

Abstract
The entry process of the avian sarcoma and leukosis virus (ASLV) family of retroviruses requires first a specific interaction between the viral surface (SU) glycoproteins and a receptor on the cell surface at a neutral pH, triggering conformational changes in the viral SU and transmembrane (TM) glycoproteins, followed by exposure to low pH to complete fusion. The ASLV TM glycoprotein has been proposed to adopt a structure similar to that of the Ebola virus GP2 protein: each contains an internal fusion peptide flanked by cysteine residues predicted to be in a disulfide bond. In a previous study, we concluded that the cysteines flanking the internal fusion peptide in ASLV TM are critical for efficient function of the ASLV viral glycoproteins in mediating entry. In this study, replication-competent ASLV mutant subgroup A [ASLV(A)] variants with these cysteine residues mutated were constructed and genetically selected for improved replication capacity in chicken fibroblasts. Viruses with single cysteine-to-serine mutations reverted to the wild-type sequence. However, viruses with both C9S and C45S (C9,45S) mutations retained both mutations and acquired a second-site mutation that significantly improved the infectivity of the genetically selected virus population. A charged-amino-acid second-site substitution in the TM internal fusion peptide at position 30 is preferred to rescue the C9,45S mutant ASLV(A). ASLV(A) envelope glycoproteins that contain the C9,45S and G30R mutations bind the Tva receptor at wild-type levels and have improved abilities to trigger conformational changes and to form stable TM oligomers compared to those of the C9,45S mutant glycoprotein.
All retroviruses have envelope glycoproteins that interact with a receptor protein on the cell surface to initiate entry (18, 36). The viral glycoprotein is synthesized as a precursor polyprotein consisting of the surface (SU) glycoprotein, which contains the domains that bind with the cellular receptor, and the transmembrane (TM) glycoprotein, which tethers the protein to the viral surface and contains the domains responsible for fusion of the viral and cellular membranes (32). After synthesis, the precursor viral glycoproteins form trimers through the interaction of the TM domains. The SU and TM domains are then cleaved by a cellular protease, forming a mature, metastable complex capable of mediating viral entry. A specific receptor protein interaction with the SU domain of the mature Env is required to initiate a conformational change in the trimer, separating the globular SU domains to allow the TM glycoproteins to form a structure that projects the fusion peptide toward the target membrane. Two domains in TM, the N-terminal heptad repeat and the C-terminal heptad repeat, are critical for the formation of the extended structure (13, 31)}. The fusion peptide is thought to interact with a target membrane irreversibly, forming an extended prehairpin TM oligomer structure anchored in both the viral and target membranes (35). The cooperation of several of these extended prehairpin TM oligomer structures is most likely required to complete fusion. The viral and target membranes are brought into close proximity when the C-terminal heptad repeats fold back into grooves formed by the N-terminal heptad repeats, forming presumably the most stable TM structure, the six-helix bundle (6HB). Fusion of the membranes proceeds through the initial mixing of the outer lipid leaflets, hemifusion, followed by initial fusion pore formation, pore widening, and the completion of fusion. The 6HB may undergo some additional structural rearrangement in order to bring the fusion peptide and membrane-spanning domain of TM into close proximity to form the final trimeric hairpin structure (22, 24, 33).
Until recently, the triggering of class I virus fusion proteins was thought to occur by one of two mechanisms (13, 35, 36). In one mechanism, the viral glycoproteins interact with receptors on the cell surface, resulting in the trafficking of the virion into an endocytic compartment, followed by the triggering of structural rearrangements in the viral glycoproteins to initiate fusion by exposure to low pH (e.g., influenza virus hemagglutinin [HA]). In a second entry mechanism, the interaction of the viral glycoproteins with receptors on the cell surface in a neutral pH environment triggers the structural rearrangements in the viral glycoproteins directly, initiating viral entry. Retroviruses predominately employ the second entry mechanism, although two cellular protein receptors may be required to complete the conformational changes in the viral glycoproteins necessary to complete entry (e.g., human immunodeficiency virus type 1). However, the entry process of the avian sarcoma and leukosis virus (ASLV) family of retroviruses demonstrates a third entry mechanism for the action of class I virus fusion proteins (25). ASLV entry requires both a specific interaction between the viral glycoproteins and receptors at the cell surface at neutral pH, triggering initial conformational changes in the viral glycoproteins, and a subsequent exposure to low pH to complete fusion (2, 3, 22-24).
The fusion peptides of ASLVs are not at the N terminus of the cleaved TM, as in all other retroviral TM proteins, but in a proposed internal loop (TM residues 22 to 37) flanked by two cysteine residues (residues C9 and C45) (Fig. 1). The ASLV TM glycoprotein has been proposed to adopt a structure similar to that of the Ebola virus GP2 protein: both contain an internal fusion peptide flanked by cysteine residues predicted to be in a disulfide bond (10). Other viruses contain internal fusion peptides also predicted to be in looped structures (35). In a study to determine if the cysteines that flank the ASLV fusion peptide are required for function, mutant ASLV Env proteins were constructed with one or both of these cysteines changed to serine (C9S, C45S, or C9S C45S [C9,45S]) (8). The mutant subgroup A ASLV [ASLV(A)] Env proteins were expressed, processed, and incorporated into virions at levels similar to those of wild-type (WT) ASLV(A) Env. The mutant and WT ASLV(A) Env proteins bound the Tva receptor with similar affinities. However, murine leukemia virus (MLV) virions pseudotyped with the mutant Envs were ∼500-fold less infectious (titer, ∼2 × 103 inclusion-forming units [IFU]/ml) than MLV virions pseudotyped with WT ASLV(A) Env (titer, ∼1 × 106 IFU/ml). The ability of the mutant Envs to mediate cell fusion was also greatly impaired compared to that of WT ASLV(A) Env in a cell-cell fusion assay. We concluded that the cysteines flanking the internal fusion peptide in ASLV TM are critical for efficient function of the ASLV viral glycoproteins in mediating entry. In a recent study, the cysteines flanking the fusion peptide region were shown to be critical for the lipid mixing stage of fusion (6).
FIG. 1.

Schematic representations of the ASLV-based RCASBP retroviral vector and the major domains of the envelope glycoproteins. The RCASBP(A)AP replication-competent vector contains a subgroup A env and a reporter gene coding for heat-stable AP. The hypervariable domains (vr1, vr2, hr1, hr2, and vr3) of the SU glycoprotein, the proteolytic cleavage site, the putative fusion peptide region (shaded box), and the membrane-spanning domain (MSD) of the TM glycoprotein are shown schematically. The first 45 residues of the TM glycoprotein are shown for wild-type subgroup A Env (WT) and for the three mutants tested in this study, with either a substitution of serine for the cysteine at position 9 in TM (C9S), a substitution of serine for the cysteine at position 45 in TM (C45S), or both substitutions (C9,45S). The complete sequence of the ASLV(A) WT TM glycoprotein is shown, with the fusion peptide region, N-terminal and C-terminal heptad repeat regions (N-alpha-helix; C-alpha helix), and membrane-spanning domain indicated.
Very little is known about the structures of fusion peptides in the context of full-length, trimeric, viral glycoproteins upon interaction with target membranes. Also, natural membrane targets contain a variety of lipid and protein compositions in an asymmetrical organization that is difficult to reproduce experimentally (27). In addition, little is known about how fusion proteins with internal fusion peptide regions interact with target membranes or the possible conformational changes that might be required to complete the fusion process (19, 20). In this study, replication-competent ASLV(A) viruses containing the C9S, C45S, or C9,45S mutations were constructed and genetically selected for improved replication in chicken fibroblasts in order to further explore the importance of these cysteines for proper TM function. Viruses with single cysteine-to-serine mutations reverted to the WT sequence. However, viruses with both the C9S and the C45S mutation retained both mutations and acquired a second-site mutation that significantly enhanced the infectivity of the genetically selected virus population. Unexpectedly, the selected second-site mutation was a charged residue located in the middle of the hydrophobic fusion peptide within TM.
MATERIALS AND METHODS
Vector constructions.
The construction of the RCASBP(A)AP retroviral vector, the ASLV-based replication-competent RCASBP vector with a subgroup A env gene and the heat-stable human placental alkaline phosphatase (AP) gene, has been described previously (9). A plasmid subclone containing the complete ASLV(A) env gene on an Asp718-to-AgeI fragment was used to produce the C9S, C45S, and C9,45S mutant env genes, as well as the amino acid substitutions in TM residue G30, by site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Stratagene). The mutations in the env genes of the mutant C9S, C45S, and C9,45S clones were verified by nucleotide sequence analysis and subcloned into the RCASBP(A)Age+(AP) proviral plasmid clone as Asp718-to-AgeI fragments replacing the WT env gene. Other mutant env genes were isolated as Asp718-to-AgeI fragments from the cloned PCR-amplified env genes in pCR2.1-TOPO (Invitrogen, Carlsbad, CA) and were cloned into the unique Asp718 and AgeI sites of the RCASBP(A)Age+AP vector.
Cloning and nucleotide sequence analysis of integrated viral DNA.
DNA was isolated from infected cells in culture using the QIAamp tissue kit (Qiagen). The entire env gene was amplified by PCR using Taq DNA polymerase (Promega, Madison, WI) with primers 5′-GGGACGAGGTTATGCCGCTG-3′ (∼50 bp upstream of the Asp718 site) and 5′-TACCACCACCCATGTACTGCC-3′ (just downstream of the env gene). Each Taq PCR mixture contained 1.25 μl 10× PCR buffer (final concentrations, 50 mM Tris-Cl [pH 8.3], 50 mM KCl, 7 mM MgCl2, 1.1 mM β-mercaptoethanol), 1.25 μl of 1.7-mg/ml bovine serum albumin, 0.5 μl of each deoxynucleoside triphosphate at 25 mM, 0.5 μl of each primer (A260, 5), 6.0 μl H2O, and 1.0 μl of DNA (genomic DNA at 1 μg/μl; plasmid DNA at ∼2 ng/μl). The reaction mixtures were heated to 90°C for 1 min, and reactions were initiated by the addition of 1.5 μl of Taq DNA polymerase diluted 1:10 (vol/vol) (0.75 U). Thirty cycles of PCR were carried out as follows: 90°C for 40 s and then 59°C for 80 s. The amplified products were separated by agarose gel electrophoresis, and the ∼2.0-kb product was purified and cloned into pCR2.1-TOPO using the TOPO TA cloning kit (Invitrogen). The nucleotide sequences of the env genes were determined by the Mayo Clinic Molecular Biology Core on an ABI Prism 377 DNA sequencer (with XL upgrade) with an ABI Prism dRhodamine Terminator cycle-sequencing ready-reaction kit and AmpliTaq DNA polymerase (all from Perkin-Elmer Applied Biosystems, Foster City, CA).
Cell culture and virus propagation.
DF-1 cells (15, 29) were grown in Dulbecco's modified Eagle's medium (Gibco/BRL) supplemented with 10% fetal bovine serum (Gibco/BRL), 100 U of penicillin per ml, and 100 μg of streptomycin per ml (Quality Biological, Inc., Gaithersburg, MD) at 39°C under 5% CO2. Virus propagation was initiated either by transfection of plasmid DNA that contained the retroviral vector in proviral form (9) or by direct infection. In standard transfections, 5 μg of purified plasmid DNA was introduced into DF-1 cells by the calcium phosphate precipitation method (21). Viral spread was monitored by assaying culture supernatants for ASLV capsid protein (CA) by enzyme-linked immunosorbent assay (ELISA) (30). Virus stocks were generated from cell supernatants cleared of cellular debris by centrifugation at 2,000 × g for 10 min at 4°C and were stored in aliquots at −80°C.
ELISA.
The ASLV CA protein was detected in culture supernatants by ELISA as described previously (30). The levels of soluble Tva (sTva)-mouse immunoglobulin G (mIgG) and chicken sTva (cksTva)-mIgG were quantitated in culture supernatants by ELISA for the mIgG tag as previously described (17). The linear range for a standard experiment was between 0.5 and 50 ng of ImmunoPure mouse IgG Fc fragment per ml.
ALV AP assay.
For AP assays, DF-1 cell cultures (∼30% confluent) were incubated with 10-fold serial dilutions of the appropriate RCASBP/AP virus stocks for ∼48 h. The assay for AP activity has been described previously (17).
SDS-PAGE and Western immunoblot analysis.
Supernatants from confluent cultures were cleared of cellular debris by centrifugation at 2,000 × g for 10 min at 4°C. Virions (10 ml of culture supernatant) were pelleted through 1 ml of a 20% sucrose pad (20% sucrose, 100 mM NaCl, 20 mM Tris·Cl [pH 7.5], 1 mM EDTA) by ultracentrifugation at 35,000 rpm in a Beckman SW41 rotor for 60 min at 4°C. The viral pellet was resuspended in 100 μl Laemmli loading buffer (2% sodium dodecyl sulfate [SDS], 10% glycerol, 50 mM Tris·Cl [pH 6.8], 5% β-mercaptoethanol, 0.1% bromophenol blue) and boiled for 5 min. Viral proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. The Western blot transfer filters were blocked in phosphate-buffered saline (PBS) with 10% nonfat dry milk (NFDM) for 1 h at 25°C. The filters were then rinsed briefly in rinse buffer (100 mM NaCl, 10 mM Tris·Cl [pH 8], 1 mM EDTA, 0.1% Tween 20) and incubated with either rabbit anti-ASLV p27 serum (Spafas, Inc., Norwich, CT) (1:5,000 dilution), rabbit anti-ASLV(A) TM peptide (1:1,000 dilution), or an anti-ASLV(A) SU mouse monoclonal antibody (28) (1:1,000) in rinse buffer containing 1% NFDM for 1 h at 25°C. The filters were washed extensively with rinse buffer and were then incubated with 50 ng/ml peroxidase-labeled goat anti-rabbit or goat anti-mouse IgG (H+L) (Kirkegaard and Perry, Gaithersburg, MD) in rinse buffer with 1% NFDM for 1 h at 25°C. After extensive washing with rinse buffer, immunodetection of the protein-antibody-peroxidase complexes was performed with the Western Blot chemiluminescence reagent (DuPont NEN, Boston, MA). The immunoblots were then exposed to Kodak X-Omat film.
SU conformational change assay.
The SU conformational change assay is a variation of assays previously described (5, 11). Briefly, pelleted virions were resuspended in HN buffer (10 mM HEPES [pH 8.0], 130 mM NaCl), and aliquots were mixed with 200 ng cksTva-mIgG on ice for 30 min, followed by incubation at 37°C for 15 min. The samples were then returned to ice; 1 M CaCl2 was added to a final concentration of 50 mM; thermolysin (Sigma) was added to a final concentration of 80 ng/μl; and the mixture was incubated on ice for 20 min. The reaction was stopped with 2× Laemmli load buffer, and the proteins were separated by SDS-PAGE and analyzed by Western immunoblotting as described above but using rabbit antisera against the N terminus of ASLV SU (1:1,000).
Virus-liposome binding assay.
Virus-liposome association assays were modified from the work of Netter et al. (26). Briefly, 40 μl virus was mixed with either chicken or quail sTva (1 μM, final concentration) (1) and allowed to associate for 30 min on ice. The sTva proteins consist of 47 amino acid residues containing the ligand binding module of either chicken or quail Tva (containing the EnvA binding regions) produced in Escherichia coli and purified as described previously (14). Next, 50 μl liposomes (a 1:1:1:1.5 mixture of phosphatidylcholine [PC], phosphatidylethanolamine [PE], sphingomyelin, and cholesterol extruded through 100-nm-pore-size filters) in HM buffer (20 mM HEPES, 20 mM MES [morpholineethanesulfonic acid], 150 mM NaCl, pH 7.8) and additional HM buffer to a 100-μl final volume were then added, and the samples were incubated at 37°C for 30 min and then returned to ice. The virus-liposome mixture was then diluted 1:1 with 80% (wt/vol) sucrose in HM, transferred to a 700-μl centrifuge tube, and overlaid first with 400 μl 25% sucrose and then with 50 μl 5% sucrose. Samples were centrifuged at 150,000 × g in a Ti55 rotor for 2 h. Four fractions, of 100, 150, and 150 μl and the remainder (approximately 200 μl), were collected from the top of the gradient. Proteins were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, probed with rabbit anti-A-tail sera, and visualized with anti-rabbit Ig conjugated to horseradish peroxidase as described above.
TM oligomerization assays.
The TM oligomerization assays were performed essentially as described by Smith et al. (31). In short, pelleted virus was resuspended in a small volume of HNC buffer (10 mM HEPES, pH 8.0, 130 mM NaCl, 1 mM CaCl2) and incubated with or without chicken sTva-mIgG (50 nM) for 20 min on ice to allow binding of Tva. The pH was adjusted with predetermined amounts of 1 M HEPES, pH 7.4 or 4.5, and the samples were incubated at 37°C for 30 min. The samples were then neutralized with 1 M Tris-HCl (pH 8.0 or 9.5), lysed with Laemmli load buffer, and incubated at 37°C for 10 min. Samples were then separated by SDS-PAGE, and analyzed by Western immunoblotting using an antibody specific for the C terminus of ASLV(A) TM. The temperature threshold that triggers TM oligomerization without a receptor was determined by incubating the purified virions in HN buffer at the indicated temperature for 30 min or boiling for 5 min (100°C) prior to lysis in SDS.
Fluorescence-activated cell sorting (FACS) analysis of envelope glycoprotein binding to the receptor.
Uninfected DF-1 cells or DF-1 cells infected with either WT or mutant ASLV viruses were removed from culture with Trypsin de Larco (Quality Biological, Inc.) and washed with Dulbecco's PBS. The cells were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min and were then washed with PBS. Approximately 1 × 106 cells in PBS supplemented with 1% calf serum (PBS-CS) were incubated with supernatant containing either chicken or quail sTva-mIgG on ice for 30 min. The stable DF-1 cell lines TF/cksTva-15 (chicken sTva-mIgG) and TF/sTva-4 (quail sTva-mIgG) were the sources of the sTva-mIgG proteins (16). The cells were then washed with PBS-CS and incubated with 5 μl of goat anti-mouse IgG (H+L) linked to phycoerythrin (Kirkegaard & Perry Laboratories, Gaithersburg, MD) in PBS-CS (1 ml, total volume) on ice for 30 min. The cell-soluble receptor-mIgG-Ig-phycoerythrin complexes were washed with PBS-CS, resuspended in 0.5 ml PBS-CS, and analyzed with a Becton Dickinson FACSCalibur instrument using CellQuest software, version 3.1.
Apparent dissociation constant (KD) calculations.
The maximum possible bound fluorescence and KD value for each data set obtained from the FACS binding assays were estimated by fitting the data via nonlinear least squares to a log logistic growth curve function: 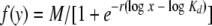 , where y is the mean fluorescence, M is the maximum fluorescence, r is the rate, x is the concentration of sTva-mIgG, and Kd is the dissociation constant defined as the concentration of sTva-mIgG at half-maximal binding [16]. The statistical significance of differences among the estimated KD values was analyzed by analysis-of-variance methods. The estimated average KD for each glycoprotein was obtained along with the associated 95% confidence interval.
, where y is the mean fluorescence, M is the maximum fluorescence, r is the rate, x is the concentration of sTva-mIgG, and Kd is the dissociation constant defined as the concentration of sTva-mIgG at half-maximal binding [16]. The statistical significance of differences among the estimated KD values was analyzed by analysis-of-variance methods. The estimated average KD for each glycoprotein was obtained along with the associated 95% confidence interval.
RESULTS
Rescue of mutant viruses.
Plasmids were constructed containing the recombinant RCASBP(A)AP proviral coding region with the C9S, C45S, or C9,45S mutations in TM (Fig. 1). DF-1 cells were transfected with no DNA (mock transfection) or plasmid DNA encoding either the WT virus or a mutant virus. The cell cultures were passaged, and virus replication was monitored by an ELISA for the ASLV CA protein (Fig. 2A). In this experiment, WT virus replicated rapidly, reaching high CA levels in 4 to 8 days posttransfection. Virus replication was observed in all three mutant-virus-infected cultures, but each mutant virus was significantly delayed in reaching high levels of CA compared to the WT. New DF-1 cells were then infected with supernatants (1 ml) from day 44 posttransfection cultures (Fig. 2A), and the infected cells were passaged. Now, both the C9S and C45S mutant virus population-infected cultures replicated at levels comparable to that of the WT virus, while the C9,45S mutant virus population was still delayed but replicated significantly faster than at passage 1 (Fig. 2B).
FIG. 2.

Selection of ASLV(A) variants that rescue the defect in replication from the C9S, C45S, or C9,45S mutations. Viral growth was monitored by ELISA for the ASLV CA protein. (A) DF-1 cells were transfected with DNA containing proviral forms of the RCASBP(A)AP virus with WT Env or with Env with the C9S, C45S, or C9,45S mutations. Transfected cells were passaged to allow virus production and spread. (B) Uninfected DF-1 cells were infected with 1 ml of day 44 culture supernatants from the viruses shown in panel A and were passaged. (C) Genomic DNA was isolated from day 8 infected cells from panel B; the ASLV(A) env genes were amplified by PCR and cloned; and the nucleotide sequences were determined from 10 to 14 individual clones. The deduced amino acid sequences were compared to that of WT RCASBP(A) Env.
To determine what mutations the C9S, C45S, and C9,45S viruses had acquired to explain the improvement in replication, the ASLV env regions were amplified by PCR from genomic DNA isolated from each infected culture. The PCR products were subcloned into plasmids, and the nucleotide sequence of the entire env gene was determined from 10 individual clones. Both of the viruses with the single cysteine mutations, C9S and C45S, reverted to coding for cysteine at both C9 and C45 in 10/10 clones. However, neither serine reverted to cysteine in the C9,45S double mutant virus. Instead, the virus acquired second-site mutations (Fig. 2C). The majority of the clones analyzed contained a glycine-to-arginine mutation at position 30 (G30R) in TM either alone (virus 29) or in combination with either an alanine-to-threonine mutation at position 33 (A33T) in TM (virus 28), A33T plus a glycine-to-serine mutation at position 123 (G123S) in TM (virus 31), or other mutations. This was unexpected, since it resulted in a charged residue in the middle of the otherwise hydrophobic fusion peptide region.
The G30R second-site mutation in the putative fusion peptide enhances viral replication in the TM C9,45S mutant.
Recombinant mutant RCASBP(A)AP clones were constructed with each of the predominate viral genotypes, virus 28, virus 29, or virus 31. DF-1 cells were transfected with plasmids carrying either WT, mutant C9,45S, or virus 28, 29, or 31 RCASBP(A)AP, and viral replication was followed by CA ELISA. All three of the C9,45S viruses containing second-site mutations replicated more efficiently than the parental C9,45S mutant virus in DF-1 cells (Fig. 3A). All four viruses contained detectable levels of envelope glycoprotein incorporated into virions as assayed by Western immunoblotting for the SU(A) glycoprotein and normalization to viral CA levels (Fig. 3B). The infectious titers of the viruses were quantitated by an assay for the AP enzyme activity of the reporter AP gene contained in the RCASBP(A)AP viruses (Fig. 1). The infectious virus titer produced by WT virus in DF-1 cells at day 17 averaged 10- to 20-fold higher than those produced by viruses 28, 29, and 31 (Fig. 3C). The rates of replication of the WT virus and viruses 28, 29, and 31 were characterized by infecting DF-1 cells at a multiplicity of infection of 0.1 and following virus replication by CA ELISA. Viruses 28, 29, and 31 replicated at a lower rate than the WT virus (Fig. 3D), most likely resulting in the consistently lower maximum titers. From these data we conclude that the G30R mutation alone is sufficient to rescue the C9,45S mutations in cultured DF-1 cells, although only 5 to 10% of the maximum titer achieved by WT virus was reached with both C9 and C45 in TM.
FIG. 3.

Viral growth of recombinant RCASBP(A) with Envs containing secondary mutations in addition to C9,45S compared to the growth of the WT. (A) DNA containing proviral forms of RCASBP(A)AP with WT or mutant (C9,45S, virus 28, virus 29, or virus 31) Env was transfected into DF-1 cells. The cells were passaged and virus propagation monitored by ELISA for ASLV CA. (B) Western immunoblot analysis of SU(A) glycoprotein and CA protein levels in purified virions. (C) Virus titers of day 17 supernatants (see panel A) determined using DF-1 cells and the AP titer assay. (D) The rates of virus replication were compared by infecting DF-1 cells at a multiplicity of infection of 0.1 and then following the virus replication for 10 days by ELISA for CA protein.
A second-site, charged amino acid in the TM internal fusion peptide at position 30 is preferred to rescue the C9,45S mutant ASLV(A).
To determine the amino acid requirement at position G30 that could rescue the replication of the C9,45S ASLV mutant, G30 was replaced with a variety of charged (lysine, histidine, glutamic acid), aromatic (phenylalanine, tyrosine), polar (glutamine, serine), and nonpolar (alanine, leucine) amino acids by site-directed mutagenesis of the ASLV(A) C9,45S mutant env gene. In addition, WT ASLV(A) env genes coding for the G30R mutation alone (WT+G30R) or deletion of G30 alone (WR+G30del) were also constructed. The integrity of all mutant env genes was verified by determining the complete nucleotide sequences (data not shown), and they were then cloned into the RCASBP(A)AP proviral plasmid, replacing the WT env gene. DF-1 cells were then transfected with each proviral plasmid, and the cultures were subsequently passaged following viral replication by CA ELISA (Fig. 4A) for 38 days. Titers of infectious virus were determined by an AP assay using culture supernatants from day 2 and day 38 (Fig. 4B). The incorporation of ASLV envelope glycoprotein into virions was determined by Western immunoblot assays for TM and CA levels, using purified virions from day 2 posttransfection (Fig. 4C). The level of virions in the day 2 supernatants can be influenced by differences in the DNA transfection efficiencies of the different plasmids encoding the proviral constructs, but they are provided to give an estimate of the level of infectious virus initially produced.
FIG. 4.

Viral growth of recombinant RCASBP(A)AP viruses with a variety of mutations in the TM glycoprotein. (A) DF-1 cells were transfected with DNA containing the proviral forms of RCASBP(A)AP viruses with either WT Env or Env with the mutations listed. The cells were passaged, and viral replication was followed by ELISA for the CA protein. (B) Viral titers were determined in supernatants from day 2 after transfection and from day 38 after the subsequent virus production and spread (see panel A) using DF-1 cells and the AP titer assay. The limit of infectious virus detection was 1 IFU/ml. (C) The levels of TM glycoprotein and CA protein in purified virions from day 2 supernatants were determined by Western immunoblot analysis. Molecular sizes (in kilodaltons) are given on the left.
Despite the unexpected genetic selection of the designed G30R secondary mutation that rescues the replication of the C9,45S mutant TM, with a positively charged amino acid in the middle of the hydrophobic internal fusion peptide region, WT+G30R TM replicated in DF-1 cells at rates and to titers comparable to those of WT virus. However, the WT+G30del virus synthesized and incorporated the mutant Env into virions but did not produce detectable infectious virus. As before, the original C9,45S mutant synthesized and incorporated the mutant Env into virions, but the production of high levels of virus was significantly delayed (∼20 days) compared to that for the WT and WT+G30R viruses. The majority of the env genes in this virus population now contained a secondary mutation encoding G30R in addition to the original C9,45S mutations (data not shown).
Of all the C9,45S mutant viruses with G30 secondary mutations, virus 29, with the genetically selected G30R secondary mutation, replicated most efficiently (5- to 7-day lag behind WT and WT+G30R viruses) and to the highest titers, 5- to 10-fold lower than those for the WT and WT+G30R viruses. The next most efficient mutant virus was the C9,45S mutant with the G30K secondary mutation, which displayed a ∼12-day replication lag and a ∼200-fold lower maximum titer compared to those of the WT and WT+G30R viruses. The third C9,45S mutant virus with a positively charged secondary mutation (G30H) did not sustain replication much over detectable levels despite high levels of incorporated mutant Env protein in virions. However, the viral CA level late in the experiment (days 33 to 38 [Fig. 4A]) indicated that an improved virus might be emerging. Further passage of this culture produced a viral population that had changed the secondary mutation in the C9,45S virus from G30H to G30R (data not shown). Apart from the C9,45S G30H mutant and the original C9,45S mutant, the viral populations of all the other viruses tested at day 38 contained the constructed, expected mutations in the viral env genes as determined by nucleotide sequencing (data not shown).
Two other C9,45S mutant viruses, with G30E or G30S secondary mutations, replicated at higher rates than the C9,45S mutant virus alone, but significantly more slowly than the WT, with a ∼20-day lag in reaching maximum CA levels and ∼2000-fold-lower infectious titers. Only two other C9,45S mutant viruses, with G30Q and G30A secondary mutations, were able to replicate to detectable levels by day 38. Three C9,45S mutant viruses, with G30Y, G30L, and G30F secondary mutations, did not produce detectable levels of CA, except during the initial transient period from plasmid transfection, or infectious virus; however, this may be due in part to the low levels of Env incorporated into virions (Fig. 4C).
Viral envelope glycoproteins that contain the G30R mutation bind Tva receptors at WT levels.
Several assays were performed in an effort to determine why virus 29 does not replicate as well as the WT or WT+G30R virus. The initial step of entry requires specific and high-affinity binding of the viral glycoproteins with their receptor to trigger an initial conformational change. The envelope glycoproteins expressed on the surfaces of DF-1 cells chronically infected with the WT virus, the WT+G30R virus, or virus 29 were assayed by FACS as described previously for binding to soluble forms of the Tva receptor (chicken or quail sTva-mIgG) that are fused to a mouse IgG domain (16, 17). The integrity of the purified sTva receptors was verified by immunoprecipitation and Western blot analysis, and the concentration of each protein stock was quantitated by ELISA for the mIgG tag (data not shown). Both chicken and quail Tva receptors were assayed, because a previous study had selected mutant ASLV(A) viruses with mutations in the SU hypervariable regions that resulted in preferential binding to chicken Tva over quail Tva (16). The three viral envelope glycoproteins bound each sTva-mIgG receptor with similar affinities, each averaging 0.1 nM affinity (data not shown).
The G30R substitution improves but does not completely rescue the ability of the ASLV(A) C9,45S mutant envelope glycoproteins to be triggered by receptor interaction.
Normally, high-affinity Tva interaction with the SU subunits of ASLV(A) glycoprotein trimers triggers a conformational change in the structure of the viral glycoproteins where the conformational change reveals a thermolysin cleavage site in SU (5, 11). Experimentally, maximum levels of the SU cleavage product (SU*) are produced by thermolysin digestion of WT glycoprotein trimers after they are bound to soluble forms of the Tva receptor and incubated at 37°C to trigger conformational changes in the viral glycoproteins (Fig. 5A). If the virion receptor complexes are kept at 4°C, significantly less SU* is produced, due to the limited ability of the receptor to induce conformational changes in the viral glycoprotein at this nonphysiological temperature. Few or no SU* cleavage products were produced from thermolysin digestion of ASLV(A) WT Env without interaction with the receptor, even at 37°C. The mutant glycoproteins that contain C9S, C45S, or C9,45S mutations display a thermolysin digestion pattern very different from that of WT glycoproteins (Fig. 5A; C9S and C45S data not shown). First, thermolysin digestion of all three mutant glycoproteins without the receptor produced low but easily detected SU* levels, perhaps indicating that the mature glycoprotein trimer structures were unstable compared to WT trimers, allowing access to thermolysin under these experimental conditions. Second, while the binding of the receptor with mutant glycoproteins resulted in a significant increase in the amount of the SU* thermolysin digestion product, approximately similar levels of SU* were produced at both 4°C and 37°C, again indicating an alteration in the overall stability of the mutant glycoprotein trimers compared to that for the WT.
FIG. 5.

Conformational properties of the mutant Env proteins compared to WT Env. The stability of the WT and mutant Env trimers and the ability of chicken sTva-mIgG receptor protein to trigger conformational changes in SU were assayed by sensitivity to thermolysin digestion. Purified virions from 2-day transfections (A) or stably infected DF-1 cultures (B) were first incubated with or without the sTva-mIgG receptor at 4°C, then incubated at 37°C to trigger possible receptor-induced conformational changes in Env, and finally incubated at 4°C with or without thermolysin (80 ng/ml). The full-length SU and digested (SU*) products were detected by Western immunoblotting using anti-SU sera.
To determine the possible effects of the G30R substitution on the structural stability of the WT and C9,45S glycoproteins and their abilities to undergo receptor-induced conformational changes, purified virions were triggered with a soluble form of the Tva receptor and then digested with thermolysin as described above. As expected, WT glycoproteins triggered by the receptor and digested with thermolysin produce the highest level of SU* at 37°C, a lower level of SU* at 4°C, and very little or no detectable SU* at 37°C without receptor-induced conformational triggering (Fig. 5B). WT+G30R glycoproteins produce SU* in a pattern similar to the WT pattern, except that SU* is routinely detectable without receptor triggering at 37°C at a level higher than that for the WT, but at a level lower than that observed with C9,45S glycoproteins (Fig. 5B). The G30R substitution in the C9,45S mutant glycoprotein appears to rescue the ability of the glycoproteins to be triggered most efficiently at 37°C after receptor-induced conformation triggering, with a lower level of SU* produced at 4°C than that for C9,45S mutant glycoproteins. The C9,45S and C9,45S+G30R glycoproteins appear to produce similar levels of SU* at 37°C in the absence of receptor triggering.
To determine the abilities of the ASLV envelope glycoproteins on virions to be triggered by binding the receptor at 37°C to form a stable extended TM oligomer that associates stably with liposomes by fusion peptide insertion, virus-liposome binding assays were performed (26). Virions were incubated with or without 1 μM chicken or quail sTva (these are 47-amino-acid residue forms of the receptor EnvA binding regions produced in E. coli [34]) at 4°C to allow receptor envelope glycoprotein binding. After liposomes were added to the virion-receptor complexes, the mixture was incubated at 37°C to initiate receptor-triggered conformational changes in Env exposing the fusion peptide of TM and enabling its interaction with target membranes (liposomes). The mixture was then separated on a sucrose step gradient: virions that were successfully primed by the receptor and are stably associated with liposomes will “float” higher in the gradient than unassociated virions. Results of a representative experiment are shown in Fig. 6. As expected, all of the WT virions not primed with receptor remained at the bottom of the gradient (fraction 4), while virtually all of the WT virions primed with either quail sTva or chicken sTva were found at the top of the gradient (fraction 1). We had shown previously that the same analysis of C9,45S mutant virions produced data indistinguishable from those for the WT (6), despite the apparently altered stability of the mutant C9,45S glycoproteins (Fig. 5A). WT+G30R virions primed with the sTva receptors associated with liposomes as well as WT virions did (Fig. 6). However, only ∼40% of C9,45S+G30R virions mixed with either sTva receptor were found in the top fraction of the gradient. Since C9,45S+G30R envelope glycoproteins bind to the sTva receptors with wild-type affinity, the glycoproteins appear to be defective in their ability to form the extended TM oligomer and/or for the fusion peptide to associate stably with a target membrane.
FIG. 6.

Representative virus-liposome binding assay. Purified virions were mixed with no receptor (none), quail sTva (qsTva), or cksTva proteins (1 μM) at 4°C; then they were incubated with liposomes at 37°C, and the mixture was separated by sucrose gradient centrifugation. Four fractions were collected (fraction 1, top; fraction 4, bottom), and the location of the TM glycoprotein was analyzed by Western immunoblotting using anti-subgroup A TM tail sera. This experiment has been repeated multiple times with similar results. S, starting material.
Smith et al. demonstrated that the ASLV(A) glycoproteins are in a metastable conformation that can be released by heat denaturation to form stable, SDS-resistant TM oligomers at neutral pH, similar to the TM oligomers formed after receptor triggering followed by low-pH exposure (31). The ability of wild-type and mutant ASLV(A) virions produced and purified from 2-day transient transfected culture supernatants to form stable TM oligomers was assayed using TM oligomerization assays based on the experimental conditions defined by Smith et al. (31). The conformation of the mature, metastable ASLV(A) glycoprotein trimer on WT virions can be triggered to form the most stable, SDS-resistant TM oligomeric forms by a high temperature alone (Fig. 7A): this occurs predominately at 55 to 60°C and is characterized by the monomer of TM (∼30 kDa) forming two oligomeric species of 70 to 80 kDa and ∼170 kDa, although due to assay procedures, some TM oligomer “background” may form inefficiently at lower temperatures. However, all three mutant glycoproteins were barely able to form detectable levels of stable TM oligomers by use of this experimental approach (Fig. 7A; C9S and C45S data not shown).
FIG. 7.

Representative TM oligomerization assays. Shown are representative experiments demonstrating the abilities of purified ASLV(A) WT and mutant Env trimers to form stable, SDS-resistant TM oligomers from 2-day transfections (A) or stably infected DF-1 cells (B and C). The C9,45S mutant glycoproteins were studied only with virions produced from transient transfections, since the virus population starts to change upon cell passage. The abilities of Env on ASLV(A) virions to be triggered to form stable TM oligomer conformations by temperature alone (A and B) and by prior binding with the receptor, different temperatures, and low-pH exposure (C) are shown. Molecular sizes (in kilodaltons) are given on the left. The separated proteins were analyzed by Western immunoblotting using anti-ASLV(A) TM peptide sera followed by horseradish peroxidase-conjugated goat anti-rabbit IgG.
The ability of envelope glycoprotein trimers on purified WT, WT+G30R, and C9,45S+G30R virions to form stable, SDS-resistant TM oligomers at a neutral pH was also analyzed using TM oligomerization assays. Viruses were purified from the supernatants of chronically infected DF-1 cell cultures. WT glycoprotein trimers were reproducibly triggered to form the 70- to 80-kDa stable TM oligomers by a temperature of ≥60°C alone (Fig. 7B), a slightly higher temperature than that for viruses purified from 2-day transient transfections (Fig. 7A). The mutant with the G30R mutation in an otherwise WT glycoprotein forms stable TM oligomers with efficiencies very similar to that of the WT, although we consistently observed the formation of low levels of WT+G30R oligomers (above “background”) at slightly lower temperatures alone (50 to 55°C) (Fig. 7B). In sharp contrast, C9,45S+G30R glycoprotein trimers have very little ability to form stable TM oligomers, even after treatment with extremely high temperatures, compared to the WT (Fig. 7B). However, we consistently observed that the C9,45S+G30R glycoproteins formed a low level of some sort of stable TM oligomer more efficiently than the C9,45S mutant glycoproteins at temperatures between 60 and 80°C (compare Fig. 7A, C9,45S, and Fig. 7B, C9,45S+G30R).
WT glycoprotein trimers do not form TM oligomers efficiently just with binding to the sTva-IgG receptor at 4°C and a neutral pH of 7.4; the inefficient formation of some “background” TM oligomers increases as the incubation temperature increases (Fig. 7C). As expected, the combination of sTva-IgG receptor triggering and exposure to a low pH of 5.0 most efficiently converts the WT glycoprotein trimers to stable TM oligomers at 37°C; pH 5.0 exposure also increases the inefficient “background” TM oligomer formation at lower temperatures, compared to pH 7.4, in this assay. The mutant with the G30R mutation in an otherwise WT glycoprotein forms stable TM oligomers even more efficiently at pH 5.0 and 4°C than the WT (Fig. 7C). Again, the C9,45S+G30R glycoprotein trimers have very little ability to form stable TM oligomers, even after sTva receptor triggering and low-pH treatment, compared to that of the WT (Fig. 7C). It is interesting that the C9,45S+G30R trimers appear to be more efficient at forming these TM oligomers, after priming with sTva-mIgG receptor, at pH 5.0 and 37°C than at lower temperatures and pH 7.4.
DISCUSSION
ASLVs and Ebola viruses contain internal fusion peptides near the N termini of the fusion glycoprotein subunits. Two cysteine residues flank the fusion peptide regions, presumably in a disulfide bond to form a loop important for the fusion process. Studies using viruses pseudotyped with ASLV or Ebola glycoproteins with mutations in these flanking cysteines have shown that these cysteine residues are required for efficient fusion with a target membrane and viral infection (8, 20). To further understand the relationship of the fusion peptide region and the flanking cysteines with the various steps of viral entry and replication, replication-competent ASLVs were constructed with the C9S, C45S, or C9,45S mutations in the TM glycoproteins and were given a chance to acquire genetic mutations to improve entry and replication in chicken DF-1 cells. As predicted by the hypothesis that these cysteine residues are required for efficient ASLV infection and replication, both the single cysteine mutants reverted to wild-type glycoproteins. Unexpectedly however, the C9,45S double mutant glycoproteins did not: the dominant mutant virus selected retained both C9S and C45S substitutions and acquired the G30R secondary mutation in the middle of the hydrophobic fusion peptide region. The G30R substitution in the C9,45S glycoproteins improved viral replication to titers within 5 to 10% of WT titers. The identical second-site substitution was obtained in three independent experiments using DF-1 cells and one experiment using chicken embryo fibroblasts (all data not shown).
In this study with replication-competent ASLVs, and in a previous study using MLV pseudotypes (8), we have shown that the mutant C9,45S glycoproteins are efficiently synthesized, processed, and incorporated into ASLV particles and are capable of producing infectious virions, although at titers ∼500- to 1,000-fold-lower than those with WT glycoproteins. Compared to WT Env glycoproteins, the mutant C9,45S glycoprotein trimers were more sensitive to thermolysin digestion at nonphysiological temperatures and in the absence of receptor triggering, perhaps indicating slightly unfolded native structures. Nevertheless, the C9,45S mutant glycoproteins were still capable of WT levels of receptor-triggered interactions between virions and target liposomes. However, the C9,45S mutant glycoproteins are unable to form stable TM oligomers in the presence of Tva receptor and low pH (data not shown) or after high-temperature denaturation (Fig. 7A). The G30R secondary mutation partially rescues the ability of C9,45S glycoproteins to form stable TM oligomers triggered by the Tva receptor most efficiently at pH 5.0 and 37°C, and with heat denaturation (Fig. 7B). The G30R secondary mutation also partially restores the ability of C9,45S glycoproteins to perform the lipid mixing step of fusion (6). The thermolysin sensitivity of the C9,45S+G30R glycoprotein trimers appeared more like that of WT glycoprotein trimers but still demonstrated some unfolded native structures (Fig. 5). However, the G30R secondary mutation reduced the ability of the C9,45S+G30R glycoproteins in virions triggered with Tva to interact with target liposomes compared to virions with C9,45S (6) and WT glycoproteins (Fig. 6). Thus, the selection of the G30R substitution appears to rescue the fusion function by improvement in the abilities of C9,45S mutant TM glycoproteins to form appropriate TM oligomeric structures and enable some degree of lipid mixing to account for the 100-fold improvement in replication.
Upon testing of a variety of other amino acids at G30 for rescue of C9,45S mutant virus entry, a positively charged residue substitution was preferred at this position. Clearly the G30R substitution in C9,45S recovered the highest level of viral replication and the highest titers. The mutant with the G30K substitution in C9,45S was the next most efficient virus, although this virus replicated to ∼20-fold-lower titers than the C9,45S+G30R mutant (Fig. 4). The C9,45S+G30H virus essentially was not viable, presumably due to the incompatibility of the large ring structure of the histidine residue. The selection of a charged residue could provide new possibilities of electrostatic interactions, i.e., a salt bridge, between G30R and other charged residues in the envelope glycoprotein trimer, which then form a looped structure similar to that of wild-type glycoproteins. Alternatively, an electrostatic interaction could be formed between G30R and the phosphates of the membrane hydrophilic head groups, which then promote the formation of stable TM oligomers and lipid mixing despite a nonoptimal overall structural presentation of the fusion peptide region without the cysteine bond. Differentiating between these models, a combination of these and/or other possible hypothetical models may have to wait until there are atomic structures of the ASLV glycoproteins.
Normally, the selection of a charged residue in the middle of the hydrophobic fusion peptide region would be expected to severely alter or even inhibit the interaction of this region with a target membrane, not to rescue function. Structural and membrane interaction properties of a synthetic peptide containing a portion of the wild-type ASLV(A) fusion peptide region (TM residues 17 to 43) have been described by Cheng et al. (4). In an aqueous environment, this synthetic ASLV fusion peptide has a beta-sheet structure N-terminal to P29, a slight kink in the structure around P29, and an alpha-helical structure C-terminal to P29. Upon interaction with a micellar medium, the beta-sheet region transforms into an alpha-helical structure with the P29 kink closest to the micellar surface. This peptide has a higher helical structure content and penetrates into the membrane at a shallower angle than the prototypic N-terminal influenza virus HA fusion peptide structure and membrane interactions (12, 33). The ASLV fusion region C-terminal to P29 is embedded more deeply than the N-terminal region, the reverse of the HA N-terminal fusion peptide interaction with a membrane. At a low pH, the synthetic ASLV peptide lies deeper in the membrane, possibly facilitating the membrane disruption necessary for the lipid mixing process leading to fusion pores, although the kinked region still resides near the membrane-water interface (4). The flexibility of the fusion peptide kinked region centered on P29 has previously been shown to be critical: mutant ASLV TM glycoproteins with replacements of P29 with amino acids predicted to restrict flexibility resulted in the loss of fusion activity (7).
The Cheng et al. model of the ASLV internal fusion peptide interaction with a target membrane as two alpha-helices connected by a kinked region of P29 G30 near the membrane-water interface would appear to tolerate the substitution of a charged residue at the G30 position. One might expect that a charged residue at position 30 would likely restrict the depth at which the ASLV fusion peptide region can move in the membrane during fusion. However, there does not seem to be much of a price in terms of replication fitness for a virus with a charged residue at this location in the fusion peptide region, since a virus with the G30R substitution in an otherwise wild-type ASLV(A) Env replicates at wild-type levels. In contrast, the deletion of G30 in an otherwise WT ASLV(A) TM completely blocked the production of infectious virus despite the synthesis of the mutant protein. These data are consistent with a model in which critical conformation changes required to complete ASLV(A) virus-cell fusion occur centered around the P29 and G30 residues: the P29 G30 region residing near the polar head groups allows the alpha-helical regions at a relatively shallow depth in the membrane to flex, perhaps in a “scissors-like motion,” to disrupt the membrane. We hypothesize that the deletion of TM residue 30 prevents sufficient flexibility between the alpha-helices of the fusion domain to appropriately destabilize the target membrane and/or to complete the fusion process.
Finally, it is noteworthy that a region of conserved, mainly hydrophobic residues is located directly upstream of the putative ASLV R21-to-R38 fusion peptide region (TM residues 13 to 20) and can be modeled as a potential alpha-helix region (data not shown). In addition, this region has another glycine-proline pair, G17 P18, implying another region of possible flexibility (Fig. 1). Given the model of Cheng et al., in which ASLV fusion peptide insertion appears to be relatively shallow, and our data showing that a charged residue can be tolerated at G30 in this region, it may be reasonable to consider that the ASLV(A) fusion peptide region may extend to include these additional N-terminal conserved hydrophobic residues. This additional hydrophobic, alpha-helical region may facilitate target membrane interaction, the deeper penetration of the peptide at a low pH, membrane-spanning segment interaction, and/or the subsequent conformational changes required to attain the 6HB and mature trimeric hairpin structures required to complete fusion pore formation.
Acknowledgments
This work was supported in part by National Institutes of Health grant AI48682 and the Mayo Clinic (to M.J.F.) and by NIH grant AI22470 (to S.E.D. and Judith M. White).
Footnotes
Published ahead of print on 10 June 2009.
REFERENCES
- 1.Balliet, J. W., J. Berson, C. M. D'Cruz, J. Huang, J. Crane, J. M. Gilbert, and P. Bates. 1999. Production and characterization of a soluble, active form of Tva, the subgroup A avian sarcoma and leukosis virus receptor. J. Virol. 733054-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnard, R. J., D. Elleder, and J. A. T. Young. 2006. Avian sarcoma and leukosis virus-receptor interactions: from classical genetics to novel insights into virus-cell membrane fusion. Virology 34425-29. [DOI] [PubMed] [Google Scholar]
- 3.Barnard, R. J., and J. A. T. Young. 2003. Alpharetrovirus envelope-receptor interactions. Curr. Top. Microbiol. Immunol. 281107-136. [DOI] [PubMed] [Google Scholar]
- 4.Cheng, S.-F., C.-W. Wu, E. A. B. Kantchev, and D.-K. Chang. 2004. Structure and membrane interaction of the internal fusion peptide of avian sarcoma and leukosis virus. Eur. J. Biochem. 2714725-4736. [DOI] [PubMed] [Google Scholar]
- 5.Damico, R., L. Rong, and P. Bates. 1999. Substitutions in the receptor-binding domain of the avian sarcoma and leukosis virus envelope uncouple receptor-triggered structural rearrangements in the surface and transmembrane subunits. J. Virol. 733087-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delos, S. E., M. B. Brecher, Z. Chen, D. C. Melder, M. J. Federspiel, and J. M. White. 2008. Cysteines flanking the internal fusion peptide are required for the avian sarcoma/leukosis virus glycoprotein to mediate the lipid mixing stage of fusion with high efficiency. J. Virol. 823131-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delos, S. E., J. M. Gilbert, and J. M. White. 2000. The central proline of an internal viral fusion peptide serves two important roles. J. Virol. 741686-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delos, S. E., and J. M. White. 2000. Critical role for the cysteines flanking the internal fusion peptide of avian sarcoma/leukosis virus envelope glycoprotein. J. Virol. 749738-9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Federspiel, M. J., and S. H. Hughes. 1997. Retroviral gene delivery. Methods Cell Biol. 52179-214. [PubMed] [Google Scholar]
- 10.Gallaher, W. R. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell 85477-478. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert, J. M., L. D. Hernandez, J. W. Balliet, P. Bates, and J. M. White. 1995. Receptor-induced conformational changes in the subgroup A avian leukosis and sarcoma virus envelope glycoprotein. J. Virol. 697410-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han, X., J. H. Bushweller, D. S. Cafiso, and L. K. Tamm. 2001. Membrane structure and fusion-triggering conformational change in the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8715-720. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez, L. D., L. R. Hoffman, T. G. Wolfsberg, and J. M. White. 1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12627-661. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez, L. D., R. J. Peters, S. E. Delos, J. A. T. Young, D. A. Agard, and J. M. White. 1997. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 1391455-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himly, M., D. N. Foster, I. Bottoli, J. S. Iacovoni, and P. K. Vogt. 1998. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248295-304. [DOI] [PubMed] [Google Scholar]
- 16.Holmen, S. L., D. C. Melder, and M. J. Federspiel. 2001. Identification of key residues in subgroup A avian leukosis virus envelope determining receptor binding affinity and infectivity of cells expressing chicken or quail Tva receptor. J. Virol. 75726-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmen, S. L., D. W. Salter, W. S. Payne, J. B. Dodgson, S. H. Hughes, and M. J. Federspiel. 1999. Soluble forms of the subgroup A avian leukosis virus [ALV(A)] receptor Tva significantly inhibit ALV(A) infection in vitro and in vivo. J. Virol. 7310051-10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter, E. 1997. Viral entry and receptors, p. 71-120. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 19.Ito, H., S. Watanabe, A. Sanchez, M. A. Whitt, and Y. Kawaoka. 1999. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J. Virol. 738907-8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffers, S. A., D. A. Sanders, and A. Sanchez. 2002. Covalent modifications of the Ebola virus glycoprotein. J. Virol. 7612463-12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kingston, R. E., C. A. Chen, and H. Okayama. 1989. Introduction of DNA into eukaryotic cells, p. 911-919. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 1. John Wiley & Sons, Inc., New York, NY. [Google Scholar]
- 22.Markosyan, R. M., P. Bates, F. S. Cohen, and G. B. Melikyan. 2004. A study of low pH-induced refolding of Env of avian sarcoma and leukosis virus into a six-helix bundle. Biophys. J. 873291-3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuyama, S., S. E. Delos, and J. M. White. 2004. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J. Virol. 788201-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melikyan, G. B., R. J. Barnard, L. G. Abrahamyan, W. Mothes, and J. A. T. Young. 2005. Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc. Natl. Acad. Sci. USA 1028728-8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. T. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103679-689. [DOI] [PubMed] [Google Scholar]
- 26.Netter, R. C., S. M. Amberg, J. W. Balliet, M. J. Biscone, A. Vermeulen, L. J. Earp, J. M. White, and P. Bates. 2004. Heptad repeat 2-based peptides inhibit avian sarcoma and leukosis virus subgroup A infection and identify a fusion intermediate. J. Virol. 7813430-13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieva, J. L., and A. Agirre. 2003. Are fusion peptides a good model to study viral cell fusion? Biochim. Biophys. Acta 1614104-115. [DOI] [PubMed] [Google Scholar]
- 28.Ochsenbauer-Jambor, C., S. E. Delos, M. A. Accavitti, J. M. White, and E. Hunter. 2002. Novel monoclonal antibody directed at the receptor binding site on the avian sarcoma and leukosis virus Env complex. J. Virol. 767518-7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer-Klein, J., I. Givol, E. V. Barsov, J. M. Whitcomb, M. VanBrocklin, D. N. Foster, M. J. Federspiel, and S. H. Hughes. 1998. The EV-0-derived cell line DF-1 supports efficient replication of avian leukosis-sarcoma viruses and vectors. Virology 248305-311. [DOI] [PubMed] [Google Scholar]
- 30.Smith, E. J., A. M. Fadly, and W. Okazaki. 1979. An enzyme-linked immunoabsorbant assay for detecting avian leukosis sarcoma viruses. Avian Dis. 23698-707. [PubMed] [Google Scholar]
- 31.Smith, J. G., W. Mothes, S. C. Blacklow, and J. M. Cunningham. 2004. The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J. Virol. 781403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swanstrom, R., and J. W. Wills. 1997. Synthesis, assembly, and processing of viral proteins, p. 263-334. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 33.Tamm, L. K. 2003. Hypothesis: spring-loaded boomerang mechanism of influenza virus hemagglutinin-mediated membrane fusion. Biochim. Biophys. Acta 161414-23. [DOI] [PubMed] [Google Scholar]
- 34.Tonelli, M., R. J. Peters, T. L. James, and D. A. Agard. 2001. The solution structure of the viral binding domain of Tva, the cellular receptor for subgroup A avian leukosis and sarcoma virus. FEBS Lett. 509161-168. [DOI] [PubMed] [Google Scholar]
- 35.White, J. M., S. E. Delos, M. Brecher, and K. Schornberg. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43189-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young, J. A. T. 2001. Virus entry and uncoating., p. 87-103. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]