented by MG132. Immunostaining of the PML dots after transfection of the PML mutants in the lysine residues susceptible to sumoylation indicates that the PML residue lysine 160 is required for the disruption of PODs induced by LANA2. Lys 160 sumoylation is essential for many properties of PML, including ICP0-induced catabolism or the proteasome-dependent degradation triggered by arsenic (8, 30). Our results suggest that LANA2 may induce POD disruption by similar molecular mechanisms. Analysis of the modifications of PML in vivo after expression of LANA2 reveals that LANA2 increases the levels of SUMO2- and ubiquitin-modified PML in a dose-response fashion. Given that PML is degraded by SUMO-dependent polyubiquitination, we speculate that LANA2 may destabilize PML, promoting the formation of ubiquitin-SUMO conjugates.
The physiological consequences of SUMO modification are typically mediated by effector proteins that recognize SUMO through SIM domains. A SUMO-binding motif has been identified in different proteins associated with PODs. GST pull-down assays using GST-SUMO1 and in vitro translated LANA2 protein demonstrate LANA2 binding to SUMO1 in a noncovalent fashion, indicating that LANA2 may interact with different sumoylated proteins through a SIM domain. The presence of a functional SIM has been found in cellular and viral proteins. Little is known regarding the biological significance of these noncovalent interactions. However, for some proteins such as HIPK2, PKM, or RNF4, the noncovalent binding of SUMO1 is needed for the induction of structural changes in PODs or for POD disruption (16, 48). Similarly, expression of a LANA2 mutant in the putative SIM domain did not disrupt PODs, confirming that the SIM in LANA2 is functional and is required for disruption of PODs.
One of the PML4 activities that contributes to its role as a cell growth and tumor suppressor is the repression of the survivin promoter. It is well known that altered expression of survivin is a common event associated with the pathogenesis of human cancer. Survivin is overexpressed in many transformed cell lines and in common cancers and directly contributes to malignant progression of PEL (3, 4, 23). Our results show that LANA2 inhibits the transcriptional repression of the survivin promoter mediated by PML, suggesting that disruption of PODs by LANA2 may contribute to cellular transformation and carcinogenesis. In addition, inhibition of the transcriptional repressor activity of PML by WT LANA2 but not the SIM LANA2 mutant implies that the transcriptional regulation by LANA2 is directly associated with its ability to disrupt PODs.
Stable expression of an shRNA targeting LANA2 in PEL cells is associated with reduced proliferation and viability, indicating that expression of LANA2 is absolutely required for the survival of KSHV-infected primary effusion lymphoma cells (52; also our unpublished observations) and hampering the demonstration of a direct role for LANA2 in the control of the PML NBs in the KSHV-infected PEL cells. Our results demonstrate an increase in PML levels after transient knockdown of LANA2 expression in PEL cells, pointing to the control of PML as required for the maintenance of PEL viability by LANA2.
Together, these results strongly suggest that, similar to other transforming and oncogenic viruses, KSHV has developed strategies to neutralize the PML tumor suppressor function, and this may represent a novel mechanism by which LANA2 contributes to the transformed phenotype mediated by KSHV.
Supplementary Material
Acknowledgments
We thank Jin-Hyun Ahn and Kun-Sang Chang for providing reagents and Manuel Collado for valuable criticism of the manuscript.
L.M.V. is supported by Comunidad de Madrid. M.C. is supported by Fundación Botín and Juan de la Cierva Program. J.G.-S. is supported by the IFARHU-SENACYT Program from Panama. P.G. is funded by the JAE CSIC Program. M.S.R. and F.L.O. are funded by the Ministerio de Educación y Ciencia grant BFU 2005-04091, Fondo de Investigaciones Sanitarias, CIBERhed. C.R. is funded by Spanish Ministry of Education and Science (BIO2005-00599 and BFU2008-03784) and Fundación de Investigación Médica Mútua Madrileña.
Footnotes
Published ahead of print on 24 June 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adamson, A. L., and S. Kenney. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 752388-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J. H., and G. S. Hayward. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 27439-55. [DOI] [PubMed] [Google Scholar]
- 3.Ambrosini, G., C. Adida, and D. C. Altieri. 1997. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3917-921. [DOI] [PubMed] [Google Scholar]
- 4.Aoki, Y., G. M. Feldman, and G. Tosato. 2003. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 1011535-1542. [DOI] [PubMed] [Google Scholar]
- 5.Bernardi, R., and P. P. Pandolfi. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 81006-1016. [DOI] [PubMed] [Google Scholar]
- 6.Bernardi, R., A. Papa, and P. P. Pandolfi. 2008. Regulation of apoptosis by PML and the PML-NBs. Oncogene 276299-6312. [DOI] [PubMed] [Google Scholar]
- 7.Boddy, M. N., K. Howe, L. D. Etkin, E. Solomon, and P. S. Freemont. 1996. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13971-982. [PubMed] [Google Scholar]
- 8.Boutell, C., A. Orr, and R. D. Everett. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 778686-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvalho, T., J. S. Seeler, K. Ohman, P. Jordan, U. Pettersson, G. Akusjarvi, M. Carmo-Fonseca, and A. Dejean. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 13145-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelbi-Alix, M. K., and H. de The. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18935-941. [DOI] [PubMed] [Google Scholar]
- 11.Cheng, J., X. Kang, S. Zhang, and E. T. Yeh. 2007. SUMO-specific protease 1 is essential for stabilization of HIF1α during hypoxia. Cell 131584-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniel, M. T., M. Koken, O. Romagne, S. Barbey, A. Bazarbachi, M. Stadler, M. C. Guillemin, L. Degos, C. Chomienne, and H. de The. 1993. PML protein expression in hematopoietic and acute promyelocytic leukemia cells. Blood 821858-1867. [PubMed] [Google Scholar]
- 13.Desbois, C., R. Rousset, F. Bantignies, and P. Jalinot. 1996. Exclusion of Int.-6 from PML nuclear bodies by binding to the HTLV-I Tax oncoprotein. Science 273951-953. [DOI] [PubMed] [Google Scholar]
- 14.Desterro, J. M., M. S. Rodriguez, and R. T. Hay. 1998. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 2233-239. [DOI] [PubMed] [Google Scholar]
- 15.Doucas, V., A. M. Ishov, A. Romo, H. Juguilon, M. D. Weitzman, R. M. Evans, and G. G. Maul. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10196-207. [DOI] [PubMed] [Google Scholar]
- 16.Engelhardt, O. G., C. Boutell, A. Orr, E. Ullrich, O. Haller, and R. D. Everett. 2003. The homeodomain-interacting kinase PKM (HIPK-2) modifies ND10 through both its kinase domain and a SUMO-1 interaction motif and alters the posttranslational modification of PML. Exp. Cell Res. 28336-50. [DOI] [PubMed] [Google Scholar]
- 17.Esteban, M., M. A. Garcia, E. Domingo-Gil, J. Arroyo, C. Nombela, and C. Rivas. 2003. The latency protein LANA2 from Kaposi's sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 841463-1470. [DOI] [PubMed] [Google Scholar]
- 18.Everett, R. D. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 207266-7273. [DOI] [PubMed] [Google Scholar]
- 19.Everett, R. D., and M. K. Chelbi-Alix. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89819-830. [DOI] [PubMed] [Google Scholar]
- 20.Hecker, C. M., M. Rabiller, K. Haglund, P. Bayer, and I. Dikic. 2006. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 28116117-16127. [DOI] [PubMed] [Google Scholar]
- 21.Hodges, M., C. Tissot, K. Howe, D. Grimwade, and P. S. Freemont. 1998. Structure, organization, and dynamics of promyelocytic leukemia protein nuclear bodies. Am. J. Hum. Genet. 63297-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishov, A. M., A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss III, and G. G. Maul. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito, T., K. Shiraki, K. Sugimoto, T. Yamanaka, K. Fujikawa, M. Ito, K. Takase, M. Moriyama, H. Kawano, M. Hayashida, T. Nakano, and A. Suzuki. 2000. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology 311080-1085. [DOI] [PubMed] [Google Scholar]
- 24.Joo, C. H., Y. C. Shin, M. Gack, L. Wu, D. Levy, and J. U. Jung. 2007. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 818282-8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katano, H., K. Ogawa-Goto, H. Hasegawa, T. Kurata, and T. Sata. 2001. Human-herpesvirus-8-encoded K8 protein colocalizes with the promyelocytic leukemia protein (PML) bodies and recruits p53 to the PML bodies. Virology 286446-455. [DOI] [PubMed] [Google Scholar]
- 26.Katano, H., Y. Sato, H. Itoh, and T. Sata. 2001. Expression of human herpesvirus 8 (HHV-8)-encoded immediate early protein, open reading frame 50, in HHV-8-associated diseases. J. Hum. Virol. 496-102. [PubMed] [Google Scholar]
- 27.Katano, H., Y. Sato, T. Kurata, S. Mori, and T. Sata. 2000. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 269335-344. [DOI] [PubMed] [Google Scholar]
- 28.Korioth, F., G. G. Maul, B. Plachter, T. Stamminger, and J. Frey. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 229155-158. [DOI] [PubMed] [Google Scholar]
- 29.Lallemand-Breitenbach, V., M. Jeanne, S. Benhenda, R. Nasr, M. Lei, L. Peres, J. Zhou, J. Zhu, B. Raught, and H. de The. 2008. Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 10547-555. [DOI] [PubMed] [Google Scholar]
- 30.Lallemand-Breitenbach, V., J. Zhu, F. Puvion, M. Koken, N. Honore, A. Doubeikovsky, E. Duprez, P. P. Pandolfi, E. Puvion, P. Freemont, and H. de The. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 1931361-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubyova, B., M. J. Kellum, A. J. Frisancho, and P. M. Pitha. 2004. Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 2797643-7654. [DOI] [PubMed] [Google Scholar]
- 32.Lubyova, B., and P. M. Pitha. 2000. Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J. Virol. 748194-8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maul, G. G. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20660-667. [DOI] [PubMed] [Google Scholar]
- 34.Maul, G. G., D. Negorev, P. Bell, and A. M. Ishov. 2000. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129278-287. [DOI] [PubMed] [Google Scholar]
- 35.Muller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 735137-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munoz-Fontela, C., M. Collado, E. Rodriguez, M. A. Garcia, A. Alvarez-Barrientos, J. Arroyo, C. Nombela, and C. Rivas. 2005. Identification of a nuclear export signal in the KSHV latent protein LANA2 mediating its export from the nucleus. Exp. Cell Res. 31196-105. [DOI] [PubMed] [Google Scholar]
- 37.Munoz-Fontela, C., L. Marcos-Villar, F. Hernandez, P. Gallego, E. Rodriguez, J. Arroyo, S. J. Gao, J. Avila, and C. Rivas. 2008. Induction of paclitaxel resistance by the Kaposi's sarcoma-associated herpesvirus latent protein LANA2. J. Virol. 821518-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz-Fontela, C., E. Rodriguez, C. Nombela, J. Arroyo, and C. Rivas. 2003. Characterization of the bipartite nuclear localization signal of protein LANA2 from Kaposi's sarcoma-associated herpesvirus. Biochem. J. 374545-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prudden, J., S. Pebernard, G. Raffa, D. A. Slavin, J. J. Perry, J. A. Tainer, C. H. McGowan, and M. N. Boddy. 2007. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 264089-4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quignon, F., F. De Bels, M. Koken, J. Feunteun, J. C. Ameisen, and H. de The. 1998. PML induces a novel caspase-independent death process. Nat. Genet. 20259-265. [DOI] [PubMed] [Google Scholar]
- 41.Regad, T., and M. K. Chelbi-Alix. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 207274-7286. [DOI] [PubMed] [Google Scholar]
- 42.Rivas, C., A. E. Thlick, C. Parravicini, P. S. Moore, and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo, T., J. Park, C. Lim, and J. Choe. 2004. Inhibition of nuclear factor κB activity by viral interferon regulatory factor 3 of Kaposi's sarcoma-associated herpesvirus. Oncogene 236146-6155. [DOI] [PubMed] [Google Scholar]
- 44.Shen, T. H., H. K. Lin, P. P. Scaglioni, T. M. Yung, and P. P. Pandolfi. 2006. The mechanisms of PML-nuclear body formation. Mol. Cell 24331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song, J., L. K. Durrin, T. A. Wilkinson, T. G. Krontiris, and Y. Chen. 2004. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 10114373-14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun, H., J. D. Leverson, and T. Hunter. 2007. Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 264102-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szekely, L., C. Kiss, K. Mattsson, E. Kashuba, K. Pokrovskaja, A. Juhasz, P. Holmvall, and G. Klein. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J. Gen. Virol. 802889-2900. [DOI] [PubMed] [Google Scholar]
- 48.Tatham, M. H., M. C. Geoffroy, L. Shen, A. Plechanovova, N. Hattersley, E. G. Jaffray, J. J. Palvimo, and R. T. Hay. 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10538-546. [DOI] [PubMed] [Google Scholar]
- 49.Uzunova, K., K. Gottsche, M. Miteva, S. R. Weisshaar, C. Glanemann, M. Schnellhardt, M. Niessen, H. Scheel, K. Hofmann, E. S. Johnson, G. J. Praefcke, and R. J. Dohmen. 2007. Ubiquitin-dependent proteolytic control of SUMO conjugates. J. Biol. Chem. 28234167-34175. [DOI] [PubMed] [Google Scholar]
- 50.Wang, Z. G., D. Ruggero, S. Ronchetti, S. Zhong, M. Gaboli, R. Rivi, and P. P. Pandolfi. 1998. PML is essential for multiple apoptotic pathways. Nat. Genet. 20266-272. [DOI] [PubMed] [Google Scholar]
- 51.Weisshaar, S. R., K. Keusekotten, A. Krause, C. Horst, H. M. Springer, K. Gottsche, R. J. Dohmen, and G. J. Praefcke. 2008. Arsenic trioxide stimulates SUMO-2/3 modification leading to RNF4-dependent proteolytic targeting of PML. FEBS Lett. 5823174-3178. [DOI] [PubMed] [Google Scholar]
- 52.Wies, E., Y. Mori, A. Hahn, E. Kremmer, M. Sturzl, B. Fleckenstein, and F. Neipel. 2008. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 111320-327. [DOI] [PubMed] [Google Scholar]
- 53.Xie, Y., O. Kerscher, M. B. Kroetz, H. F. McConchie, P. Sung, and M. Hochstrasser. 2007. The yeast Hex3 · Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J. Biol. Chem. 28234176-34184. [DOI] [PubMed] [Google Scholar]
- 54.Xu, Z. X., R. X. Zhao, T. Ding, T. T. Tran, W. Zhang, P. P. Pandolfi, and K. S. Chang. 2004. Promyelocytic leukemia protein 4 induces apoptosis by inhibition of survivin expression. J. Biol. Chem. 2791838-1844. [DOI] [PubMed] [Google Scholar]
- 55.Zhao, J., T. Tenev, L. M. Martins, J. Downward, and N. R. Lemoine. 2000. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J. Cell Sci. 1134363-4371. [DOI] [PubMed] [Google Scholar]
- 56.Zhong, S., S. Muller, S. Ronchetti, P. S. Freemont, A. Dejean, and P. P. Pandolfi. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 952748-2752. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.