

Abstract
A poliovirus (PV) mutant (termed GG), which is incapable of producing 3AB, VPg, and 3CD proteins due to a defective cleavage site between the 3B and 3C proteins, replicated, producing 3BC-linked RNA rather than the VPg-linked RNA produced by the wild type (WT). GG PV RNA is quasi-infectious. The yield of infectious GG PV relative to replicated RNA is reduced by almost 5 logs relative to that of WT PV. Proteolytic activity required for polyprotein processing is normal for the GG mutant. 3BC-linked RNA can be encapsidated as efficiently as VPg-linked RNA. However, a step after genome replication but preceding virus assembly that is dependent on 3CD and/or 3AB proteins limits production of infectious GG PV. This step may involve release of replicated genomes from replication complexes. A pseudorevertant (termed EG) partially restored cleavage at the 3B-3C cleavage site. The reduced rate of formation of 3AB and 3CD caused corresponding reductions in the observed rate of genome replication and infectious virus production by EG PV without impacting the final yield of replicated RNA or infectious virus relative to that of WT PV. Using EG PV, we showed that genome replication and encapsidation were distinct steps in the multiplication cycle. Ectopic expression of 3CD protein reversed the genome replication phenotype without alleviating the infectious-virus production phenotype. This is the first report of a trans-complementable function for 3CD for any picornavirus. This observation supports an interaction between 3CD protein and viral and/or host factors that is critical for genome replication, perhaps formation of replication complexes.
Poliovirus (PV) is the most extensively studied member of the picornavirus family and serves as a paradigm not only for picornaviruses but also for many of the nonretroviral positive strand RNA viruses (74). A schematic of the ∼7,500-nucleotide PV genome is shown in Fig. 1A. The 5′ end is linked covalently to a 22-amino-acid peptide termed VPg (virion protein genome linked) that is encoded by the 3B region of the genome. VPg and 3B are therefore used interchangeably. The 3′ end of the genome is terminated by a poly(rA) tail. Upon release of the genome into the host cell cytoplasm, genome translation is initiated by using the internal ribosome entry site. An ∼3,000-amino-acid polyprotein is produced. Complete cleavage of the polyprotein by virus-encoded proteases yields 10 proteins. The polyprotein can be divided further into three smaller polyproteins: P1, P2, and P3. P1 contains capsid proteins: VP0, VP3, and VP1. VP0 undergoes autocatalytic cleavage after genome encapsidation to produce VP4 and VP2 proteins. P2 performs host interaction functions required for robust virus multiplication, for example, shutoff of host cell translation and induction of vesicles employed for genome replication, the so-called replication complexes (RCs). P3 contains proteins that function most directly in genome replication, including the RNA-dependent RNA polymerase. Translation induces RCs, leading to genome replication. Early during infection, replicated genomes are employed as templates for translation, leading to an exponential amplification of RCs and replicated RNA. Ultimately, production of viral proteins ceases and replicated genomes are packaged. The use of RCs provides a barrier to genetic complementation; all proteins must be provided in cis, that is, produced from the RNA that they replicate.
FIG. 1.

PV genome organization and P3 processing pathway. (A) Schematic of the PV genome. The 5′ end of the genome is covalently linked to a peptide (VPg) encoded by the 3B region of the genome. The 3′ end contains a poly(rA) tail. Three cis-acting replication elements are known. oriL is located in 5′ NTR. oriR is located in the 3′ NTR. oriI is located in 2C-coding sequence for PV; the position of this element is virus dependent. oriI is the template for VPg uridylylation. Translation initiation employs an internal ribosome entry site (IRES). The single open reading frame encodes a polyprotein. P1 produces virion structural proteins as indicated. P2 produces proteins thought to participate in virus-host interactions required for genome replication. P3 produces proteins thought to participate directly in genome replication. Polyprotein processing is mediated by protease activity residing in 2A, 3C, and/or 3CD proteins. (B) Processing of the P3 precursor occurs by two independent pathways (60). There are major (I) and minor (II) pathways. In pathway I, processing between 3B and 3C yields 3AB and 3CD. In pathway II, processing between 3A and 3B yields 3A and 3BCD. 3BCD processing yields 3BC and 3D; 3BC processing yields 3B and 3C. Pathway II is proposed to function in genome replication and is not perturbed in the GG mutant.
In addition to P3 proteins, genome replication requires three cis-acting replication elements (CREs): a cloverleaf structure located in the 5′ nontranslated region (NTR), termed oriL (left) (1, 5); a stem-loop structure located in 2C-coding sequence, termed oriI (internal) (30, 61); and a pseudoknot structure located in the 3′ NTR, termed oriR (right) (1, 40). The first step of genome replication is diuridylylation of VPg or a VPg-containing protein primer (62, 74). This reaction is templated by oriI but also requires oriL in a cell-free reaction and is catalyzed by the viral RNA-dependent RNA polymerase 3Dpol (4, 5, 11, 30, 61). In addition to the four terminal P3 cleavage products (3A, 3B, 3C, and 3D proteins) and the uncleaved P3 polyprotein, several “intermediates” are observed in infected cells (3AB, 3CD, and 3BCD proteins) (Fig. 1B) (43, 57, 73). The major P3 cleavage pathway (I) produces 3AB and 3CD proteins; the minor P3 cleavage pathway (II) produces 3A and 3BCD proteins (Fig. 1B) (60). In some cases, the intermediates have unique activities, specificities, and/or functions relative to their corresponding terminal cleavage products.
Over the past 8 years much has been learned about oriI-templated VPg uridylylation in vitro for a variety of picornaviruses (28, 49, 53, 77, 92). However, it is still unclear whether or not VPg, 3BC(D), or 3AB is used in vivo to initiate genome replication. The VPg peptide can be uridylylated in vitro (62); however, VPg-pUpU does not chase efficiently into full-length RNA (81). 3BC(D) is uridylylated more efficiently than VPg in vitro, leading to the possibility that this precursor could be used in vivo (60). To date, 3AB has been uridylylated in vitro only in the presence of Mn2+ (66). In order to begin to probe the origin of VPg that is linked to picornaviral RNA, we created a PV mutant in which the cleavage site between 3B and 3C was changed from Gln-Gly to Gly-Gly (60). We refer to this mutant as GG. The GG mutation should be lethal for genome replication if use of the processed VPg peptide is absolutely required for genome replication. For the GG mutant, products of the major P3 cleavage pathway were no longer 3AB and 3CD but were now 3ABC and 3D instead. The kinetics of genome replication were reduced for the GG mutant relative to those for the wild type (WT). Surprisingly, the yield of replicated GG RNA was within an order of magnitude of that observed for WT RNA. Replicated GG RNA was then linked covalently to 3BC instead of VPg, as observed for WT PV. In spite of the substantial yield of replicated RNA, infectious virus was not recovered.
We have performed a molecular characterization of the GG mutant. GG PV RNA is quasi-infectious. The low yield of virus recovery relative to replicated RNA reflects a block at a step in the PV multiplication cycle positioned after genome replication but prior to virus assembly. The existence of this step in the PV life cycle was suggested previously by Baltimore (8). Surprisingly, none of the defects associated with GG PV could be attributed to the absence of 3CD protease activity, suggesting that precursors larger than 3CD may be the primary proteases employed in vivo. All of the observed defects in GG PV multiplication were ameliorated in a pseudorevertant in which the 3B-3C cleavage site was changed from Gly-Gly to Glu-Gly. This mutant is referred to as EG. Molecular characterization of EG PV revealed for the first time a trans-complementable function for 3CD in genome replication. This observation supports a role for 3CD at a step preceding genome replication within RCs, perhaps RC formation. Our studies of EG PV confirmed the existence of a step between genome replication and virus assembly that requires 3CD and/or 3AB, thus providing compelling evidence for genome replication and genome encapsidation as distinct steps in the multiplication cycle. This study highlights the utility of polyprotein cleavage site mutants for evaluation of the viral multiplication cycle.
MATERIALS AND METHODS
Materials.
Restriction enzymes, Deep Vent DNA polymerase, and Moloney murine leukemia virus reverse transcriptase were from New England BioLabs, Inc.; T4 DNA ligase was from Invitrogen Life Technologies; shrimp alkaline phosphatase was from USB; Qiaex was from Qiagen; Difco-NZCYM was from BD Biosciences; cell culture media and supplements were from Invitrogen Life Technologies (Gibco); DE-81 filter paper was from Whatman; nitrocellulose membranes, ECL Western blot detection reagent, and nucleotide solutions were from GE Healthcare; hygromycin B was from Cellgro; protein labeling mix (NEG072; 1,175 Ci/mmol), [α-32P]UTP (6,000 Ci/mmol), and [α-32P]dATP (1 mCi/ml; 3,000 Ci/mmol) were from PerkinElmer Life Sciences, Inc.; Sephadex G-25 was from Sigma; RNase inhibitor was from Promega; cacodylate and Epon were from Electron Microscopy Sciences; general chemicals and other lab ware were from VWR, Fisher Scientific, or Sigma, unless otherwise indicated.
Construction of 3B-3C Gly-Gly and Glu-Gly mutant replicons and viral cDNA; construction of a retrovirus plasmid containing PV 3CD.
In order to mutate the Gln-Gly (WT) junction between 3B and 3C to Glu-Gly (EG) in the PV cDNA and the PV subgenomic replicon, QuikChange mutagenesis (Stratagene) was used by following the manufacturer's protocol using oligonucleotides 1 and 2 (Table 1 lists all oligonucleotides used in this study; oligonucleotides were from Invitrogen Life Technologies or Integrated DNA Technologies, Inc.) and the pUC18-3A-3C-HpaI-SacII plasmid, which contains a majority of the 3A-3C coding region (76). The resulting pUC18-3A-3C-HpaI-SacII clone containing the EG mutation was digested with HpaI and SacII, and this fragment was ligated into pMoRA-HpaI-SacII and pRLuc-HpaI-SacII plasmids, both of which were digested with unique HpaI and SacII sites (76). The resultant clones contained an EG mutation between 3B and 3C. A mutation of the Gln-Gly cleavage site between 3B and 3C to Gly-Gly (GG) in the subgenomic replicon was described previously (60). The GG mutation in the viral cDNA was achieved by overlap extension PCR amplification using oligonucleotides 3, 4, 5, and 6 (Table 1) and the pMoRA plasmid as the template. The amplified fragment was cloned into the pMoRA plasmid using SnaBI and BglII sites. In order to construct the Y3F subgenomic replicon, QuikChange mutagenesis was also performed using oligonucleotides 7 and 8 (Table 1) and the pUC18-3A-3C-HpaI-SacII plasmid as a template. The resulting pUC18-3A-3C-HpaI-SacII clone containing the Y3F mutation was digested with HpaI and SacII, and this fragment was ligated into the pMoRA-HpaI-SacII and pRLuc-HpaI-SacII plasmids, which were digested with unique HpaI and SacII sites. The resultant clone encoded the Y3F mutation in 3B.
TABLE 1.
Oligonucleotides used in this study
| No. | Name | Sequence (5′-3′)a |
|---|---|---|
| 1 | PV-3B-3C-Glu-Gly-for | CGG ACA GCA AAG GTA GAA GGA CCA GGG TTC G |
| 2 | PV-3B-3C-Glu-Gly-rev | CGA ACC CTG GTC CTT CTA CCT TTG CTG TCC G |
| 3 | 3B-3C-QGtoGG-f | CGG ACA GCA AAG GTA GGA GGA CCA GGG TTC GAT TAC |
| 4 | 3B-3C-QGtoGG-r | GTA ATC GAA CCC TGG TCC TCC TAC CTT TGC TGT CCG |
| 5 | pRLuc-BglII-r | CGC AGA TCT CCA CTT CTT TGC CA |
| 6 | pMoRA-SnaBI-f | TTA TGT ACG TAC CAC CAGGAGCTC CAG TGC CCG AG |
| 7 | pRLucRA-3B-Y3F-f | GGA CAC CAG GGA GCA TTC ACT GGT TTA CC |
| 8 | pRLucRA-3B-Y3F-r | GGT AAA CCA GTG AAT GCT CCC TGG TGT CC |
| 9 | pLP-Rev-TRE BamHI 3CD for | GCG GGA TCC GCC ACC ATG GGA CCA GGG TTC GAT TAC |
| 10 | pLp-RevTRe-3CD-SphI-rev | ACG CGC ATG CTT ACT AAA ATG AGT C |
| 11 | 3A-SacII-f | GCG GAA TTC CCG CGG TGG AGG ACC ACT CCA GTA T |
| 12 | 3D seq 1085 Rev | CTC CCA TGT GAC TGT TTC AAA TG |
| 13 | 3C 1 seq | GGA CCA GGG TTC GAT TAC G |
| 14 | 3D seq 138 Rev | GTC TGT CTT AAG CCT GGG ATC G |
| 15 | 3D seq 100 | GTT TGA AGG GGT GAA GGA A |
Restriction sites are shown in boldface; codons containing nucleotide changes are italicized.
PCR was used to amplify the 3CD region using oligonucleotides 9 and 10 (Table 1) and pET26Ub-3BCD-C147G-His as the template (60). This plasmid encodes a protease-inactive 3C domain (C147G mutation in the 3C gene). The PCR product was cloned into pLP-RevTRE (BD Biosciences Clontech) using BamHI and SphI sites to obtain pLP-RevTRE-3CD-C147G. Sequencing at the Pennsylvania State University Nucleic Acid Facility was used to confirm all clones.
RNA transcription.
The pMo- and pRLuc- plasmids were linearized with EcoRI and ApaI, respectively, and purified with Qiaex II suspension (Qiagen) by following the manufacturer's protocol. RNA was then transcribed from the linearized plasmid DNAs in a 20-μl reaction mixture containing 350 mM HEPES, pH 7.5, 32 mM magnesium acetate, 40 mM dithiothreitol (DTT), 2 mM spermidine, 28 mM nucleoside triphosphates, 0.025 μg/μl linearized DNA, and 0.025 μg/μl T7 RNA polymerase as described previously (60). The reaction mixture was incubated for 3 h at 37°C, and magnesium pyrophosphate was removed by centrifugation for 2 min. The supernatant was transferred to a new tube, and RQ1 DNase (Promega) was used to remove the template. The RNA concentration was determined by measuring absorbance at 260 nm, assuming that an A260 of 1 was equivalent to 40 μg/ml, and the RNA quality was verified by 0.8% agarose gel electrophoresis.
Isolation of viral RNA from plaques.
HeLa cells (1.2 × 106) transfected with 5 μg of in vitro-transcribed pMo-Gly-Gly (GG) RNA were placed onto a monolayer of HeLa cells (0.5 × 106 cells seeded 1 day in advance) in a six-well plate. An agarose overlay (0.5%) was applied to the cells, and the plates were then incubated at 34°C for 4 days. On day 4, another overlay consisting of 0.5% agarose and 0.01% neutral red in phosphate-buffered saline (PBS) was placed on top of the original existing overlay. The plates were then incubated at 34°C overnight to allow the neutral red to diffuse to the monolayer of cells. The plaques were clearly visible the next morning without having to remove the agarose overlays. Following neutral red staining, a Pasteur pipette was used to remove the agarose directly over the visible plaque area. Five plaques were picked. The agarose was placed in Dulbecco's modified Eagle medium (DMEM)-F12 medium (1 ml) containing no serum. After overnight incubation in the medium at 4°C, the samples were passed through a centrifuge filter (Costar; SpinX) to remove the agarose. Viral RNA was then isolated from the pass-through using a NucleoSpin virus midiprep kit (BD Biosciences) by following the manufacturer's instructions. Reverse transcription was performed using oligonucleotide 5, and PCR was performed using oligonucleotides 5 and 11 (Table 1). The PCR products were gel purified and sequenced using oligonucleotide 5 at the Pennsylvania State University Nucleic Acid Facility. Five reverse transcription-PCR (RT-PCR) products corresponding to the five individual picked plaques were sequenced.
Infectious center assay.
Infectious center assays were performed as previously described (59) with the following modifications. For each transfection, 1.2 × 106 cells were centrifuged at 1,000 rpm for 4 min and resuspended in 400 μl of PBS. Five micrograms of viral RNA was mixed with the 400 μl of cell suspension. The RNA-cell mixture was transferred to an electroporation cuvette (0.2-cm gap width; VWR), and an electric pulse was applied at 500 μF and 130 V using a Gene Pulser system (Bio-Rad). Electroporated cells were serially diluted as described in the figure legends. One hundred microliters of undiluted and diluted electroporated HeLa cells was placed on top of a HeLa cell monolayer (0.5 × 106 cells seeded 1 day in advance) in a six-well plate. The plates were incubated for 1 h at 37°C to allow the electroporated cells to adsorb to the monolayer. The cells were overlaid with 1% low-melting-point agarose (EMD). The overlay was allowed to solidify for 20 min at room temperature, and the plate was incubated for 2 days at 37°C. Infections centers were visualized by crystal violet staining.
Establishment of a 3CD-expressing stable cell line.
RetroPack PT67 packaging cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), streptomycin-penicillin (100 U/ml), l-glutamine (4 mM), and high glucose (4.5 g/liter) at 37°C with 5% CO2. One day before transfection, 2 × 106 cells were seeded to 100-mm plates and pLP-RevTRE-3CD-C147G was transfected into cells using Effectene (Qiagen) by following the manufacturer's protocol. Briefly, 2 μg of DNA was diluted into 192 μl of buffer EC (Qiagen), 8 μl of enhancer was then added, and the mixture was incubated for 5 min at room temperature. Effectene (16 μl) was added, and the mixture was incubated for 10 min at room temperature. During this incubation, RetroPack PT67 cells were washed with 4 ml of PBS and suspended in 9 ml of fresh DMEM. The DNA-Effectene mixture was diluted in 1 ml of DMEM (without any supplement) and added to the RetroPack PT67 cells. Transfected RetroPack PT67 cells were held at 37°C with 5% CO2 for 24 h to release the 3CD gene-containing retrovirus into the media. HeLa Tet-off cells (Clontech Laboratories, Inc.) were maintained in DMEM supplemented with 10% FBS and streptomycin-penicillin (100 U/ml). One day prior to the first infection, 1 × 105 HeLa Tet-off cells were seeded in a 100-mm plate. After a 24-h incubation of the RetroPack PT67 cells, the retrovirus-containing medium was harvested and filtered through a 0.45-μm cellulose acetate syringe top filter. The filtered medium was diluted two times with fresh medium supplemented with Polybrene (8 μg/ml) and doxycycline (Dox) (1 μg/ml). In order to infect the HeLa Tet-off cells, old medium was replaced with 10 ml of the filtered retrovirus-containing medium and the cells were held at 37°C with 5% CO2 until the next infection. Infection was repeated every 16 to 20 h up to three times prior to antibiotic-based selection. After three infections, infected cells were selected in antibiotic-containing (hygromycin B [0.2 mg/ml], G418 [0.2 mg/ml], and Dox [1 μg/ml]) medium at 37°C with 5% CO2. Every 3 days, medium was replaced with fresh medium supplemented with hygromycin B, G418, and Dox. Uninfected HeLa Tet-off cells were also treated with the same concentrations of hygromycin B, G418, and Dox until all the cells were dead as a control. Fourteen days from the initial treatment with antibiotics, individual large colonies (96 colonies) were isolated and transferred to 12-well plates by using cylinder-aided trypsinization. From this point, each isolated colony was expanded with hygromycin B (0.1 mg/ml), G418 (0.1 mg/ml), and Dox (1 μg/ml).
In order to induce 3CD expression, each isolated HeLa-3CD cell line was washed three times with PBS and passaged to a new plate. Passaged HeLa-3CD cells were incubated with fresh medium in the presence of hygromycin B and G418 (0.1 mg/ml, each) but in the absence of Dox. Next day, the cells were washed three times with fresh medium for 3 h, followed by additional incubation for 36 h at 37°C. 3CD expression was confirmed by Western blot analysis using PV 3D antiserum. Total RNA was purified by using Trizol reagent in accordance with the manufacturer's protocol. Reverse transcription was performed with oligonucleotide 12, followed by PCR with oligonucleotides 13 and 14 (Table 1). The DNA sequence was confirmed by sequencing the PCR fragment at the Pennsylvania State University Nucleic Acid Facility.
Subgenomic luciferase replicon assay.
Subgenomic luciferase assays were performed as described previously (60) with the following modifications. Subgenomic replicon RNA (5 μg of in vitro-transcribed RNA) was electroporated into either HeLa cells, uninduced HeLa-3CD cells, or induced HeLa-3CD cells. The cells were incubated in normal growth medium (DMEM-F12 supplemented with 10% FBS, 1% penicillin-streptomycin, and 5 ml of cells [1 × 106 cells]), and 1 × 105 cells were harvested and lysed using 100 μl of 1× cell culture lysis reagent (Promega) postelectroporation at various times as described in the figure legends. In some cases, cordycepin (3-deoxyadenosine; Sigma) was added to the growth medium at various concentrations (0 to 400 μM) to inhibit RNA synthesis. Luciferase assays were performed by mixing an equal volume of firefly luciferase assay substrate (Promega) with the lysate. The reaction mixture was applied to a Junior LB 9509 luminometer (Berthold Technologies), and the relative light units (RLU) were quantified for 10 s. RLU were then normalized based on the total protein concentration determined by Bio-Rad protein assay reagent for each sample.
One-step growth curve assay.
One day prior to infection, HeLa cells, uninduced HeLa-3CD cells, or induced HeLa-3CD cells were propagated and seeded at 5 × 105 cells per well in a six-well plate. On the day of the infection, the prepared HeLa monolayers were washed and infected at a multiplicity of infection (MOI) of 10 PFU per cell with WT or EG PV for 30 min at room temperature. Infected cells were washed once with PBS to remove unattached virus and then incubated at 37°C. At various times postinfection, virus was harvested from the cells and media by three cycles of freeze-thaw with vortexing in between. The isolated virus was titered by plaque assay as described above.
Virus purification.
HeLa cell monolayers were prepared in 100-mm tissue plates by seeding 3.5 × 106 cells 1 day before RNA transfection. WT or EG PV genomic RNA (5 μg of in vitro-transcribed RNA) was electroporated into 1.2 × 106 cells, followed by seeding onto the HeLa monolayer and incubation at 37°C until cytopathic effect (CPE) appeared (48 h). The cells and media were collected, and PV particles were released from cell debris by three cycles of freeze-thaw with vortexing in between. The supernatant was collected after centrifugation at 12,000 × g for 10 min. PV particles were then precipitated in precipitation buffer (0.3 M NaCl, 10% polyethylene glycol [PEG] 8000, pH 7.2 to 7.5) for 16 h at 4°C (78). The precipitated PV particles were harvested by centrifugation at 12,000 × g for 30 min at 15°C. The PV pellet was resuspended in 1 ml of TN buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl). In order to remove residual cellular debris, the resuspended PV was centrifuged on top of 3 virus volumes through a 30% sucrose cushion (0.1 mM NaCl in 30 mM Tris-HCl, pH 8.0) at 190,000 × g for 16 h (S55-S; Beckman). The pellet was resuspended in 1 ml of TND buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl, and 2% deoxycholic acid). The resuspended sample was dialyzed against TN buffer (1 liter) using Spectra/Por dialysis tubing with a molecular mass cutoff of 50,000 Da overnight at 4°C. The dialyzed virus was centrifuged through a 30% sucrose cushion at 190,000 × g for 16 h. The final pellet was suspended in 0.1 ml of TN.
For GG virus particle purification, 350 μg of transcribed GG RNA was electroporated into 1.2 × 107 HeLa cells (35 μg per 1.2 × 106 cells) and cells were incubated in 30 ml of prewarmed DMEM-F12 (supplemented with 10% FBS and 1% penicillin-streptomycin). The transfected cells were incubated for 20 h at 34°C; virus was harvested and purified as described above.
Negative staining.
The purified WT, EG, or GG PV was negatively stained with aqueous uranyl acetate (UA). Briefly, 10 μl of the purified PV was placed on a 200- to 400-mesh Formvar copper grid and incubated for 3 min at room temperature. Residual PV was wicked off with a wedge of filter paper without disturbing the grid. Aqueous UA (10 μl of 0.5 to 2%) was overlaid on the grid to cover the absorbed PV; excessive UA was wicked off immediately. The grid was air dried completely prior to transmission electron microscopy (Jeol; JEM 1200 EXII).
Preparation of HeLa cytoplasmic extracts and in vitro translation reaction with HeLa cell extracts.
Cytoplasmic extracts from HeLa S3 cells were prepared as previously described (20, 51) with slight modifications. Harvested HeLa cells (1.37 × 109) were washed with 40 ml of ice-cold isotonic buffer (35 mM HEPES, pH 7.5, 146 mM NaCl, and 11 mM glucose) three times and suspended in an equal volume of hypotonic buffer (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM Mg acetate, and 1 mM DTT). The cells were swollen in ice for 10 min and transferred to a prechilled Dounce homogenizer (Belco type). The cells were homogenized with 10 to 50 strokes until 90% lysis observed by trypan blue staining. Freshly made 10× buffer (0.1 volume) (200 mM HEPES, pH 7.5, 1,200 mM K acetate, 40 mM Mg acetate, 50 mM DTT) was added to the lysed cells. The extract was transferred to a prechilled tube and centrifuged for 10 min at 2,000 rpm. The supernatant was collected in a prechilled tube and centrifuged again at 9,000 rpm for 10 min. The new supernatant was collected and combined with the first supernatant. The supernatant was dialyzed (12,000- to 14,000-Da cutoff) in 1 liter of dialysis buffer (40 mM HEPES-free acid, pH 8.0, 120 mM K acetate, 5.5 mM Mg acetate, 10 mM KCl, and 6 mM DTT) for 2 h with one buffer exchange. The viscous supernatant was frozen at −80°C overnight. Next day, the supernatant was thawed and 1/1,000 volume of 1 M CaCl2 (final concentration, 1 mM) was added, followed by addition of micrococcal nuclease (final concentration, 75 units/ml; Fermentas) and gentle mixing. The mixture was incubated at room temperature for 15 min, and 1/100 volume of 200 mM EGTA was added (final concentration, 2 mM), followed by centrifugation at 9,000 rpm for 10 min in 4°C. The supernatant was aliquoted (100 μl each) and stored at −80°C until it was used. The translation reaction was performed as described previously (24) with the following modifications. Ten percent rabbit reticulocyte lysate (Promega) was used as an initiation factor and radiolabeling was performed with 1 μCi of [35S]methionine (GE Healthcare) per reaction at 30°C for various times.
Generation and purification of polyclonal PV VPg and 3D antiserum.
Generation and purification of PV VPg, 3C, and hepatitis C virus (HCV) nonstructural protein 5A (NS5A) polyclonal antibodies were described previously (37, 60). Purified PV 3D was used to raise polyclonal antiserum in rabbits (Covance Research Products, Denver, PA). Anti-3D antiserum was purified by using ammonium sulfate precipitation and DEAE-Affi-gel Blue gel (Bio-Rad) column purification as described previously (60) with slight modifications. Initially, 5 ml of anti-3D polyclonal antiserum (PA 473) was precleared by centrifugation at 16,000 × g at 4°C for 15 min. Ammonium sulfate was added over 30 min up to 33% saturation while the precleared antiserum was stirred at 4°C. After 2 h of additional stirring, precipitated antiserum was collected by centrifugation at 70,000 × g at 4°C. The antiserum pellet was resuspended in 5 ml of loading buffer (20 mM Tris-HCl, pH 8.0, and 50 mM NaCl), followed by dialysis against 1 liter of loading buffer for 12 h with one exchange. Dialyzed antiserum was loaded onto a DEAE-Affi-Gel Blue (Bio-Rad) column at 0.16 ml/min. A bed volume of 1 ml resin per 5 mg of total protein was packaged. DEAE-Affi-Gel Blue was washed with 5 column volumes of prewashing buffer (100 mM acetic acid, 1.4 M NaCl, and 40% isopropyl alcohol) and 10 column volumes of loading buffer continuously. The pass-through samples were collected (1 ml per fraction), and protein concentration was determined by using Bio-Rad protein assay reagent. Fractions containing immunoglobulin Gs were pooled and concentrated using Vivaspin (30,000-Da cutoff; Sartorius). Glycerol and sodium azide were added to concentrated antiserum (50% and 0.1%, respectively). The antiserum was aliquoted and stored at −80°C.
Western blot analysis of replicon proteins.
Western blot analysis was performed as described previously (60). Briefly, HeLa cells were transfected with transcribed RNA as described above. The transfected cells (1.2 × 106) were incubated at 34°C for the times indicated in the figure legends and harvested by centrifugation at 14,000 × g for 2 min. The cells were lysed in 1× cell culture lysis reagent (100 μl; Promega), and luciferase activity was measured as described above. The remainder of lysed cells were mixed with an equal volume of 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading dye (225 mM Tris, pH 6.8, 5% SDS, 50% glycerol, 5% β-mercaptoethanol, and 0.05% bromophenol blue) and boiled for 10 min. Proteins were separated by 8, 10, or 15% SDS-PAGE and transferred to a nitrocellulose membrane (GE Healthcare) using the Genie transfer unit (Idea Scientific Company) for 1 h at 24 V in transfer buffer (25 mM Tris-glycine, 3 mM SDS, and 20% [vol/vol] methanol). The membrane was blocked in 5% (wt/vol) skim milk in TBS-T (137 mM NaCl, 0.1% Tween 20, and 20 mM Tris, pH 7.6) for 1 h at room temperature. Polyclonal antisera used in this study against synthesized (VPg) or purified recombinant PV proteins (3C-His, 3D, and NS5A-His) were produced in rabbits (Covance Research Products, Inc.). The membrane was incubated in TBS-T containing diluted purified (anti-VPg and anti-3C) or unpurified (anti-3D) polyclonal antisera (dilutions: anti-VPg, 1:2,500; anti-3C, 1:5,000; anti-3D, 1:10,000) for 1 h at room temperature. The membrane was incubated with secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit secondary antibody; Santa Cruz Biotechnology, Inc.) in TBS-T at 1:10,000 for 1 h at room temperature. Immunocomplexes were detected by using the ECL system (GE Healthcare) and Kodak Biomax MR film.
Immunofluorescence.
Induced or uninduced HeLa-3CD cells were fixed with fixation buffer (3.7% formaldehyde) for 20 min at room temperature. After the cells were washed with buffer A (PBS [20 mM phosphate and 150 mM NaCl, pH 7.4], 0.2% saponin) three times, blocking was performed in buffer B (3% bovine serum albumin in buffer A) for 30 min at room temperature. 3D and actin were probed with polyclonal anti-3D antiserum (PA 473, 1:1,000 dilution) and anti-β-actin (Abcam; 1:15,000 dilution) in buffer B for 30 min at room temperature, followed by incubation with donkey anti-rabbit Alexa 647 (Red, Molecular Probes) and goat anti-mouse Alexa 488 (dilution, 1:1,000) secondary antibodies for another 30 min. The cells and proteins were visualized by using Olympus Fluoview 300 at the Cytometry Facility at Pennsylvania State University.
Transmission electron microscopy (TEM).
Uninduced or induced HeLa-3CD cells (3 × 106) were harvested and washed with prewarmed 1× PBS and fixed with 1% glutaraldehyde in 1× PBS for 15 min at room temperature. An additional fixation was performed with fresh 1% glutaraldehyde in 1× PBS for 60 min at 4°C. The next few procedures were all done on ice. Once the cells turned into a firm, yellow pellet, 0.1 M cacodylate (sodium dimethyl arsenate; Electron Microscopy Sciences) was used twice to wash the pellet for 5 min. The pellet was incubated in 1% reduced osmium tetroxide containing 1% potassium ferricyanide in 0.1 M cacodylate for 60 min in the dark with one exchange. The pellet was briefly washed two times with 0.1 M cacodylate. En bloc staining was performed with 3% UA in 50% ethanol for 30 to 60 min in the dark. Extra UA was removed by three washes with 50% ethanol for 5 min. Dehydration was carried out with different concentrations of ethanol (70 and 95%) for 10 min. From this point forward, all the embedding procedures were done at room temperature. The pellet was washed with 100% ethanol for 10 min, followed by three washes with 100% acetonitrile. The pellet then was treated with 1:1 acetonitrile-Epon (Electron Microscopy Sciences) for 1 h while being rotated. One hundred percent of the Epon was replaced twice for every hour prior to embedding. Embedding was performed overnight with 100% Epon at 65°C. The embedded sample was sectioned with a diamond knife (DiATOME) to slice at 60- to 90-nm thickness by using an ultramicrotome (Reichart-Jung). The sectioned sample was placed on a 300-mesh copper grid (Electron Microscopy Sciences) and dried prior to staining. UA-lead citrate staining was carried out as described below. On the day of staining, deionized H2O was boiled to remove CO2 and cooled down to room temperature. Two percent UA in 50% ethanol was filtered (0.2-μm syringe filter), and a drop of UA was placed on a wax dent. The grid faced down to allow a section to contact UA, and it was incubated for 16 min in a dark container. The grid was quickly immersed in the boiled H2O with forceps and washed by agitating vertically for 1 min. Excess H2O was removed with a wedge of filter paper, and the grid was dried completely. In order to do lead staining, NaOH pellets were placed around the edge of a staining dish and the lid was closed until staining. A drop of filtered lead staining solution (4.35 to 4.65 mg lead citrate with 1.35 to 11.85 μl of 10 N NaOH [fresh] in 1.2 ml) was placed in the staining dish, and the grid was placed section side down on the drop for 12 min. During the lead citrate staining, the staining dish was closed securely to prevent lead precipitation. The grid was agitated vertically for 30 s in 40 ml of boiled H2O (2 drops of 10 N NaOH were added). The grid was quickly transferred and agitated for 30 s in 40 ml of boiled water (1 drop of 10 N NaOH was added), followed by a 1-min rinse in 40 ml of boiled H2O as above. Excess H2O was removed with the wedge of filter paper, and the grid was dried completely. The image was obtained by using a Jeol JEM 1200 EXII in the Electron Microscopy Facility at Pennsylvania State University.
Virus assembly and encapsidation assay in the presence of hydantoin.
WT or EG PV was adsorbed to a HeLa cell monolayer at an MOI of 1 for 30 min in the presence of 50 μg/ml of 5-(3,4-dichlorophenyl)-methylhydantoin (referred to as hydantoin). Hydantoin was a gift from Marie Chow (University of Arkansas for Medical Sciences). The infected cells were incubated with hydantoin (50 μg/ml) at 37°C for 6 or 8 h until WT or EG PV replication was completed, respectively, and hydantoin was removed by washing cells with PBS. Prewarmed fresh medium was added to the cells, and the cells were kept at 37°C until they were harvested. At various time points, virus was isolated and titered as described above.
Immunoprecipitation of RNA.
HeLa cells (6 × 106) were transfected with 175 μg of transcribed RNA (35 μg/1.2 × 106 cells) as described above. Total RNA was prepared by using an RNAqueous-4PCR kit (Ambion). The transfected cells were lysed with 0.5 ml lysis/binding buffer, followed by addition of an equal amount of 64% ethanol and mix buffer in the kit. The lysate-ethanol mixture was passed through the provided filter cartridge for 30 s at 12,000 rpm in a tabletop centrifuge. The filter cartridge was washed with 0.7 ml of wash solution 1 once and 0.5 ml of wash solution 2/3 twice. Total RNA was eluted two times with 50 μl of prewarmed (70°C) elution buffer, which is provided. The RNA quantity was determined by using NanoDrop 1000 (Thermo Fisher Scientific), and the quality was evaluated by using 0.8% agarose gel. Total RNA was normalized based on the 18S band. Immunoprecipitation was performed with total RNAs prepared from WT- and EG and Y3F mutant-transfected HeLa cells as described previously (60). Total RNA was aliquoted equally into four microcentrifuge tubes (20 μg each) containing 500 μl of IP buffer (50 mM Tris, pH 7.4, 0.5 M NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS). Thirteen micrograms of purified polyclonal anti-VPg, anti-3C, anti-3D, or anti-NS5A was added to the total RNA, and the mixtures were rotated at room temperature for 45 min. For each RNA-antiserum mixture, 35 μl of protein A magnetic beads (New England Biolabs) was preconditioned by two washes with IP buffer (1 ml for each wash) and resuspended in 50 μl of IP buffer. The preconditioned protein A magnetic beads were added to the RNA-antiserum mixtures and rotated for 1.5 h at room temperature. The beads were collected by using a magnetic stand (Dynal, Oslo, Norway). The beads were washed four times with IP buffer and twice with wash buffer (50 mM Tris, pH 7.4, and 150 mM NaCl) and resuspended in 8 μl of water, and 16 μl of formaldehyde gel loading mix (20 mM MOPS [morpholinepropanesulfonic acid], 63.3% formamide, 23.3% formaldehyde, 5 mM sodium acetate, 4.3 mM EDTA, 0.066% bromophenol blue, 0.066% xylene cyanol) was added to immunoprecipitated RNA for Northern blot analysis. RNAs isolated from purified virus particles were also immunoprecipitated as described above. Two of the HeLa cell monolayers were prepared in 100-mm tissue plates by seeding 3.5 × 106 cells 1 day before infection. On the day of the infection, the prepared HeLa monolayers were washed and infected at an MOI of 10 with WT or EG PV and incubated at 37°C until CPE appeared (48 h). The cells and media were collected to purify PV particles, and viral RNA was purified by using the RNAqueous-4PCR kit (Ambion) as described above.
Northern blot analysis.
The RNA was separated on a 0.6% denaturing agarose gel (0.8 M formaldehyde in 1× MOPS) by running at 120V for 2 h until the bromophenol blue band migrated through three-fourths of the gel. The gel was washed in water for 30 min twice, followed by soaking in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) for 30 min. RNA was transferred to a nylon membrane (Hybond XL; GE Healthcare) using capillary blotting with 20× SSC for 16 h at room temperature. The membrane was slightly dried, and RNA was cross-linked to the membrane using a UV cross-linker (Stratalinker 2400; Stratagene). The membrane was washed twice with wash buffer (1× SSC and 0.1% SDS) at 65°C for 30 min each. The membrane was prehybridized in 100 ml of modified Church's buffer (0.5 M sodium phosphate, pH 7.2, 7% SDS, and 1 mM EDTA) for 4 h at 65°C. The hybridization probe was denatured for 5 min at 95°C and chilled on ice for 1 min. Hybridization was performed in modified Church's buffer for 16 h at 65°C. The membrane was washed in wash buffer (1× SSC and 0.1% SDS) for 20 min at 65°C twice and once at room temperature. RNA was visualized by exposing the membrane to a phosphor screen followed by scanning the screen on a Typhoon 8600 scanner (Promega) in the phosphor mode.
The hybridization probe was made by PCR using oligonucleotides 12 and 15 (Table 1) and pRLuc RA as a template. In the reaction, [α-32P]dATP (1 mCi/ml, 3,000 Ci/mmol; GE Healthcare) was used with cold deoxynucleoside triphosphates (300 μM each for dCTP, dGTP, and dTTP and 10 μM for dATP) in a 100-μl reaction mixture. The quality of the PCR product was confirmed by agarose gel electrophoresis, and the number of cpm was determined by using a scintillation counter (LKB Wallac; 1217 Rackbeta liquid scintillation counter).
RESULTS
Characterization of a PV mutant with a defective 3B-3C cleavage site (GG PV) and isolation of a pseudorevertant (EG PV).
We recently suggested that the VPg uridylylation reaction of picornaviruses may employ a VPg-containing precursor rather than the processed VPg peptide (60). Our approach to address this question was the creation of a PV subgenomic replicon that prevented production of the VPg peptide by mutating the sequence encoding the Gln-Gly cleavage site between 3B and 3C to encode a Gly-Gly cleavage site. This mutant is referred to as GG. We observed replication of the GG subgenomic replicon, albeit with reduced kinetics relative to those of the corresponding WT replicon (60). The major P3 precursor processing program (pathway I in Fig. 1B) was altered such that 3ABC and 3D were produced instead of 3AB and 3CD (60). In addition, 3BC was covalently linked to the 5′ end of GG subgenomic RNA (60). Given these phenotypes, it was not surprising that infectious virus could not be produced from GG genomic RNA under conditions in which WT genomic RNA yielded high-titer virus.
Specific infectivity of genomic RNA.
This study was motivated by a desire to determine the molecular basis for the apparent inability of GG genomic RNA to yield virus. First, we measured the specific infectivity of WT and GG genomic RNAs. RNA was produced by in vitro transcription of the WT or mutated cDNA and transfected into HeLa cells by electroporation. The pool of transfected cells was placed on a monolayer of HeLa cells, overlaid with agarose, and incubated at 34°C for 5 days. The overlay was removed, and the cells were stained with crystal violet to reveal infectious centers. Infectious centers were produced by WT genomic RNA at both 1,000- and 10,000-fold dilutions (Fig. 2A), yielding a specific infectivity of ∼70,000 infectious centers/μg RNA (Table 2). For GG genomic RNA, infectious centers were apparent only in the undiluted pool of transfected cells (Fig. 2A), yielding a specific infectivity of 1 infectious center/μg RNA. Therefore, GG genomic RNA is ∼70,000-fold less infectious than WT genomic RNA.
FIG. 2.
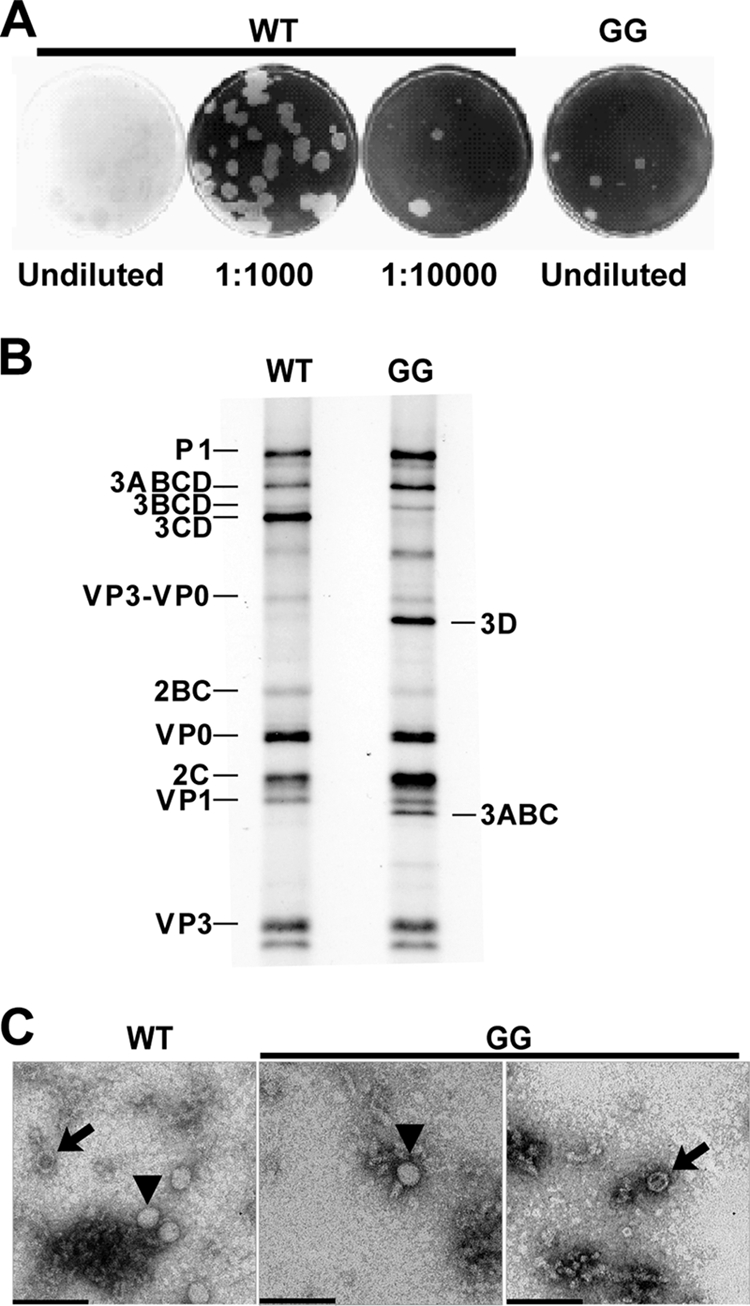
Changing the 3B-3C cleavage site from QG to GG produces a quasi-infectious virus. (A) Specific infectivity of GG mutant and WT PV RNA. HeLa cells were transfected with GG mutant or WT PV RNA, diluted, added to HeLa cell monolayers, overlaid with agarose, and held at 34°C for 5 days, at which time the agarose overlay was removed and cells were stained with crystal violet. From each of the five plaques obtained for the GG mutant RNA transfection, virus was plaque purified and viral RNA was extracted, reverse transcribed, and sequenced. Sequencing identified a single mutation at the 3B-3C cleavage site that converted GG to EG. (B) Processing evaluated by cell-free translation. HeLa cell-free translation extracts containing [35S]methionine and [35S]cysteine were programmed with WT or GG mutant RNA. Radiolabeled proteins were separated by 15% SDS-PAGE and detected by phosphorimaging. The bands corresponding to the different precursor and processed proteins expected for the WT are indicated on the left. The identities of bands unique to the GG mutant are indicated on the right. (C) TEM of WT and GG virus particles. HeLa cells were transfected with GG mutant or WT PV RNA and incubated at 34°C for 20 h, at which time cells were harvested and lysed with three freeze-thaw cycles. WT and GG mutant PV particles were then purified by precipitation using 10% PEG 8000 followed by centrifugation through a 30% sucrose cushion. Purified virus particles were negatively stained with 2% UA and observed by TEM. Bars = 0.1 μm. Representative virus particles that are empty (arrows) or contain packaged RNA (▾) are indicated.
TABLE 2.
Characterization of virus particles produced by WT, GG, and EG genomic RNA
| Virus | Specific infectivitya (infectious centers/μg RNA) | No. of particlesb | % Empty particlesc |
|---|---|---|---|
| WT | 6.7 × 104 | 1.4 × 106 | 32 ± 3 |
| GG | 1 | 1.3 × 103 | 46 ± 5 |
| EG | 1.4 × 104 | 1.3 × 106 | 39 ± 4 |
Specific infectivity was measured by a virus infectious center assay (Fig. 3). The error associated with this measurement was less than 20%.
Equal volumes of virus precipitated from genomic-RNA-transfected cells was evaluated by TEM. The indicated values represent the average numbers of particles per TEM grid, which has an area of 7.2 × 1011 nm2. For the WT and EG, the value was calculated by evaluating the number of particles in three regions of the grid. Each region was 3.4 × 106 nm2. The number of particles observed in each region varied by less than 20%. For GG, four regions were evaluated. Each region was 1.8 × 109 nm2. These data should be used qualitatively because we have not controlled for virion loss during the isolation procedure.
Empty particles observed by TEM were scored as indicated in the legend to Fig. 2C.
Polyprotein processing.
It is generally believed that PV 3C and 3CD proteins are responsible for most cleavage events within the polyprotein (47). However, there is some evidence that 3CD cleaves the P1 precursor more efficiently than 3C and may require a cofactor (14, 41). Therefore, it was surprising to observe robust cleavage of P2 and P3 precursors produced by translation of GG subgenomic replicon RNA because these precursors fail to produce both 3C and 3CD (60). It was possible that precursor forms of 3C other than 3CD might be incapable of cleaving the P1 precursor, giving rise to the reduced infectivity of GG PV. In order to test this possibility, WT and GG genomic RNAs were translated in HeLa-based cell extracts in the presence of [35S]methionine. After 4 h of incubation at 30°C, samples were processed for electrophoresis to resolve labeled cleavage products. These products were visualized by phosphorimaging. Consistent with results obtained previously by using subgenomic RNA, P2 processing and P3 processing were similar for both WT and GG polyproteins (Fig. 2B). The major pathway for P3 processing was altered for the GG polyprotein, producing 3ABC and 3D instead of 3AB and 3CD (Fig. 2B). The GG polyprotein appeared to yield higher levels of 2C than the WT polyprotein (Fig. 2B). This difference was not observed previously in our analysis of GG P2-P3 polyprotein produced by using subgenomic replicon RNA (60). Therefore, the identity of this protein cannot be assigned unambiguously. Interestingly, the P1 precursor from the GG polyprotein produced normal levels of VP3-VP0, VP0, VP1, and VP3 proteins (Fig. 2B). This observation suggests that P1 processing can be achieved by precursor forms of 3C other than 3CD.
Virus particle analysis.
We have shown that RNA produced by replication of GG subgenomic replicon RNA is linked covalently to 3BC protein. There has been some suggestion that 3B may be a signal for encapsidation (65); 3BC may not be able to serve this function. Direct analysis of virus particles by electron microscopy would not only provide information on the extent to which encapsidation occurred but would also provide an alternative perspective on virus particle concentration. HeLa cells were transfected with WT or GG genomic RNA and incubated under conditions optimal for RNA synthesis. Virus was released from cells by multiple freeze-thaw cycles and concentrated by PEG precipitation. Virus was purified further by centrifugation through a sucrose cushion. A portion of the virus pellet was used to prepare cDNA for sequencing, which confirmed that the isolated particles were indeed GG PV instead of a revertant (data not shown). The remainder of the virus pellet was suspended and processed for visualization by TEM after UA staining. Both full and empty virus particles were observed in preparations from both WT and GG genomic RNA-transfected cells (Fig. 2C). The amount of GG PV per grid was reduced by at least 3 logs relative to that of WT PV (Fig. 2C and Table 2). The fraction of empty GG particles was elevated by only 40% relative to WT particles (Table 2). Because the yield of virus particles is at least 10-fold lower than the amount of RNA synthesized without a substantial reduction in encapsidation efficiency, we conclude that GG PV is also defective for virus particle assembly. The absence of a substantial encapsidation defect would suggest that determinants of 3B required for encapsidation are accessible in 3BC or that 3B lacks a role in encapsidation.
Isolation of a pseudorevertant of GG PV.
The quasi-infectious nature of GG PV permitted us to use the power of PV evolution and selection to identify suppressors of one or more of the GG PV phenotypes, facilitating a deeper insight into the molecular basis for the observed phenotypes. Two approaches were pursued. First, infectious center assays were performed as described above; however, phenol red staining was used to isolate agarose plugs containing virus. Virus extracted from these plugs was divided into two fractions. One fraction was used for cDNA synthesis and amplification by RT-PCR for sequencing; the other fraction was used as an inoculum to amplify the virus. This amplified virus pool was also processed for sequencing. Second, we used medium fractions from HeLa cells transfected with GG genomic RNA as an inoculum to initiate a series of blind passages that led to the recovery of a rapidly growing, cytolytic virus population. This virus population was also processed for cDNA synthesis and sequencing. In all cases, the only missense mutation observed changed the Gly-Gly cleavage site between 3B and 3C to Glu-Gly. The mutant is referred to as EG.
Reduced specific infectivity of EG genomic RNA.
In order to prove that the missense mutation observed in the viral population caused the increase in virus multiplication efficiency, site-directed mutagenesis was employed to create the EG cDNA. The nucleotide substitutions made were designed to limit reversion to the WT codon by requiring a transversion for reversion, a low-frequency event for PV polymerase (7, 25). EG-specific infectivity was measured by infectious center assay as described above. Infectious centers produced by EG genomic RNA were smaller than infectious centers produced by WT genomic RNA (Fig. 3A). The specific infectivity of EG genomic RNA was ∼10,000 infectious centers/μg, a value ∼10,000-fold higher than that observed for GG genomic RNA but still ∼7-fold lower than that observed for WT genomic RNA (Fig. 3B and Table 2). Together, these observations suggest that EG PV is at least partially defective relative to WT PV but robust enough in cell culture relative to GG PV to be used as a tool to obtain insight into the mechanistic basis for the phenotype observed for GG PV.
FIG. 3.

Changing the 3B-3C cleavage site from GG to EG restores infectivity. (A) Infectious center assay for EG mutant and WT PV RNA. HeLa cells were transfected with in vitro-transcribed EG mutant or WT PV RNA, diluted, added to HeLa cell monolayers, overlaid with agarose, and held at 37°C for 2 days, at which time the agarose overlay was removed and cells were stained with crystal violet. (B) Quantitation of data shown in panel A. The specific infectivities for WT and EG mutant RNA are 68,000 ± 10,000 and 14,000 ± 1,000 PFU/μg RNA, respectively.
Characterization of EG PV.
The observation that the specific infectivity of EG genomic RNA was reduced relative to that of WT genomic RNA encouraged detailed characterization of the EG PV multiplication cycle.
One-step growth analysis.
The infectious center assay requires virus not only to be produced but also to spread. Therefore, reduced specific infectivity could be caused by defects after virus assembly. In order to rule out this possibility, HeLa cells were infected with EG PV at an MOI of 10. At various times postinfection, the titer of virus was determined by plaque assay. Comparison of the one-step growth curve for EG PV to that for WT PV revealed two major differences. First, EG PV exhibited an ∼1-h lag in appearance of infectious virus relative to WT PV (Fig. 4A). Second, the fast rate of infectious virus production observed for WT PV between 4 and 6 hours postinfection (5 × 107 PFU/ml/h) was reduced by ∼20-fold. Interestingly, the final yield of EG PV (3 × 108 PFU/ml) was very similar to that of WT PV (4 × 108 PFU/ml). These data would suggest that the specific infectivity as determined by using the infectious center assay reports on changes in the kinetics of infectious virus production instead of overall virus yield. This observation may reflect competition between degradation and translation/replication of the transfected RNA that produces the inoculum that spreads, causing the cell death that is monitored.
FIG. 4.
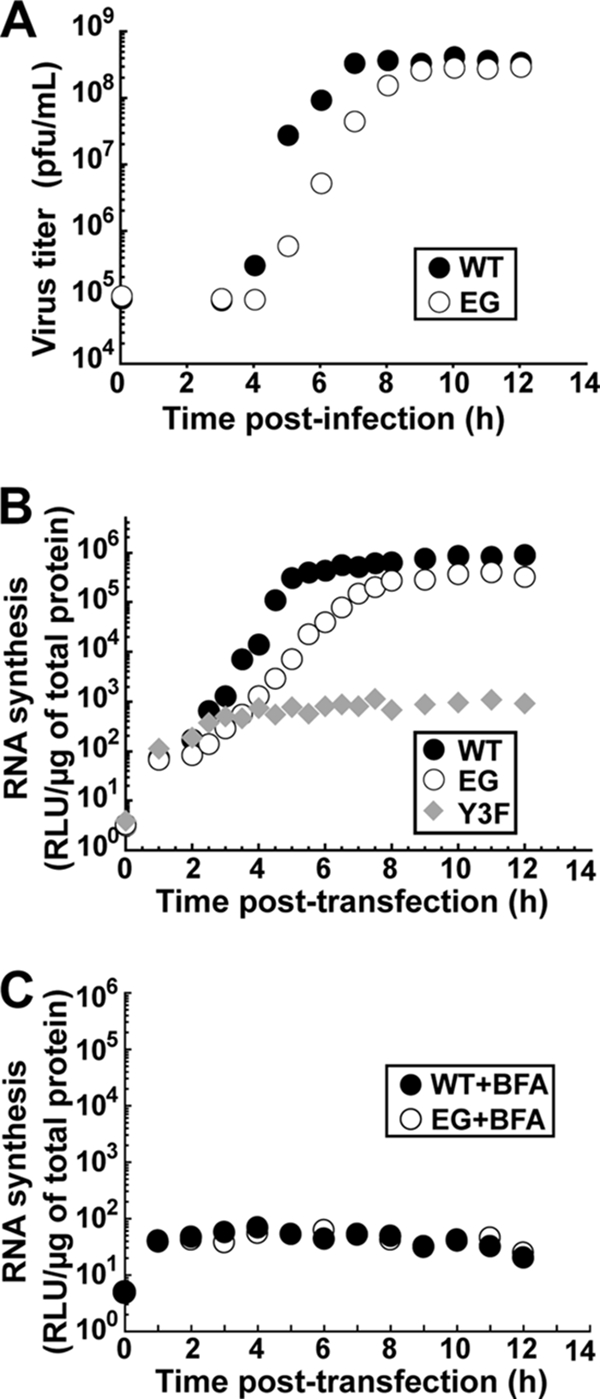
EG PV exhibits a decreased rate of RNA synthesis and virus production. (A) Kinetics of virus production by WT and EG PV. HeLa cells were infected with WT or EG PV (MOI, 10) and held at 37°C. At the indicated times postinfection, cells were harvested and lysed by freeze-thawing and virus was titered. (B) Kinetics of RNA synthesis by WT, EG mutant, and Y3F mutant subgenomic replicon RNA. The Y3F replicon encodes a 3B with a change of tyrosine-3 to phenylalanine. Y-3 is the nucleophile employed to form VPg-pU. Products formed in this background would be irrelevant. HeLa cells were transfected with in vitro-transcribed replicon RNA and held at 37°C, and luciferase activity was monitored for 12 h posttransfection. (C) Kinetics of translation by WT and EG mutant subgenomic replicon RNA in the presence of brefeldin A (BFA). HeLa cells were transfected with in vitro-transcribed replicon RNA, incubated in the presence of brefeldin A (2 μg/ml), and held at 37°C, and luciferase activity was monitored for 12 h posttransfection.
Replicon analysis.
The reduced rate of infectious virus production observed by the one-step growth analysis could be attributed to any step preceding production of infectious virus, for example, genome replication or virus particle assembly or maturation. First, we evaluated RNA synthesis indirectly by using a subgenomic replicon that has capsid-coding sequence replaced with a luciferase reporter, thus allowing luciferase activity to provide an indication of RNA level (36, 59, 60). EG replicon RNA was compared not only to WT replicon RNA but also to a replicon RNA containing a mutation that changes Tyr-3 of 3B (VPg) to Phe. This mutant is referred to as Y3F. Y3F replicon RNA can only be translated, providing the background level of luciferase activity derived from translation of transfected RNA. In cells transfected with WT replicon RNA, the fast rate of RNA synthesis begins at 3 hours posttransfection (Fig. 4B), 1 hour prior to the first appearance of virus (Fig. 4A). From 3 to 5 hours after transfection of WT replicon RNA, luciferase activity increased at a rate of 1.5 × 105 RLU/μg lysate/h). During this same time frame, luciferase activity increased at a rate of 3 × 103 RLU/μg lysate/h in cells transfected with EG replicon RNA. The final yields of luciferase activity were 8 × 105 RLU/μg lysate and 3 × 105 RLU/μg lysate for WT and EG replicon RNAs, respectively. The amount of luciferase produced by EG replicon RNA in the presence of brefeldin A, an inhibitor of genome replication (38, 50), was the same as that produced by WT replicon RNA under the same conditions (Fig. 4C). We conclude that changes in translational efficiency and/or stability of EG replicon RNA are not responsible for the reduced rate of genome replication. Thus, EG PV replicates RNA 50-fold slower than WT PV from 3 to 5 h posttransfection, with only a minor reduction in the final yield of replicated RNA. These observations parallel observations made by evaluating infectious virus production, suggesting that the reduced rate of genome replication for EG PV is the cause of the reduced rate of infectious EG virus production.
Virus assembly and encapsidation.
The direct correlation between reduced kinetics of RNA synthesis and infectious virus production does not prove that steps after RNA synthesis, for example, virus assembly, are normal. In spite of substantial accumulation of GG RNA in cells (60), a virus assembly defect is suggested by the 3-log reduction in particles isolated from GG genomic RNA-transfected cells relative to particles isolated from cells with WT genomic RNA (Table 2). It is possible to separate genome replication from virion production by using hydantoin (84, 87). Hydantoin targets the PV 2C protein and blocks production of infectious virus without affecting genome replication (84). The experimental design is illustrated in Fig. 5A. Cells are infected with virus at an MOI of 1 in the presence of hydantoin. After a time sufficient for completion of genome replication (6 h for WT PV [Fig. 5B]; 8 h for EG PV [Fig. 5C]), hydantoin is removed and total infectious virus produced was quantified by plaque assay. In the presence of hydantoin, neither WT nor EG PV produces infectious virus (Fig. 5B and C). However, by 30 min after hydantoin removal, a 4-log burst of infectious virus can be observed for WT PV (Fig. 5B and C). Interestingly, both the burst and subsequent kinetics of infectious virus production by EG PV are reduced relative to those for WT PV when hydantoin is removed at 6 h postinfection (Fig. 5B) because EG PV genome replication is incomplete at this time (Fig. 4B). When hydantoin is removed at 8 h postinfection, the burst and subsequent kinetics of infectious virus production by EG PV are equivalent to those for WT PV (Fig. 5C). In both cases, equivalent amounts of infectious WT and EG PVs were made (Fig. 5B and C). Consistent with this observation, EG PV was indistinguishable from WT PV (Fig. 5D) and contained equivalent numbers of empty particles (Table 2). The electron microscopy analysis should be used qualitatively because we have not controlled for virion loss during the isolation procedure. These studies suggest that genome replication and virus assembly may be uncoupled.
FIG. 5.

EG PV exhibits a post-genome replication, pre-virus assembly defect. (A) Experimental design. HeLa cells were infected with WT or EG PV (MOI, 1) in the presence of hydantoin and held at 37°C for 6 or 8 h, at which time the medium containing hydantoin was removed, cells were washed with PBS, fresh medium was added, and the infected cells were returned to 37°C. At the indicated times postinfection, cells were harvested and lysed by multiple freeze-thaw cycles and virus was titered. (B and C) Virus titer for WT and EG PV from the experiment described in panel A when hydantoin was removed at 6 (B) or 8 (C) h postinfection. (D) TEM of WT and EG virus particles. WT and EG PV particles were purified by precipitation using 10% PEG 8000 followed by centrifugation through a 30% sucrose cushion. Purified virus particles were negatively stained with 2% UA and observed by TEM. Representative virus particles that are empty (arrow) or contain packaged RNA (▾) are indicated. Bars = 0.1 μm.
Although the rate of RNA synthesis in the exponential phase of genome replication appears much too slow for new RNA synthesis to account for infectious virus produced after removal of hydantoin, it was important to formally exclude this possibility. Cordycepin is a potent inhibitor of RNA viruses (35, 52, 85, 86, 90). It is converted to the triphosphate, is incorporated by the viral polymerase into nascent RNA, and prematurely terminates RNA synthesis (7, 58, 88). Concentrations of cordycepin greater than 200 μM were sufficient to prevent replication of subgenomic replicon RNA (Fig. 6A). Addition of cordycepin at a concentration of 200 μM at 3 h posttransfection was sufficient to shut down RNA synthesis in 30 to 60 min and maintain the block for as long as 8 h posttransfection (Fig. 6B). Therefore, we revised our hydantoin experiment to include cordycepin as shown in Fig. 6C. HeLa cells were infected in the presence of hydantoin, cordycepin was added at 6 h (WT PV) or 8 h (EG PV) postinfection, and hydantoin was removed 30 min later, but the cordycepin block was maintained. Virus was harvested at various times after removal of hydantoin and titered. The yields of virus in the presence and absence of cordycepin were the same for both WT PV (Fig. 6D) and EG PV (Fig. 6E). We conclude that genome replication and virus assembly are distinct steps in the virus life cycle that need not be coupled.
FIG. 6.

EG PV produced in the presence of hydantoin does not require new RNA synthesis. (A) Cordycepin blocks new RNA synthesis. HeLa cells were transfected with in vitro-transcribed WT subgenomic replicon RNA, incubated with cordycepin at the indicated concentrations (μM), and held at 37°C, and luciferase activity was monitored for 8 h posttransfection. (B) Cordycepin blocks ongoing RNA synthesis. HeLa cells were transfected with in vitro-transcribed WT subgenomic replicon RNA and held at 37°C. At 0 or 3 h posttransfection, 200 μM cordycepin was added and luciferase activity was monitored. A control experiment was performed in the absence of cordycepin. (C) The hydantoin experiment in the presence and absence of cordycepin. HeLa cells were infected with WT or EG PV (MOI, 1) in the presence of hydantoin and held at 37°C for 6 or 8 h, at which time either 0 or 300 μM cordycepin was added. At 6.5 or 8.5 h, the medium was removed, cells were washed with PBS, fresh medium containing either no or 300 μM cordycepin was added, and the cells were returned to 37°C. At the indicated times postinfection, cells were harvested, lysed by freeze-thawing, and virus titered. (D) Kinetics of WT PV production after removal of hydantoin (Hyd) is not altered by the cordycepin block. The experiment was performed as described for panel C. Data shown were obtained in the presence or absence of cordycepin (Cor). (E) Kinetics of EG PV production after removal of hydantoin is not altered by the cordycepin block. The experiment was performed as described for panel C. Data shown were obtained in the presence or absence of cordycepin.
Our studies clearly define a hydantoin-sensitive step that occurs after genome replication and before virus assembly. The number of steps between the genome replication and hydantoin-sensitive steps or the hydantoin-sensitive and virus assembly steps, if any, is not known. Defects between genome replication and hydantoin-sensitive steps would be masked by hydantoin treatment, but defects between the hydantoin-sensitive and virus assembly steps would not.
Polyprotein processing.
GG PV fails to produce 3AB and 3CD. The absence of one or both of these proteins likely contributes to the phenotypes for this mutant. Substrates for the protease activity of 3C (and its precursors) contain a Gln-Gly cleavage site. Therefore, the phenotypes observed for EG PV may result from the Glu-Gly cleavage site. We transfected cells with WT, GG, or EG subgenomic replicon RNAs and evaluated lysates 20 h posttransfection by Western blotting with antiserum raised against PV VPg (3B), 3C, or 3D proteins. Cells transfected with WT replicon RNA produced 3AB (Fig. 7A, lane 1) and 3CD (Fig. 7A, lane 7). As expected, cells transfected with GG replicon RNA produced neither of these proteins (Fig. 7A, lanes 2 and 8) and instead produced 3ABC and 3BC (Fig. 7A, lanes 2 and 5), as well as 3D and its 3ABCD and 3BCD precursors (Fig. 7A, lane 8). Cells transfected with EG replicon RNA exhibited an intermediate phenotype: 3AB and 3CD were produced (Fig. 7A, lanes 3, 6, and 9), and cleavage intermediates such as 3BC were also still apparent 20 h posttransfection (Fig. 7A, lanes 3 and 6). These data show the EG PV restored cleavage at the 3B-3C junction but suggest that cleavage at this site is slower than that for the WT, leading to lower steady-state levels of 3AB and 3CD.
FIG. 7.

EG PV exhibits delayed kinetics of P3 processing. (A) EG restores P3 processing. Processing was evaluated by Western blotting. HeLa cells were transfected with WT, GG, or EG subgenomic replicon RNA, held at 34°C, and harvested 20 h posttransfection. Extracts were prepared and processed for Western blotting as described in Materials and Methods. Antisera against VPg, 3C, and 3D were employed. The bands corresponding to the different precursor and processed proteins are indicated. (B) Kinetics of P3 processing for EG is delayed. Processing was evaluated by Western blotting. HeLa cells were transfected with WT or EG mutant subgenomic replicon RNA, held at 34°C, and harvested at 6, 8, and 20 h posttransfection. Extracts were prepared and processed for Western blotting as described in Materials and Methods. Antisera against VPg and 3D were employed. The bands corresponding to the different precursor and processed proteins are indicated. Purified 3CD and 3D (M1) or 3ABC and 3BC (M2) were loaded in lanes 8 and 17 and 9 and 18 as positive controls. Uninfected cells were used as negative controls (lanes 1 and 10). (C) Processing was evaluated by cell-free translation. HeLa cell-free translation extracts containing [35S]methionine and [35S]cysteine were programmed with WT or EG mutant RNA for 0.25, 1, and 2 h. Radiolabeled proteins were separated by 15% SDS-PAGE and detected by phosphorimaging. The bands corresponding to the different precursor and processed proteins are indicated.
In order to evaluate the kinetics of 3B-3C cleavage and obtain some qualitative information on the steady-state levels of 3AB and 3CD, we repeated the experiment described above. However, lysates were evaluated at earlier times (6 and 8 h) posttransfection by Western blotting with antisera raised against VPg and 3D (Fig. 7B). WT replicon replication is complete by 6 h posttransfection; EG replicon replication is complete by 8 h posttransfection (Fig. 4B). Cells expressing the WT P3 polyprotein show complete cleavage of 3ABCD and 3BCD by 20 h posttransfection (compare lanes 2 and 4 to lane 6 of Fig. 7B). The identity of the band below 3BCD in lanes 2 to 7 of Fig. 7B is not known. This band represents a viral protein, as it is not present in control cells (Fig. 7B, compare lanes 2 to 7 to lane 1). Cells expressing the EG P3 polyprotein fail to completely process both 3ABCD and 3BCD, even by 20 h posttransfection (Fig. 7B, lane 7). At 8 h posttransfection, cells expressing the P3 polyprotein also showed evidence for delayed processing relative to the WT (Fig. 7B, compare lanes 5 and 4). At no time during or after genome replication does the level of 3CD observed in EG replicon RNA-transfected cells exceed that observed in WT replicon RNA-transfected cells (compare the following pairs of lanes of Fig. 7B: 12 to 11, 14 to 13, and 16 to 15). We conclude that the rate of cleavage of the 3B-3C site in EG PV is slower than that in WT PV, leading to a decrease in the steady-state level of 3CD and, by inference, 3AB.
In order to determine whether or not the delayed cleavage of the 3B-3C cleavage site in EG PV caused a global change in polyprotein processing, translation of EG replicon RNA was compared to that of WT replicon RNA in cell-free translation experiments as described above (Fig. 7C). In extracts translating WT replicon RNA, 3CD was clearly visible by the 2-h time point (Fig. 7C, compare lanes 4 and 3). In contrast, 3CD was still absent at this time point in extracts translating EG replicon RNA (Fig. 7C, compare lanes 7 and 4). Importantly, none of the other detectable P2- or P3-generated proteins exhibited an observable difference in cleavage kinetics or abundance when EG replicon RNA translation was compared to that of the WT (Fig. 7C).
Composition of protein-linked RNA.
Inactivation of the 3B-3C cleavage site (GG mutant) leads to production of RNAs containing 3BC covalently linked to their 5′ ends (60). Whether or not 3BC linkage occurs normally or is an off-path intermediate unique to the GG mutant remains to be proven. The EG mutant represents a unique tool to approach this question. Robust genome replication occurs, but the kinetics of 3B-3C processing are sufficiently slow relative to those for the WT to possibly permit detection of intermediates involving 3BC(D) precursors that would never accumulate during WT genome replication. HeLa cells were transfected with WT, EG, or Y3F replicon RNA. Eight hours posttransfection, total RNA was isolated. The nature of the protein(s) covalently linked to the RNA was determined by immunoprecipitation with antiserum raised against PV VPg, 3C, 3D, or HCV NS5A. HCV NS5A served as a negative control. Immunoprecipitated RNA was interrogated by Northern analysis using a 995-bp, 32P-labeled probe directed to the 3D-coding sequence to ensure that full-length RNA was being evaluated. Only anti-VPg serum precipitated RNA from cells transfected with WT replicon RNA (Fig. 8A, lane 1), consistent with WT PV producing VPg-linked RNA (60). This serum did not precipitate RNA from cells transfected with Y3F replicon RNA (Fig. 8A, lane 9), thus confirming that the hybridization signal observed does not arise from contamination of the total RNA preparation with in vitro-transcribed RNA used to launch the multiplication cycle. Interestingly, RNA from cells transfected with EG replicon RNA was precipitated by sera against not only VPg (Fig. 8A, lane 5) but also 3C (Fig. 8A, lane 6) and 3D (Fig. 8A, lane 7). Replicated EG RNA was not precipitated by anti-NS5A serum (Fig. 8A, lane 8), and transfected Y3F replicon RNA was not precipitated by anti-3C or anti-3D serum (Fig. 8A, lanes 10 and 11, respectively), thus confirming the specificity of the experiment. Because replication in this system requires Tyr-3 of 3B/VPg (Fig. 4B) (69), covalent linkage of 3C must really imply 3BC and covalent linkage of 3D must really imply 3BCD.
FIG. 8.

3B precursor-linked RNA is produced by EG PV and converted to VPg-linked RNA prior to the hydantoin-sensitive step. Interrogation of protein linkage to WT and EG mutant RNA was analyzed by RNA immunoprecipitation and Northern blotting. (A) RNA isolated from replicon RNA-transfected cells. HeLa cells were transfected with WT, EG, or Y3F RNA and held at 37°C for 10 h. Total RNA was isolated from transfected HeLa cells and immunoprecipitated using antibodies against VPg, 3C, 3D, or HCV NS5A (as a negative control). The immunoprecipitated RNA was separated on a 0.6% agarose gel containing 0.8 M formaldehyde, transferred to a nylon membrane, and hybridized with a 32P-labeled DNA probe. The hybridized DNA probe was visualized by phosphorimaging. Shown is a phosphorimage after a 1-day exposure. In vitro-transcribed RNA is shown as a reference. (B) RNA isolated at early times postinfection. HeLa cells were infected with WT PV (MOI, 10) and held at 37°C for 0, 4, or 5 h. As a positive control, EG PV (MOI, 10)-infected HeLa cells were incubated at 37°C for 10 h. Total RNA was isolated from infected HeLa cells and immunoprecipitated using antibodies against VPg, 3C, or HCV NS5A. The immunoprecipitated RNA was separated as described above. In vitro-transcribed RNA is shown as a reference. (C) RNA isolated from HeLa cells infected with WT PV or EG PV (MOI, 10) in the presence (+Hyd) or absence (−Hyd) of hydantoin at 37°C for 6 h (WT PV) or 8 h (EG PV). Total RNA was immunoprecipitated using antibodies against VPg and 3C. The immunoprecipitated RNA was separated as described above. WT or EG PV RNA used for immunoprecipitation is shown in lanes 1 to 4. (D) RNA isolated from purified virus particles. HeLa cells were infected with WT or EG PV and held at 37°C until CPE. Cells were harvested, and WT and EG PV was purified by precipitation using 8 to 10% PEG 8000 followed by centrifugation through a 30% sucrose cushion. Viral RNA was isolated from purified viruses and immunoprecipitated using antibodies against VPg, 3C, 3D, or HCV NS5A. The immunoprecipitated RNA was separated as described above. Total RNA purified from Y3F mutant RNA-transfected cells was used as a negative control.
These data are consistent with recent models suggesting that precursors, such as 3BCD, are used for initiation of and elongation during genome replication, with processing of 3BCD-linked RNA to VPg-linked RNA occurring during or after production of full-length RNA (60). However, it is possible that precursor-linked RNA is unique to EG and GG PVs. All attempts to trap precursors on RNA during genome replication by WT PV failed. First, we interrogated earlier time points during the multiplication cycle of WT PV with the hope that intermediates might be present (Fig. 8B, lanes 1 to 9). Second, we interrogated WT PV RNA from hydantoin-treated cells with the thought that processing might occur after the hydantoin-sensitive step (Fig. 8C, lanes 5 to 8). Antiserum raised against PV 3C did not immunoprecipitate WT PV RNA under any condition (Fig. 8B, lanes 5 and 8, and C, lanes 6 and 8). Surprisingly, the presence of hydantoin prevented the accumulation of anti-3C-immunoprecipitable EG PV RNA (Fig. 8C, compare lanes 10 and 12). An increase in the level of VPg-linked EG PV RNA was evident (Fig. 8C, compare lanes 11 and 9), suggesting that 3B precursor-linked EG PV RNA is a direct precursor of VPg-linked EG PV RNA. These data are consistent with a model in which 3BC-linked EG PV RNA is processed before the hydantoin-sensitive step, RNA traversal through this step is not inhibited by the presence of a precursor like 3BC, and processing of precursor-linked RNA does not occur beyond the hydantoin-sensitive step. Although this sequence of events may be unique to EG PV and its RNA, a similar precursor RNA-to-VPg RNA relationship for WT PV should not be excluded. Clearly, the processing efficiency of the 3B-3C cleavage site is substantially higher and faster for WT PV than for EG PV (Fig. 7). The only way to trap precursors onto WT RNA would be to reduce processing (as done here) or increase RNA traversal through the hydantoin-sensitive step, which is not currently possible.
Our experiments with the GG mutant suggested that 3BC-linked RNA could be packaged essentially as efficiently as for the WT based on the percentage of empty particles (Table 2). In order to test further the specificity of packaging, the RNA immunoprecipitation experiment was repeated using RNA isolated from purified virus particles (Fig. 8D). Only VPg-linked RNA was detected in WT particles (Fig. 8D, lane 5), and only VPg- and 3BC-linked RNA was detected in EG particles (Fig. 8D, lanes 1 and 2, respectively). The inability to detect 3BCD-linked RNA in EG particles reflects either the inability of 3BCD-linked RNA to be packaged or an insufficient limit of detection. Additional experiments will be necessary to distinguish between these possibilities. We conclude that processing between 3B and 3C is not essential for packaging of 3BC-linked RNA.
A trans-complementable function for PV 3CD in genome replication.
Either loss of (GG PV) or delay in (EG PV) cleavage at the 3B-3C junction causes two phenotypes: (i) a lag in the onset of genome replication and a reduced rate of genome replication (Fig. 4B) or (ii) a reduced rate of infectious virus production (Fig. 4A). It should be noted that it is possible that the observed lag in the onset of genome replication could be caused by an even slower rate of genome replication that maintains the signal below the limit of detection. We conclude that the rate of genome replication is sensitive to the concentration of 3AB and/or 3CD. PV 3CD protein alters the subcellular distribution of proteins involved in vesicular trafficking, like the Arf GTPase, which may be a prerequisite to formation of RCs (12). PV 3CD protein binds to all of the cis-acting replication elements of the virus that are required for genome replication (5, 34, 61, 91, 93). Finally, PV 3CD protein increases the yield of infectious virus recovered from cell extracts by modulating a post-genome replication step, for example, virion maturation, although genome encapsidation cannot be ruled out (24). Impairment of the genome replication functions for the PV 3CD protein could explain the phenotypes observed for GG and EG PVs.
It is generally believed that all functions for P3-encoded proteins are required in cis, thus explaining the inability to complement defective proteins in trans, at least in a cellular context (29, 83). However, interactions between 3CD proteins and/or between 3CD and viral proteins required prior to formation of RCs may be complemented in trans. In addition, defects in host-interaction functions of PV 3CD required for genome replication should be complemented in trans. Finally, functions in virion maturation that occur outside of RCs may also be complemented in trans. Therefore, we constructed HeLa cell lines capable of regulated expression of PV 3CD protein. Recombinant, defective Moloney murine leukemia virus harboring genes encoding 3CD proteins with or without an active protease was used to infect a HeLa cell line expressing the tetracycline repressor protein. The 3CD gene is under the control of a minimal cytomegalovirus promoter that is repressed by the binding of the tetracycline repressor. Cells containing an integrated provirus were selected with hygromycin in the presence of Dox to limit 3CD expression. Lysates from clones grown in the presence or absence of Dox were screened by Western blotting for regulated expression of PV 3CD (Fig. 9A). Multiple clones were obtained for both protease-active and protease-inactive forms of PV 3CD. Cell viability was reduced dramatically by induction of the protease-active form of PV 3CD; therefore, these studies employed a cell line expressing the protease-inactive form of PV 3CD. Induction of PV 3CD did not cause a change in cell morphology based on differential interference contrast microscopy (Fig. 9B, compare +Dox to −Dox in the DIC column). Subcellular localization of PV 3CD was determined by fluorescence microscopy. PV 3CD accumulated to the highest level in the nucleus rather than the cytoplasm (Fig. 9B, −Dox in the FLUOR column), as observed for other picornavirus 3CD proteins (3, 27, 75). However, we fully expected some level of 3CD to be found in the cytoplasm. TEM showed that expression of PV 3CD did not cause any alterations to cellular ultrastructure (Fig. 9C). We refer to this cell line as HeLa-3CD.
FIG. 9.

Establishment of a HeLa cell line capable of regulated expression of PV 3CD. HeLa-3CD cell lines were incubated in the absence (−) or presence (+) of Dox (1 μg/ml) for 36 h at 37°C as described in Materials and Methods. (A) Dox removal induces 3CD expression. 3CD induction was confirmed by Western blot of total cellular protein using anti-3D antisera. (B) Expressed 3CD localizes to the nucleus. Immunofluorescence of HeLa-3CD cell lines using anti-3D (red) and antiactin (green) labeled antibodies. (C) 3CD expression is not cytopathic. HeLa-3CD cells were fixed and visualized by electron microscopy. Bars = 2 μm. HeLa-3CD cells appear normal both in the absence (−) and presence (+) of Dox.
Kinetics of and end points for genome replication (Fig. 10A, +Dox) and infectious virus production (Fig. 10B, +Dox) by the WT in uninduced HeLa-3CD cells were the same as those observed in our standard HeLa cells (Fig. 4). Importantly, the EG phenotype was observed in the HeLa-3CD cells as well (Fig. 10A and B, +Dox). Expression of 3CD did not change the kinetics or end points for WT genome replication (Fig. 10A, compare −Dox to +Dox) or infectious virus production (Fig. 10B, compare −Dox to +Dox). In contrast, the EG genome replication phenotype was suppressed by expression of 3CD (Fig. 10A, compare −Dox to +Dox) without changing the EG infectious virus production phenotype (Fig. 10B, compare −Dox to +Dox). Semiquantitative analysis of the data showed that ectopic expression of 3CD increased the rate of EG RNA synthesis fourfold to a rate on par with WT RNA synthesis in the absence of 3CD, with no greater than a 25% increase in the rate of infectious virus production (Fig. 10C). To our knowledge, this is the first example of trans complementation of a genome replication defect in a cellular context by ectopic expression of 3CD. The inability of ectopic expression of 3CD to complement the EG infectious virus production phenotype suggests that a step after genome replication but prior to virus assembly was affected. This step either cannot be trans-complemented by the level of 3CD produced in HeLa-3CD cells or requires 3AB. A more comprehensive evaluation of the ability of P3-derived proteins to complement the EG infectious virus production phenotype may help to clarify this issue. We conclude that reducing the efficiency of cleavage between 3B and 3C produces two distinct effects on virus multiplication: one occurs at a pre-genome replication step that requires 3CD; the other occurs at a post-genome replication/pre-virus assembly step that requires 3CD in cis, higher concentrations of 3CD, and/or 3AB.
FIG. 10.

Ectopic expression of 3CD enhances the kinetics of RNA synthesis of EG PV without impacting the kinetics of virus production. (A) Kinetics of RNA synthesis by WT and EG mutant subgenomic replicon RNA in the absence (+Dox, −3CD) or presence (−Dox, +3CD) of ectopic expression of 3CD. HeLa-3CD cells incubated in the presence or absence of Dox were transfected with either WT or EG in vitro-transcribed replicon RNA and held at 37°C, and luciferase activity was monitored for 12 h posttransfection. The kinetics of RNA synthesis for EG expressing 3CD (○) are essentially identical to that of the WT (□). There was no difference in the kinetics of RNA synthesis for WT replicon in the presence or absence of 3CD. (B) Kinetics of virus production by WT and EG PV in the absence or presence of ectopic expression of 3CD. HeLa-3CD cells incubated in the presence or absence of Dox were infected with WT or EG PV (MOI, 10) and held at 37°C. At the indicated times postinfection, cells were harvested and lysed by freeze-thawing and virus was titered. There was no difference in the kinetics of EG mutant virus production in the absence (•) or presence (○) of 3CD. (C) Average rates of RNA synthesis (RNA) and infectious virus production (virus) in the absence or presence of ectopic expression of 3CD for WT and EG. Rates shown were calculated from data obtained between 3 and 6 h, inclusive, posttransfection or postinfection.
DISCUSSION
Many positive-strand RNA viruses produce polyproteins that are co- and posttranslationally cleaved by virus-encoded proteases (46, 47, 79). In all cases, polyprotein cleavage is essential for production of infectious virus (6, 19, 47, 64, 79). Therefore, viral proteases have represented an important target for antiviral drug development (15, 18, 67). For decades, many laboratories have investigated two questions regarding polyprotein processing: (i) is cleavage ordered or stochastic and (ii) do cleavage intermediates expand the proteome by creating unique functions? In most cases, a reverse-genetic approach has been used to address these questions. Picornavirologists have used the same approach (19, 31, 48, 74). Most often, investigators have reduced or inhibited cleavage (6, 19, 31, 45, 48); sometimes, however, cleavage efficiency has been increased (19). Regardless of the phenotype, infectious virus production is usually reduced significantly or inhibited altogether (6, 19, 31, 45). Unfortunately, pursuit of suppressors or detailed studies of the multiplication cycle of these mutants are, at best, rare (19, 31). We recently reported a PV mutant (GG) that prevented cleavage between the 3B and 3C proteins in order to determine whether the processed VPg peptide was essential for genome replication (60). Inactivation of this cleavage site was not lethal for genome replication, consistent with observations made for encephalomyocarditis virus (31). We have performed a detailed molecular characterization of GG PV and its pseudorevertant (EG PV). This study provides unique insight into the following: possible functions of unique precursor proteins in well-established steps of the multiplication cycle, new functions for well-established proteins in well-established steps of the multiplication cycle, and the existence of a post-genome replication/pre-virus assembly step of the multiplication cycle.
The intracellular steps of the PV multiplication cycle after genome uncoating are shown in Fig. 11. VPg is removed from the genomic RNA (55). Genomic RNA is translated to produce a polyprotein that is co- and posttranslationally cleaved to produce proteins required for converting the host cell into a virus production factory (P2 proteins in Fig. 1A) (2, 10, 80), genome replication (P3 proteins in Fig. 1A) (44, 89), and virus assembly (P1 proteins in Fig. 1A) (68). Genome replication occurs within clusters of vesicles termed RCs. Formation of functional RCs requires P2 proteins, in particular 2B, 2C, and/or 2BC (2, 10, 80). The origin of the membranes employed for RCs is not clear but may be pirated from vesicles functioning in normal cellular processes, including anterograde and/or retrograde trafficking between the endoplasmic reticulum and Golgi apparatus (21, 70, 71) and autophagy (39, 72, 80). Genome replication occurs, producing VPg-linked, genomic RNA. Early during infection, replicated RNA is used for translation. Replicated RNA is thought to be released from RCs for translation to occur (16). Later during infection, replicated RNA is encapsidated to produce immature virions that mature via a process requiring cleavage of the VP0 capsid precursor into VP2 and VP4, an autocatalytic process that appears to occur only in particles containing genomic RNA. Hydantoin inhibits production of infectious virus at the virus assembly step by inhibiting either encapsidation or maturation (84, 87). The switch from translation of replicated RNA to encapsidation of replicated RNA occurs at a point in the life cycle when RNA amplification enters the exponential phase, ∼3 h postinfection (9). At this stage of the multiplication cycle, infectious virus production no longer exhibits sensitivity to cycloheximide (9). The general aspects of this model apply to all picornaviruses (74).
FIG. 11.
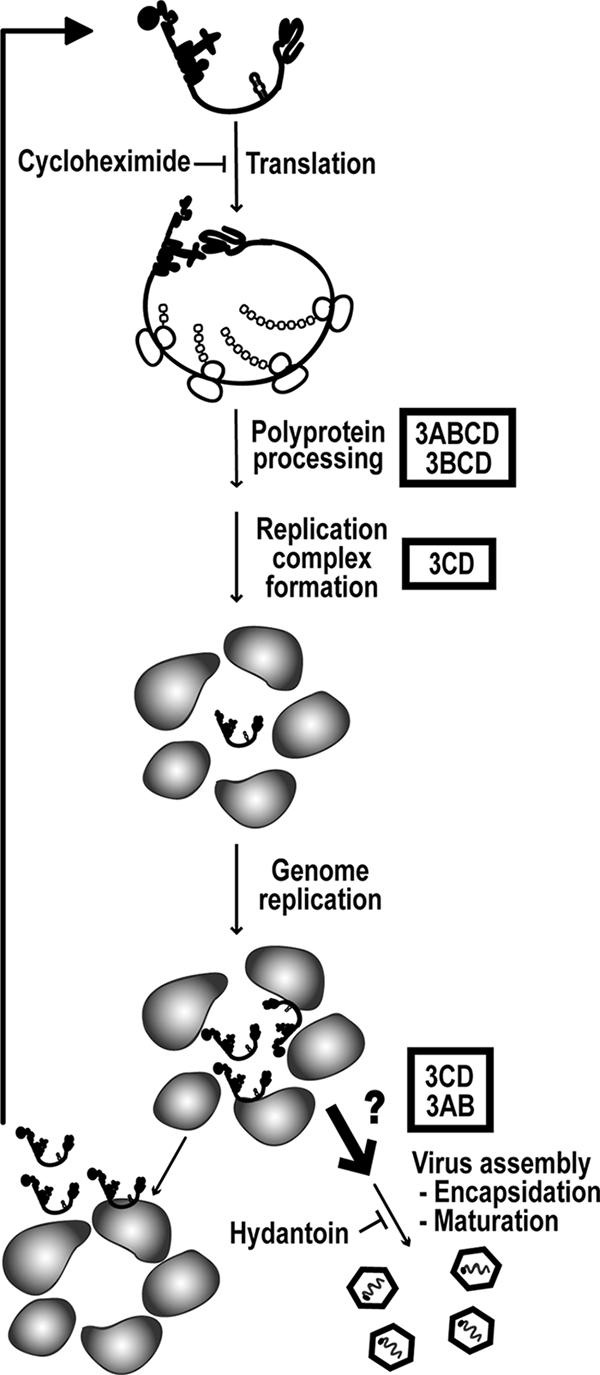
Implications of this study presented in the context of the PV multiplication cycle. Roles for P3 proteins suggested by this study appear in boxes, and the additional step appears as a thick arrow. Genomic RNA is first translated to produce the PV polyprotein. Translation is inhibited by cycloheximide. The polyprotein is co- and posttranslationally processed to produce the various precursors and processed proteins that are needed for PV multiplication. This study suggests that 3ABCD and/or 3BCD efficiently cleaves the PV polyprotein. RCs are formed, followed by genome replication. This study implicates 3CD in a step prior to genome replication. We have assigned this function of 3CD to RC formation. However, additional studies will be required to confirm this possibility. Early during infection, replicated RNA enters the pool of transfected RNA. This study implicates 3AB or 3CD in a step after genome replication but prior to virus assembly. We propose that this step permits transit of replicated RNA from RCs to virus particles. Finally, virus assembly, i.e., encapsidation and maturation, occurs. Hydantoin can inhibit production of infectious virus at the virus assembly step.
Most picornavirus polyprotein cleavage events require the thiol protease active site associated with the 3C domain of the P3 precursor protein (79). The most abundant 3C-containing protein found in cells infected with WT PV is the 3CD protein (Fig. 2B) (60); the 3CD protein is produced by the major processing pathway (Fig. 1B) (60). The abundance of 3CD protein relative to 3C protein is consistent with the use of 3CD protein for proteolysis in vivo (26). In addition, the 3CD protein cleaves the capsid precursor better than the 3C protein (14, 41). Our study forces us to consider the possibility that a 3C-containing protein other than the 3CD protein may function in vivo. First, the GG polyprotein produced the 3ABC protein instead of the 3CD protein, without a significant change in the yield of cleavage products, including capsid proteins (Fig. 2B and 7A) (60). Second, the kinetics of processing of the EG polyprotein appears normal in the absence of detectable levels of 3CD (Fig. 7C). Finally, most 3CD protein produced in cells traffics to the nucleus (Fig. 9B) (3, 27, 75). The 3C-containing precursors that WT, GG, and EG PVs produce at similar levels are 3ABCD and 3BCD. It is possible that one or both of these precursors are responsible for polyprotein processing in vivo (Fig. 11). The use of precursor proteins larger than the 3CD protein might explain the very low protease activity observed for 3C and 3CD proteins in vitro (32, 33, 54, 94), although the absence of a protein or RNA cofactor would also explain the reduced activity in vitro (14).
All picornaviruses perform genome replication in association with membranes (74, 82). A hallmark of picornavirus-infected cells is clusters of vesicles that have been termed RCs (13, 17). Formation of these complexes occurs either before or concomitant with genome replication and is thought to be induced by proteins encoded by the P2 region of the genome (17, 22). Replication of both GG and EG RNAs exhibits a lag and reduced kinetics of RNA synthesis (Fig. 4B) (60). Interestingly, both the lag and slow kinetics of RNA synthesis observed during replication of the EG subgenomic replicon can be complemented in trans by expression of 3CD protein (Fig. 10A). This observation required the use of a HeLa cell line capable of regulated expression of 3CD (Fig. 9). This cell line has been designated HeLa-3CD. GG subgenomic replicon RNA synthesis is also stimulated in HeLa-3CD cells, but the substantial reduction in the kinetics of RNA synthesis is really not reversed (data not shown). To our knowledge, this study provides the first example of suppression of a replication defect caused by a mutation in the P3-coding sequence by ectopic expression of a 3CD protein in a cellular context. It is well known that P3 proteins required for genome replication function in cis (23, 29, 42, 83). Any trans complementation that has been observed in a cellular context in the past has required, minimally, expression of the complete P3 protein (42, 83). We suggest that the 3CD protein has a function prior to genome replication, perhaps in formation of RCs (Fig. 11). Belov and colleagues have shown that PV 3CD is responsible for maintenance of Arf GTPase in its GTP-bound form and stabilization on membranes, providing a provocative link between PV 3CD and RC formation (12). Activities of PV proteins that involve interaction with the host should be trans-complementable. Of course, trans-complementable interactions with viral proteins should not be excluded.
It is well known that picornavirus 3CD proteins function directly in genome replication by binding to at least three CREs: the cloverleaf structure at the 5′ end (oriL), the template for VPg uridylylation (oriI), and the complex structure located in the 3′ NTR (oriR) (1, 5, 30, 40, 61). We have shown that precursors like 3BCD also retain CRE-binding activity (60); therefore, the slow rate of 3CD production in the EG mutant (Fig. 7) should not preclude this aspect of the genome replication process. The ability of the genome replication defect observed for the EG mutant to be complemented by ectopic expression of 3CD also suggests that the cis function of 3CD in establishing replication-competent templates is normal (Fig. 10B).
Once replication-competent templates are produced, initiation of RNA synthesis occurs. There is consensus that initiation requires formation of a ribonucleoprotein complex at oriI and that this complex adds two uridylate residues to a tyrosine of VPg or some precursor thereof (61, 63). The prevailing model suggests that the 3AB protein serves as the source of VPg, which, as a processed peptide, is used as the primer for initiation (74). Our data do not formally rule out this model. However, our data do permit us to make the very provocative suggestion that a precursor other than 3AB may serve as the VPg donor. The EG mutant produced RNA covalently linked not only to VPg but also 3BC and/or 3BCD (Fig. 8A). The ability to trap these particular precursor-linked forms of RNA in a mutant that produces nearly normal quantities of 3AB and no 3ABC (Fig. 7A and B) provides some support for the use of 3BCD as the form of VPg utilized in vivo. However, 3BC(D)-linked RNA could be unique to GG and EG PVs. This aspect of picornaviral genome replication merits additional attention by us and others.
The observation of 3BC(D)-linked RNA intermediates suggests that formation of VPg-linked RNA may occur by using a postsynthetic processing mechanism. Consistent with this possibility was the finding that 3BC(D)-linked RNA could be converted essentially quantitatively to VPg-linked RNA by inhibiting encapsidation and/or virus assembly with hydantoin (Fig. 8C). Such a postsynthetic processing mechanism could also be exploited by WT PV to convert 3AB-linked RNA to VPg-linked RNA.
During the late stages of infection, replicated RNA is packaged into virus particles. Very little is known about the encapsidation process for any picornavirus. There has been some suggestion that VPg may constitute part of the packaging signal (55). The observation that 3BC-linked RNA produced by the GG mutant (Fig. 2C and Table 2) and EG mutant (Fig. 8D) is packaged is not inconsistent with this possibility but may indicate flexibility in the context in which this putative signal can be presented.
It is clear that only replicated RNA is packaged (9, 56). However, it is not absolutely clear to what extent genome replication and genome encapsidation are linked. The rate of infectious virus production by the EG mutant is slow relative to that for the WT (Fig. 4A and 10B). This phenotype is not linked to the kinetics of genome replication. When EG genome replication is slower than WT (Fig. 10A, +Dox) and when EG genome replication is equivalent to WT (Fig. 10A, −Dox), production of infectious EG PV occurs at the same rate (Fig. 10B). These data suggest that genome replication and genome encapsidation are discrete steps in the multiplication cycle. If genome encapsidation occurred immediately following genome replication, then the kinetics of infectious EG PV production should remain slower than observed for WT PV under conditions in which genome replication can reach completion prior to monitoring infectious virus production. Synchronization of infectious virus production can be achieved by using hydantoin (84, 87). When hydantoin is removed from EG PV-infected cells prior to completion of genome replication, the rate of infectious EG PV production is slower than observed for WT PV (Fig. 5B). In contrast, when hydantoin is removed from EG PV-infected cells after completion of genome replication, the rate of infectious EG PV production is equal to that observed for WT PV (Fig. 5C). Infectious virus produced after hydantoin removal was not dependent on new RNA synthesis (Fig. 6). These data reinforce the notion that genome replication and genome encapsidation are discrete steps. In addition, the ability of hydantoin to suppress the observed post-genome replication defect in infectious EG PV production suggests that this defect occurs prior to the step blocked by hydantoin. Thus, a post-genome replication/pre-virus assembly step in the PV multiplication cycle is revealed (Fig. 11). The existence of a step such as this one is not new, as Baltimore described such a step 40 years ago (8). It is likely that the low titer of GG PV (Table 2) is related to a block at this step.
In conclusion, the EG mutant clearly shows that the kinetics of polyprotein cleavage are optimized to produce the appropriate precursors in the appropriate place at the appropriate time and has permitted assignment of a new function to precursor and processed forms of PV proteins, as well as further elaboration of the PV multiplication cycle. The change of 3B-3C cleavage site from Gln-Gly to Glu-Gly is not lethal in this context. Perhaps this change can be applied at other picornavirus cleavage sites to unmask more intermediates of the multiplication cycle. Glu-Gly substitutions were introduced into all of the PV P2 polyprotein cleavage sites (19). All of these mutants were at least quasi-infectious. Lessons that these mutants can teach us about the picornavirus multiplication cycle await molecular characterization.
Acknowledgments
We thank Greg Ning for training us to do TEM and Marie Chow for providing us with hydantoin. We thank Marie Chow, George Belov, Ellie Ehrenfeld, Aniko Paul, and Spencer Weeks for comments on a draft of the manuscript.
This work was supported in part by a grant AI053531 (C.E.C.) from NIAID/NIH.
Footnotes
Published ahead of print on 8 July 2009.
REFERENCES
- 1.Agol, V. I., A. V. Paul, and E. Wimmer. 1999. Paradoxes of the replication of picornaviral genomes. Virus Res. 62:129-147. [DOI] [PubMed] [Google Scholar]
- 2.Aldabe, R., and L. Carrasco. 1995. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 206:64-76. [DOI] [PubMed] [Google Scholar]
- 3.Amineva, S. P., A. G. Aminev, A. C. Palmenberg, and J. E. Gern. 2004. Rhinovirus 3C protease precursors 3CD and 3CD′ localize to the nuclei of infected cells. J. Gen. Virol. 85:2969-2979. [DOI] [PubMed] [Google Scholar]
- 4.Andino, R., G. E. Rieckhof, P. L. Achacoso, and D. Baltimore. 1993. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 12:3587-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andino, R., G. E. Rieckhof, and D. Baltimore. 1990. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 63:369-380. [DOI] [PubMed] [Google Scholar]
- 6.Ansardi, D. C., and C. D. Morrow. 1995. Amino acid substitutions in the poliovirus maturation cleavage site affect assembly and result in accumulation of provirions. J. Virol. 69:1540-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnold, J. J., and C. E. Cameron. 2004. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry 43:5126-5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baltimore, D. 1969. The replication of picornavirus, p. 101-176. In H. B. Levy (ed.), The biochemistry of viruses. Marcel Dekker, Inc., New York, NY.
- 9.Baltimore, D., M. Girard, and J. E. Darnell. 1966. Aspects of the synthesis of poliovirus RNA and the formation of virus particles. Virology 29:179-189. [DOI] [PubMed] [Google Scholar]
- 10.Barco, A., and L. Carrasco. 1995. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 14:3349-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barton, D. J., B. J. O'Donnell, and J. B. Flanegan. 2001. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 20:1439-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belov, G. A., M. H. Fogg, and E. Ehrenfeld. 2005. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J. Virol. 79:7207-7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bienz, K., D. Egger, M. Troxler, and L. Pasamontes. 1990. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J. Virol. 64:1156-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blair, W. S., X. Li, and B. L. Semler. 1993. A cellular cofactor facilitates efficient 3CD cleavage of the poliovirus P1 precursor. J. Virol. 67:2336-2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bottcher, J., A. Blum, S. Dorr, A. Heine, W. E. Diederich, and G. Klebe. 2008. Targeting the open-flap conformation of HIV-1 protease with pyrrolidine-based inhibitors. Chem. Med. Chem. 3:1337-1344. [DOI] [PubMed] [Google Scholar]
- 16.Caliguiri, L. A., and I. Tamm. 1969. Membranous structures associated with translation and transcription of poliovirus RNA. Science 166:885-886. [DOI] [PubMed] [Google Scholar]
- 17.Cho, M. W., N. Teterina, D. Egger, K. Bienz, and E. Ehrenfeld. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129-145. [DOI] [PubMed] [Google Scholar]
- 18.Coen, D. M., and D. D. Richman. 2007. Antiviral agents, p. 447-538. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 19.Cohen, L., K. M. Kean, M. Girard, and S. Van der Werf. 1996. Effects of P2 cleavage site mutations on poliovirus polyprotein processing. Virology 224:34-42. [DOI] [PubMed] [Google Scholar]
- 20.Cuconati, A., A. Molla, and E. Wimmer. 1998. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 72:6456-6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doedens, J. R., T. H. Giddings, Jr., and K. Kirkegaard. 1997. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J. Virol. 71:9054-9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egger, D., and K. Bienz. 2005. Intracellular location and translocation of silent and active poliovirus replication complexes. J. Gen. Virol. 86:707-718. [DOI] [PubMed] [Google Scholar]
- 23.Egger, D., N. Teterina, E. Ehrenfeld, and K. Bienz. 2000. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 74:6570-6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco, D., H. B. Pathak, C. E. Cameron, B. Rombaut, E. Wimmer, and A. V. Paul. 2005. Stimulation of poliovirus synthesis in a HeLa cell-free in vitro translation-RNA replication system by viral protein 3CDpro. J. Virol. 79:6358-6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freistadt, M. S., J. A. Vaccaro, and K. E. Eberle. 2007. Biochemical characterization of the fidelity of poliovirus RNA-dependent RNA polymerase. Virol. J. 4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gamarnik, A. V., and R. Andino. 2000. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J. Virol. 74:2219-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Briones, M., M. F. Rosas, M. Gonzalez-Magaldi, M. A. Martin-Acebes, F. Sobrino, and R. Armas-Portela. 2006. Differential distribution of non-structural proteins of foot-and-mouth disease virus in BHK-21 cells. Virology 349:409-421. [DOI] [PubMed] [Google Scholar]
- 28.Gerber, K., E. Wimmer, and A. V. Paul. 2001. Biochemical and genetic studies of the initiation of human rhinovirus 2 RNA replication: purification and enzymatic analysis of the RNA-dependent RNA polymerase 3Dpol. J. Virol. 75:10969-10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giachetti, C., S. S. Hwang, and B. L. Semler. 1992. cis-acting lesions targeted to the hydrophobic domain of a poliovirus membrane protein involved in RNA replication. J. Virol. 66:6045-6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodfellow, I., Y. Chaudhry, A. Richardson, J. Meredith, J. W. Almond, W. Barclay, and D. J. Evans. 2000. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 74:4590-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall, D. J., and A. C. Palmenberg. 1996. Cleavage site mutations in the encephalomyocarditis virus P3 region lethally abrogate the normal processing cascade. J. Virol. 70:5954-5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammerle, T., C. U. Hellen, and E. Wimmer. 1991. Site-directed mutagenesis of the putative catalytic triad of poliovirus 3C proteinase. J. Biol. Chem. 266:5412-5416. [PubMed] [Google Scholar]
- 33.Harris, K. S., S. R. Reddigari, M. J. Nicklin, T. Hammerle, and E. Wimmer. 1992. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J. Virol. 66:7481-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris, K. S., W. Xiang, L. Alexander, W. S. Lane, A. V. Paul, and E. Wimmer. 1994. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 269:27004-27014. [PubMed] [Google Scholar]
- 35.Hashimoto, K., and B. Simizu. 1976. Effect of cordycepin on the replication of western equine encephalitis virus. Arch. Virol. 52:341-345. [DOI] [PubMed] [Google Scholar]
- 36.Herold, J., and R. Andino. 2000. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 74:6394-6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang, L., E. V. Sineva, M. R. Hargittai, S. D. Sharma, M. Suthar, K. D. Raney, and C. E. Cameron. 2004. Purification and characterization of hepatitis C virus non-structural protein 5A expressed in Escherichia coli. Protein Expr. Purif. 37:144-153. [DOI] [PubMed] [Google Scholar]
- 38.Irurzun, A., L. Perez, and L. Carrasco. 1992. Involvement of membrane traffic in the replication of poliovirus genomes: effects of brefeldin A. Virology 191:166-175. [DOI] [PubMed] [Google Scholar]
- 39.Jackson, W. T., T. H. Giddings, Jr., M. P. Taylor, S. Mulinyawe, M. Rabinovitch, R. R. Kopito, and K. Kirkegaard. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobson, S. J., D. A. Konings, and P. Sarnow. 1993. Biochemical and genetic evidence for a pseudoknot structure at the 3′ terminus of the poliovirus RNA genome and its role in viral RNA amplification. J. Virol. 67:2961-2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jore, J., B. De Geus, R. J. Jackson, P. H. Pouwels, and B. E. Enger-Valk. 1988. Poliovirus protein 3CD is the active protease for processing of the precursor protein P1 in vitro. J. Gen. Virol. 69:1627-1636. [DOI] [PubMed] [Google Scholar]
- 42.Jurgens, C., and J. B. Flanegan. 2003. Initiation of poliovirus negative-strand RNA synthesis requires precursor forms of p2 proteins. J. Virol. 77:1075-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch, F., and G. Koch. 1985. The molecular biology of poliovirus. Springer-Verlag, New York, NY.
- 44.Kuhn, R. J., H. Tada, M. F. Ypma-Wong, B. L. Semler, and E. Wimmer. 1988. Mutational analysis of the genome-linked protein VPg of poliovirus. J. Virol. 62:4207-4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusov, Y., and V. Gauss-Muller. 1999. Improving proteolytic cleavage at the 3A/3B site of the hepatitis A virus polyprotein impairs processing and particle formation, and the impairment can be complemented in trans by 3AB and 3ABC. J. Virol. 73:9867-9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemon, S. M., C. Walker, M. J. Alter, and M. Yi. 2007. Hepatitis C virus, p. 1253-1304. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 47.Leong, L. E., C. Cornell, and B. L. Semler. 2002. Processing determinants and functions of cleavage products of picornavirus polyproteins, p. 187-198. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 48.Liu, Y., D. Franco, A. V. Paul, and E. Wimmer. 2007. Tyrosine 3 of poliovirus terminal peptide VPg (3B) has an essential function in RNA replication in the context of its precursor protein, 3AB. J. Virol. 81:5669-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mason, P. W., S. V. Bezborodova, and T. M. Henry. 2002. Identification and characterization of a cis-acting replication element (cre) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. J. Virol. 76:9686-9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maynell, L. A., K. Kirkegaard, and M. W. Klymkowsky. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 66:1985-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molla, A., A. V. Paul, and E. Wimmer. 1991. Cell-free, de novo synthesis of poliovirus. Science 254:1647-1651. [DOI] [PubMed] [Google Scholar]
- 52.Nair, C. N., and D. L. Panicali. 1976. Polyadenylate sequences of human rhinovirus and poliovirus RNA and cordycepin sensitivity of virus replication. J. Virol. 20:170-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nayak, A., I. G. Goodfellow, and G. J. Belsham. 2005. Factors required for the uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3Dpol) in vitro. J. Virol. 79:7698-7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nicklin, M. J., K. S. Harris, P. V. Pallai, and E. Wimmer. 1988. Poliovirus proteinase 3C: large-scale expression, purification, and specific cleavage activity on natural and synthetic substrates in vitro. J. Virol. 62:4586-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nomoto, A., N. Kitamura, F. Golini, and E. Wimmer. 1977. The 5′-terminal structures of poliovirion RNA and poliovirus mRNA differ only in the genome-linked protein VPg. Proc. Natl. Acad. Sci. USA 74:5345-5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nugent, C. I., K. L. Johnson, P. Sarnow, and K. Kirkegaard. 1999. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 73:427-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pallansch, M. A., O. M. Kew, B. L. Semler, D. R. Omilianowski, C. W. Anderson, E. Wimmer, and R. R. Rueckert. 1984. Protein processing map of poliovirus. J. Virol. 49:873-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panicali, D. L., and C. N. Nair. 1978. Effect of cordycepin triphosphate on in vitro RNA synthesis by picornavirus polymerase complexes. J. Virol. 25:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pathak, H. B., S. K. Ghosh, A. W. Roberts, S. D. Sharma, J. D. Yoder, J. J. Arnold, D. W. Gohara, D. J. Barton, A. V. Paul, and C. E. Cameron. 2002. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J. Biol. Chem. 277:31551-31562. [DOI] [PubMed] [Google Scholar]
- 60.Pathak, H. B., H. S. Oh, I. G. Goodfellow, J. J. Arnold, and C. E. Cameron. 2008. Picornavirus genome replication: roles of precursor proteins and rate-limiting steps in oriI-dependent VPg uridylylation. J. Biol. Chem. 283:30677-30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paul, A. V., E. Rieder, D. W. Kim, J. H. van Boom, and E. Wimmer. 2000. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 74:10359-10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paul, A. V., J. H. van Boom, D. Filippov, and E. Wimmer. 1998. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 393:280-284. [DOI] [PubMed] [Google Scholar]
- 63.Paul, A. V., J. Yin, J. Mugavero, E. Rieder, Y. Liu, and E. Wimmer. 2003. A “slide-back” mechanism for the initiation of protein-primed RNA synthesis by the RNA polymerase of poliovirus. J. Biol. Chem. 278:43951-43960. [DOI] [PubMed] [Google Scholar]
- 64.Racaniello, V. R. 2007. Picornaviridae: the viruses and their replication, p. 795-838. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 65.Reuer, Q., R. J. Kuhn, and E. Wimmer. 1990. Characterization of poliovirus clones containing lethal and nonlethal mutations in the genome-linked protein VPg. J. Virol. 64:2967-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richards, O. C., J. F. Spagnolo, J. M. Lyle, S. E. Vleck, R. D. Kuchta, and K. Kirkegaard. 2006. Intramolecular and intermolecular uridylylation by poliovirus RNA-dependent RNA polymerase. J. Virol. 80:7405-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ronn, R., and A. Sandstrom. 2008. New developments in the discovery of agents to treat hepatitis C. Curr. Top. Med. Chem. 8:533-562. [DOI] [PubMed] [Google Scholar]
- 68.Rossmann, M. G. 2002. Picornavirus structure overview, p. 27-38. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 69.Rothberg, P. G., T. J. Harris, A. Nomoto, and E. Wimmer. 1978. O4-(5′-uridylyl)tyrosine is the bond between the genome-linked protein and the RNA of poliovirus. Proc. Natl. Acad. Sci. USA 75:4868-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rust, R. C., L. Landmann, R. Gosert, B. L. Tang, W. Hong, H. P. Hauri, D. Egger, and K. Bienz. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75:9808-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sandoval, I. V., and L. Carrasco. 1997. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J. Virol. 71:4679-4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlegel, A., T. H. Giddings, Jr., M. S. Ladinsky, and K. Kirkegaard. 1996. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70:6576-6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Semler, B. L., C. W. Anderson, N. Kitamura, P. G. Rothberg, W. L. Wishart, and E. Wimmer. 1981. Poliovirus replication proteins: RNA sequence encoding P3-1b and the sites of proteolytic processing. Proc. Natl. Acad. Sci. USA 78:3464-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Semler, B. L., and E. Wimmer (ed.). 2002. Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 75.Sharma, R., S. Raychaudhuri, and A. Dasgupta. 2004. Nuclear entry of poliovirus protease-polymerase precursor 3CD: implications for host cell transcription shut-off. Virology 320:195-205. [DOI] [PubMed] [Google Scholar]
- 76.Shen, M., Z. J. Reitman, Y. Zhao, I. Moustafa, Q. Wang, J. J. Arnold, H. B. Pathak, and C. E. Cameron. 2008. Picornavirus genome replication. Identification of the surface of the poliovirus (PV) 3C dimer that interacts with PV 3Dpol during VPg uridylylation and construction of a structural model for the PV 3C2-3Dpol complex. J. Biol. Chem. 283:875-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen, M., Q. Wang, Y. Yang, H. B. Pathak, J. J. Arnold, C. Castro, S. M. Lemon, and C. E. Cameron. 2007. Human rhinovirus type 14 gain-of-function mutants for oriI utilization define residues of 3C(D) and 3Dpol that contribute to assembly and stability of the picornavirus VPg uridylylation complex. J. Virol. 81:12485-12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shieh, Y. C., K. R. Calci, and R. S. Baric. 1999. A method to detect low levels of enteric viruses in contaminated oysters. Appl. Environ. Microbiol. 65:4709-4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skern, T., B. Hampolz, A. Guarne, I. Fita, E. Bergmann, J. Petersen, and M. N. G. James. 2002. Structure and function of picornavirus proteinases, p. 199-212. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 80.Suhy, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takeda, N., R. J. Kuhn, C. F. Yang, T. Takegami, and E. Wimmer. 1986. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J. Virol. 60:43-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takegami, T., B. L. Semler, C. W. Anderson, and E. Wimmer. 1983. Membrane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virology 128:33-47. [DOI] [PubMed] [Google Scholar]
- 83.Towner, J. S., M. M. Mazanet, and B. L. Semler. 1998. Rescue of defective poliovirus RNA replication by 3AB-containing precursor polyproteins. J. Virol. 72:7191-7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vance, L. M., N. Moscufo, M. Chow, and B. A. Heinz. 1997. Poliovirus 2C region functions during encapsidation of viral RNA. J. Virol. 71:8759-8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Ooij, M. J., C. Polacek, D. H. Glaudemans, J. Kuijpers, F. J. van Kuppeveld, R. Andino, V. I. Agol, and W. J. Melchers. 2006. Polyadenylation of genomic RNA and initiation of antigenomic RNA in a positive-strand RNA virus are controlled by the same cis-element. Nucleic Acids Res. 34:2953-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Varennes, A. D., and A. J. Maule. 1985. Independent replication of cowpea mosaic virus bottom component RNA: in vivo instability of the viral RNAs. Virology 144:495-501. [DOI] [PubMed] [Google Scholar]
- 87.Verlinden, Y., A. Cuconati, E. Wimmer, and B. Rombaut. 2000. The antiviral compound 5-(3,4-dichlorophenyl) methylhydantoin inhibits the post-synthetic cleavages and the assembly of poliovirus in a cell-free system. Antivir. Res. 48:61-69. [DOI] [PubMed] [Google Scholar]
- 88.White, J. L., and W. O. Dawson. 1979. Effect of cordycepin triphosphate on in vitro RNA synthesis by plant viral replicases. J. Virol. 29:811-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wimmer, E., C. U. Hellen, and X. Cao. 1993. Genetics of poliovirus. Annu. Rev. Genet. 27:353-436. [DOI] [PubMed] [Google Scholar]
- 90.Wittek, R., H. Koblet, A. Menna, and R. Wyler. 1977. The effect of cordycepin on the multiplication of Semliki Forest virus and on polyadenylation of viral RNA. Arch. Virol. 54:95-106. [DOI] [PubMed] [Google Scholar]
- 91.Xiang, W., K. S. Harris, L. Alexander, and E. Wimmer. 1995. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol. 69:3658-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang, Y., R. Rijnbrand, K. L. McKnight, E. Wimmer, A. Paul, A. Martin, and S. M. Lemon. 2002. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J. Virol. 76:7485-7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang, Y., R. Rijnbrand, S. Watowich, and S. M. Lemon. 2004. Genetic evidence for an interaction between a picornaviral cis-acting RNA replication element and 3CD protein. J. Biol. Chem. 279:12659-12667. [DOI] [PubMed] [Google Scholar]
- 94.Ypma-Wong, M. F., P. G. Dewalt, V. H. Johnson, J. G. Lamb, and B. L. Semler. 1988. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166:265-270. [DOI] [PubMed] [Google Scholar]