

Abstract
The bromodomain protein Brd4 plays critical roles in cellular proliferation and cell cycle progression. In this study, we investigated the involvement of Brd4 in cell cycle regulation and observed aberrant chromosome segregation and failures in cytokinesis in cancer cells as well as in primary keratinocytes in which Brd4 has been knocked down by RNA interference. Suppression of Brd4 protein levels in proliferating cells decreased Aurora B protein and transcript levels and abolished its chromosomal distribution. In contrast, exogenous Brd4 expression stimulated Aurora B promoter reporter activity and upregulated endogenous Aurora B expression. Aurora B kinase is a chromosomal passenger protein that is essential for chromosome segregation and cytokinesis. Either overexpression of Aurora B or its inactivation can induce defects in centrosome function, spindle assembly, chromosome alignment, and cytokinesis in various cancer cells. The impaired regulation of Aurora B expression in human cells by Brd4 knockdown or overexpression coincided with mitotic catastrophe and multinucleation that are typically observed when Aurora B is inactivated or overexpressed. Overall, our data suggest that Brd4 is essential for the maintenance of the cell cycle progression mediated at least in part through the control of transcription of the Aurora B kinase cell cycle regulatory gene.
Our previous work identified the cellular bromodomain protein Brd4 as a major binding protein for bovine papillomavirus (BPV) type 1 E2 (51). Brd4 tethers the E2/viral genome complex to mitotic chromosomes (51, 52), providing a molecular mechanism for BPV-1 E2-mediated papillomavirus maintenance in latently infected cells. Brd4 interacts with the E2 proteins from many different types of human and animal papillomaviruses (1, 3, 6, 16, 26, 27, 40, 51) as well as the Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen, which is required for KSHV episome maintenance during latent infection (33, 53). The EBNA1 protein of Epstein-Barr virus also functionally interacts with Brd4 (23), as does the orf73 protein of murine herpesvirus 68 (34). Besides these DNA tumor viruses, Brd4 has also been implicated in the regulation of human immunodeficiency virus transcription (54) and human cytomegalovirus immediate-early transcription (19).
Brd4 is a member of the BET family proteins that contain double bromodomains, which are conserved sequence motifs involved in chromatin targeting (11). It associates with mitotic chromosomes and has been shown to bind to acetylated chromatin, with preferential binding for acetylated histone H3 and H4 through its bromodomains (10). Brd4 plays an important role in both G1/S and G2/M cell cycle progression (11, 24, 30, 31, 33, 49). Previous in vivo studies suggested an important role for Brd4 in cellular growth control (15, 24). In mice, Brd4 knockout results in early embryonic lethality, and heterozygosity for Brd4 leads to pre- and postnatal growth defects that are associated with reduced proliferation (15, 24). In humans, the Brd4 gene, located on chromosome 19, is the target of translocation t(15;19)(q13;p13.1), which defines a highly lethal upper respiratory tract carcinoma in young people (12). Ectopic expression of Brd4 in mice represses both tumor growth and metastasis (9). In addition, Brd4 activation in human breast carcinomas induces a gene expression signature that robustly predicts progression and survival in multiple human breast cancer data sets. These studies suggest that Brd4 is a critical tumor suppressor playing a dominant role in breast cancer metastasis and that dysregulation of Brd4-associated pathways may also contribute to breast cancer progression (9).
Brd4 becomes associated with mitotic chromosomes at a time when most transcription factors are displaced from chromatin (10). It has thus been implicated in marking actively transcribed regions of the genome during mitosis to ensure the resumption of properly controlled gene expression in newly divided cells. Brd4 interacts with cyclin T1 and Cdk9, which constitute core positive transcription elongation factor b (P-TEFb) (5, 17, 50). Brd4 binding reconstitutes the active form of P-TEFb (17, 50), which phosphorylates the C-terminal domain of RNA polymerase II and stimulates RNA polymerase II transcriptional elongation (17, 50). Brd4-P-TEFb interaction increases dramatically in cells progressing from late mitosis to early G1 (49). This interaction recruits P-TEFb to mitotic chromosomes to stimulate the expression of key G1 and growth-associated genes and promotes progression to S phase (30, 49), providing a mechanism for Brd4 in transmitting transcriptional memory across cell division. The P-TEFb and Brd4 complex also contributes to expression of human immunodeficiency virus type 1 and human T-lymphotropic virus type 1 genomes (5, 8). In addition, Brd4 plays an important role in papillomavirus E2-mediated viral transcriptional activation and repression (16, 39, 48). However, the molecular mechanisms by which Brd4 regulates cellular proliferation and tumor suppression are largely not known.
The various important roles of Brd4 in transcription and in viral pathogenesis prompted us to investigate further its cellular functions using RNA interference. In this study, we demonstrated that Brd4 knockdown in human cancer cells results in an abnormal nuclear structure, including a lobulated chromatin structure and lagging/bridging chromosomes, which eventually lead to catastrophic mitotic defects and chromosomal instability. In primary human foreskin keratinocytes (HFKs), Brd4 knockdown caused severe cytokinesis defects and abnormal centrosome accumulation. Using a candidate approach, we discovered that Brd4 suppression led to the downregulation of Aurora B expression, whereas exogenous Brd4 expression stimulated the Aurora B promoter reporter gene expression and enhanced the expression of endogenous Aurora B. Aurora B, previously known as AIM-1, is a conserved eukaryotic mitotic protein kinase. In mammals, this kinase plays an essential role in chromosomal segregation processes, including chromosome condensation, alignment, control of spindle checkpoints, chromosome segregation, and cytokinesis (28). Inhibition of Aurora B leads to abnormal chromosome segregation and failed cytokinesis (18). Our studies demonstrate that Brd4 regulates Aurora B expression in mammalian cells to maintain the proper chromosome segregation and cell cycle progression.
MATERIALS AND METHODS
Cell culture, transfection, and retrovirus infection.
C33A, HEK293T, T98G, and U2OS cells were maintained in monolayer culture in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS). Monolayer cultures of primary HFKs were prepared from neonatal foreskins by using a modified version of the protocol of Rheinwald and Green (37). Keratinocytes were isolated by using dispase incubation of foreskin tissue to allow for dermal-epidermal separation. Epidermal specimens were trypsinized, and keratinocytes released from the tissue were maintained in serum-free keratinocyte growth medium (GIBCO/BRL) at 37°C in 5% CO2 (37). At about 80% confluence, the cells were frozen. The frozen cells were subsequently thawed in complete serum-free medium (GIBCO/BRL) supplemented with human recombinant epidermal growth factor (GIBCO/BRL) and bovine pituitary extract (GIBCO/BRL). Plasmid DNAs were introduced into cells using Fugene 6 transfection reagent (Roche). Recombinant retroviruses were prepared using standard methods, and infections of the monolayer cells were performed as described previously (51). Green fluorescent protein (GFP)-H2B retrovirus was generously provided by Randall King (Harvard Medical School) (41).
Recombinant plasmids.
Plasmids containing the full-length human Brd4 (pcDNA4C-Brd4-FL) and mouse pCMV2-F-mBrd4 plasmids have been described previously (11, 51). The Aurora B promoter-luciferase constructs, pGL3-1879, and pGL3-185, were generously provided by Yukio Okano (21).
Brd4 RNA interference.
For knockdown of Brd4 expression, the pRETRO vectors encoding Brd4 short hairpin RNAs (shRNAs) and control shRNA have been described previously (39). Four Brd4 small interfering RNAs (siRNAs) targeting different regions of the gene were purchased from Dharmacon. They are Si1 (catalog no. D-004937-02), Si2 (catalog no. D-004937-03), Si3 (catalog no. D-004937-04), and Si4 (catalog no. D-004937-05). The Dharmacon siGENOME nontargeting siRNA 1 (catalog no. D-001210-01) was used as a control siRNA in this study. The siRNAs were transfected into monolayer cultures using the manufacturer's protocol.
BrdU incorporation assay.
The bromodeoxyuridine (BrdU) incorporation assay was performed as recommended by the manufacturer (BrdU cell proliferation enzyme-linked immunosorbent assay; Roche). Briefly, C33A cells were split into groups of 5 × 102, 2 × 103, 6 × 103, and 14 × 103 cells per 96-well plate. Cells were incubated for 2 h with BrdU, followed by fixation for 30 min and incubation for 1 h with anti-BrdU peroxidase-conjugated antibody. After the addition of luminol (substrate), chemiluminescence was measured with a luminometer and expressed as a function of cell number.
Immunofluorescent staining.
Cells transfected with Brd4 siRNAs or infected with retroviruses encoding Brd4 shRNAs were plated onto coverslips and fixed with 3% paraformaldehyde in phosphate-buffered saline 24 h later. Cells were incubated in blocking/permeabilization buffer (0.5% Triton X-100 and 3% bovine serum albumin in PBS) for 20 min at room temperature and stained with anti-Brd4 rabbit polyclonal antibody (39) at room temperature for 60 min. After incubation, cells were washed three times with blocking/permeabilization buffer and incubated with an Alexa Fluor 594-conjugated goat anti-rabbit immunoglobulin G (IgG) (Molecular Probes) for an additional 60 min. Cells were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) and examined with either a Zeiss LSM 510 UV upright confocal microscope or a Leica DMLB epifluorescence microscope.
Western blot analysis.
For Brd4 RNA interference, C33A, 293T, or U2OS cells were transfected with either a Brd4 siRNA or a nontargeting control siRNA from Dharmacon and harvested at 72 h posttransfection. For transient protein expression in C33A cells, 40% to 80% confluent cells growing in 10-cm dishes were transfected with 16 μg of plasmid DNA using Fugene 6 transfection reagent (Roche). Cells were harvested at 48 h posttransfection. Cytoplasmic and nuclear extracts were prepared as described previously (38). Twenty micrograms of nuclear extracts was resolved on a sodium dodecyl sulfate-polyacrylamide gel. Proteins were transferred to Immobilon-P (Millipore) and blotted with specific antibodies to detect the proteins of interest (ECL detection; GE Healthcare). Antibodies employed in the Western blot analysis were as follows: the rabbit polyclonal antibody against Brd4 has been described previously (39), the cyclin B1 and cyclin A antibodies were obtained from Santa Cruz, the Aurora B and Aurora A antibodies were obtained from BD Biosciences, and the securin and Plk1 antibodies were obtained from Zymed Laboratories.
Reporter assays.
A total of 1.5 × 105 293T cells were cotransfected with 0.3 μg reporter plasmid and either 1.5 μg pcDNA4C or 1.5 μg pcDNA4C-Brd4-FL plasmid using the calcium phosphate transfection method. To monitor transfection efficiencies, 0.2 μg pEGFP-C1 plasmid was cotransfected. The transfection efficiencies were nearly 100% as determined by examining the GFP signal under a fluorescence microscope. At 24 h posttransfection, cells were lysed and luciferase activities were measured according to the manufacturer's protocol (luciferase assay system; Promega). Results were normalized to the protein concentration in the lysates. Luciferase activity is expressed as the mean ± standard deviation of triplicate luciferase measurements.
RT-PCR.
C33A cells were transfected with either a Brd4 siRNA or a control siRNA. Total RNA was isolated at 72 h posttransfection using a NucleoSpin RNA purification kit (Clontech) according to the manufacturer's manual. Reverse transcription-PCR (RT-PCR) was performed in 20 μl of first-strand buffer containing 0.2 μg of total RNA and 100 ng of oligo(dT) primer using 0.2 μl of PowerScript (Clontech) for 90 min at 42°C, followed by heat inactivation for 5 min at 99°C. PCR using gene-specific primers was performed using 1 μl of RT-PCR product in a 50-μl reaction mixture containing 0.4 μM of each primer, 0.2 mM of deoxynucleoside triphosphate, 1× Pfu polymerase buffer, and 2 U of Pfu DNA polymerase (Stratagene). PCR was carried out for 27 cycles of 94°C (1 min), 56°C (1 min), and 68°C (1 min) for Aurora B; 94°C (1 min), 56°C (1 min), and 68°C (1 min) for Aurora A; and 94°C (1 min), 59°C (1 min), and 68°C (1 min) for β-actin. The PCR products were separated on a 2% agarose gel and detected with ethidium bromide. Primer sequences are available on request.
RESULTS
Brd4 knockdown in cancer cells inhibits cellular proliferation and leads to abnormal chromosome segregation.
We have previously developed shRNAs in the pRETRO retrovirus vector to knock down Brd4 in cultured cells (39). Using Brd4 immunofluorescent staining and Western blotting, we were able to detect efficient Brd4 knockdown in cells transiently transfected with these constructs at 72 to 96 h posttransfection or in cells infected with the retroviruses encoding these shRNAs after 4 to 5 days of selection in puromycin. However, we were not able to establish any stable cell lines with stable knockdown of Brd4. Selection of the retrovirus-infected cells in puromycin for longer periods (>10 days) resulted in colonies that expressed normal Brd4 levels, suggesting that cells with efficient Brd4 knockdown had been outgrown by the cells with more normal levels of Brd4. The data suggested that Brd4 knockdown may have a detrimental effect on cellular proliferation, consistent with the fact that Brd4 knockout is embryonically lethal in mice (15). To further examine the Brd4 knockdown effect on cellular proliferation, human papillomavirus-negative cervical carcinoma C33A cells were transfected with either a control vector or a Brd4 shRNA(NT) plasmid and a plasmid encoding the cell surface ILR2 marker. The transfected cells were enriched by affinity sorting using anti-ILR2 antibodies. Equal numbers of cells were analyzed by BrdU cell proliferation assay. The data showed that Brd4 knockdown significantly inhibited cellular proliferation (Fig. 1A). To examine the knockdown effect in single cells, we infected C33A cells with retrovirus encoding either Brd4shRNA(NT), Brd4shRNA(CT), or a control shRNA. Brd4shRNA(NT) and Brd4shRNA(CT) each target two different regions of Brd4. Cells expressing the shRNAs were then selected using the puromycin resistance marker for 4 days. The selected cells were examined by a combination of anti-Brd4 immunofluorescent staining and DAPI DNA staining. As shown in Fig. 1B, compared to the control shRNA-treated cells (top panels), cells with Brd4 knockdown by either shRNA showed much larger nuclei or a lobulated abnormal nuclear structure (Fig. 1B, middle and bottom panels). To confirm that the abnormal nuclear changes were due to the Brd4 knockdown and to control for any potential off-target effect, we also analyzed the effect of Brd4 knockdown in C33A cells using a set of four Brd4 siRNAs (Si1 to -4) from Dharmacon. We observed efficient knockdown of Brd4 with each of the four siRNAs by anti-Brd4 Western blot analysis (Fig. 2A). Compared to the control siRNA-treated cells, the Brd4 knockdown cells frequently had increased cell size and manifested abnormal nuclear and chromosome structures, including enlarged, lobulated nuclei; micronuclei; and lagging/bridging chromosomes at mitosis (Fig. 2B). These nuclear and chromosomal abnormalities were observed in cells using each of the four Brd4 siRNAs (Si1 to Si4) (Fig. 2C). We also observed that the severity of the mitotic defect correlated with the level of Brd4 knockdown in Brd4 siRNA-treated cells; cells with less Brd4 knockdown appeared to have a more normal nuclear structure (Fig. 2B and C).
FIG. 1.

Brd4 knockdown by shRNA inhibits cellular proliferation and leads to abnormal chromatin structure. (A) C33A cells were cotransfected with either a control or Brd4 shRNA(NT) plasmid and a plasmid encoding the ILR2 marker. At 48 h posttransfection, the transfected cells were purified by affinity sorting using anti-ILR2 antibodies immobilized on protein A-Sepharose. Two thousand purified cells from each sample were seeded into a 96-well plate and analyzed by BrdU cell proliferation assay. Results are expressed as relative light units (Rlu) per second. Error bars indicate standard deviations. (B) C33A cells were infected with retrovirus encoding either a control shRNA or a Brd4 shRNA and a puromycin resistance marker. After 6 days of selection in puromycin, cells were fixed and stained with an anti-Brd4 rabbit polyclonal antibody. The staining was detected by incubation with an Alexa Fluor 594-conjugated goat anti-rabbit IgG. Cells were also counterstained with DAPI to identify nuclei and mitotic chromosomes. Cells were examined under a Zeiss LSM 510 UV upright confocal microscope. Shown are the cells infected with retrovirus encoding Brd4 shRNA (NT) (middle panel) and Brd4 shRNA(CT) (lower panel).
FIG. 2.

Brd4 knockdown by siRNAs leads to chromosome missegregation. (A) C33A cells were transfected with either one of the four Brd4 siRNAs (Si1 to -4) or a control siRNA (CO) or were mock transfected. At 72 h posttransfection, protein lysates were harvested. Western blotting was performed using Brd4 and actin antibodies. (B) C33A cells were transfected with either Brd4 Si1 or a control siRNA. Cells were fixed and subjected to anti-Brd4 immunofluorescent staining. Cells were counterstained with DAPI to identify nuclei and mitotic chromosomes. Cells were examined under a Zeiss LSM 510 UV upright confocal microscope. Arrows indicate the Brd4 knockdown cells with abnormal chromatin structure. The experiment shown is representative of over 10 experiments. (C) C33A cells were transfected with either a control siRNA or a Brd4 siRNA as indicated. Cells were stained as for panel B and examined under a Leica DMLB epifluorescence microscope equipped with a Leica DC 500 digital camera.
To understand the process that led to the formation of lobulated nuclei and small micronuclei in the Brd4 knockdown cells, we performed time-lapse imaging of mitosis after Brd4 knockdown. C33A cells were transduced by retrovirus to establish cells stably expressing GFP-H2B, which allows the visualization of mitotic chromosomes and interphase nuclei in live cells. Upon Brd4 knockdown with Brd4 siRNAs, the C33A/GFP-H2B cells displayed enlarged/lobulated nuclei and micronuclei as shown by the DAPI staining (data not shown). The time-lapse image analysis demonstrated that the lagging chromosomes round up as micronuclei after mitosis, whereas the bridging chromosomes prevented furrow regression from completing resulting in failure of cytokinesis and binucleated/polynucleated cells. Continual failures in the completion of furrow regression in multiple cycles of mitosis eventually resulted in enlarged/lobulated nuclei. To rule out a cell type-specific effect, we examined the impact of Brd4 suppression in several other cell lines, including 293T and U2OS. Similar chromosomal defects were observed in each of these different cell lines (see Fig. 5 and data not shown). The phenotypes observed using multiple shRNAs and siRNAs, each targeted to different regions of Brd4, indicated that Brd4 has a role in maintaining accurate chromosome segregation.
FIG. 5.

Aurora B protein level and chromosomal localization after Brd4 knockdown. 293T cells were transfected with either Brd4 Si1 (KD) or a control siRNA (CO). At 72 h posttransfection, cells were fixed and double stained with anti-Brd4 and anti-Aurora B antibodies. The staining was detected by incubation with an Alexa Fluor 594-conjugated goat anti-rabbit IgG (red) and an Alexa Fluor 488-conjugated goat anti-mouse IgG (green), respectively. Cells were also counterstained with DAPI and examined under a Zeiss LSM 510 UV upright confocal microscope.
Brd4 knockdown in primary cells leads to abnormal chromosome segregation.
To rule out the possibility that the abnormal effects we observed were cancer cell specific, we next investigated the effect of Brd4 knockdown in primary HFKs. HFKs were transfected with either Brd4 Si1 or a control siRNA, and at 72 h posttransfection, the cells were examined using Brd4 immunostaining and DAPI counterstaining. In primary HFKs, Brd4 knockdown led to a high frequency of large cells containing binuclear tetraploid (2 × 2N) and binuclear octoploid (2 × 4N) nuclei (Fig. 3A). Quantitation revealed that the number of binucleated and polynucleated cells was much higher in the Brd4 knockdown cells than in the control cells (Fig. 3B). The data clearly indicated a cytokinesis defect in the Brd4 knockdown HFKs. We also noted the abnormal accumulation of centrosomes in Brd4 knockdown cells (data not shown). This result confirmed Brd4's role in chromosome segregation. Interestingly, unlike the cancer cells, primary HFK cells did not show any abnormal nuclear structure such as lobulated nuclei, macronuclei, or micronuclei. The difference in the nuclear morphologies of Brd4 knockdown cancer cells and HFKs may be due to a lack of a centrosome-clustering mechanism in the cancer cells (36). It has been suggested that existence of the centrosome-clustering system in primary cells prevents the amplified centrosomes from causing spindle multipolarity, while the lack of centrosome clustering in the cancer cells leads to lobulated macro- or micronuclei (36) (see Discussion).
FIG. 3.

Brd4 knockdown leads to abnormal chromosome segregation in HFKs. (A) Primary HFKs were transfected with either Brd4 siRNA Si1 or a control siRNA. Cells were fixed and subjected to anti-Brd4 immunofluorescent staining and DAPI nuclear staining. Arrows mark the enlarged Brd4 knockdown cells with binucleated or polynucleated structure. (B) Cells with abnormal nuclei were quantitated from 100 cells. Three independent experiments were performed. The averages ± standard deviations are shown.
Brd4 suppression downregulates the Aurora B pathway.
Previous studies have pointed to a role of Brd4 in both the G1/S and G2/M transitions (11, 24, 30, 31, 33, 49). To gain mechanistic insight into how Brd4 knockdown induced mitotic defects, we examined the effects of Brd4 knockdown on a number of mitotic checkpoint regulators, including securin, cyclin B1, cyclin A, Aurora A, Aurora B, Plk1, APC2, Cdc2, and Cdc27. C33A cells were transfected with either Brd4 Si1 or the control siRNA and protein levels determined by Western blotting at 72 h posttransfection. Brd4 was efficiently knocked down in the Brd4 siRNA-treated cells (Fig. 4A, Brd4 blot). Among the factors tested, most showed similar protein levels in the Brd4 knockdown cells and the control cells. Only the Aurora B protein level was significantly decreased (by 69%) in the Brd4 knockdown cells (Fig. 4A). In a separate experiment, we showed that the Aurora B protein level was decreased by 56% in 293T cells, 58% in U2OS cells, and 81% in C33A cells (Fig. 4B and C). In addition, all four Brd4 siRNAs (Si1 to Si4) that target different regions of Brd4 triggered a significant reduction of Aurora B protein levels, indicating that the reduction was a consequence of the Brd4 knockdown and not due to an off-target effect of an individual siRNA used (data not shown). Aurora B encodes a protein kinase that localizes to inner centromeres at metaphase and to the spindle midzone in anaphase. It is required for proper chromosome segregation and cytokinesis (46). To further examine the impact of Brd4 knockdown on Aurora B expression and localization, 293T cells were transfected with either Brd4 Si1 or the control siRNA and examined by immunofluorescent double staining of Brd4 and Aurora B. In the control cells, Aurora B associates with condensing chromatin, localizes to centromeres during prometaphase, and relocates to the equatorial region along the midzone microtubules after onset of anaphase. However, the Aurora B signals were dramatically reduced in both nuclear locations in Brd4 knockdown cells (Fig. 5, left panel, Aurora B staining). This result confirmed the observation of a reduction in protein level made by anti-Aurora B Western blot analysis (Fig. 4). Notably, the catastrophic mitotic defects observed in Brd4 knockdown 293T cells an lacking Aurora B signal closely resembled the nuclear morphology of cells in which Aurora B is either inhibited or knocked down (Fig. 5, left panel) (44).
FIG. 4.

Brd4 knockdown reduces Aurora B protein levels in cells. (A) C33A cells were transfected with either Brd4 Si1 (KD) or a control siRNA (CO). Cells were harvested at 72 h posttransfection. Protein lysates were analyzed by Western blotting using specific antibodies as indicated. Only the Aurora B protein level was significantly decreased (by 69%) in the Brd4 knockdown cells (arrows). (B) HEK293T, U2OS, and C33A cells were transfected as for panel A and analyzed by anti-Brd4 Western blotting. (C) Anti-Aurora B Western blot of lysates prepared for panel B. Actin is shown as a loading control.
Brd4 regulates Aurora B gene expression.
To test whether Aurora B is a downstream target gene of Brd4, we examined the level of Aurora B transcript in Brd4 knockdown cells. C33A cells were mock transfected or transfected with either Brd4 Si1 or a control siRNA. After verification of Brd4 knockdown by Western blot analysis (data not shown), total RNA was isolated from the cells and subjected to RT-PCR analysis. Compared to the control siRNA-treated cells and mock-transfected cells, transfection by Brd4 siRNA significantly decreased Brd4 RNA levels. More importantly, the Aurora B RNA level was also significantly reduced (by 76%) in the Brd4 siRNA-treated cells compared to the control cells (Fig. 6A). In contrast, the RNA levels of Aurora A, a member of the same family of serine/threonine protein kinases as Aurora B, remained unchanged upon Brd4 knockdown. In addition, the RNA levels of several other genes tested, such as Cdc27 and actin, were not significantly affected by the Brd4 siRNA. These data established that Aurora B RNA was specifically reduced upon Brd4 suppression, supporting the hypothesis that Aurora B is a downstream target gene of Brd4.
FIG. 6.
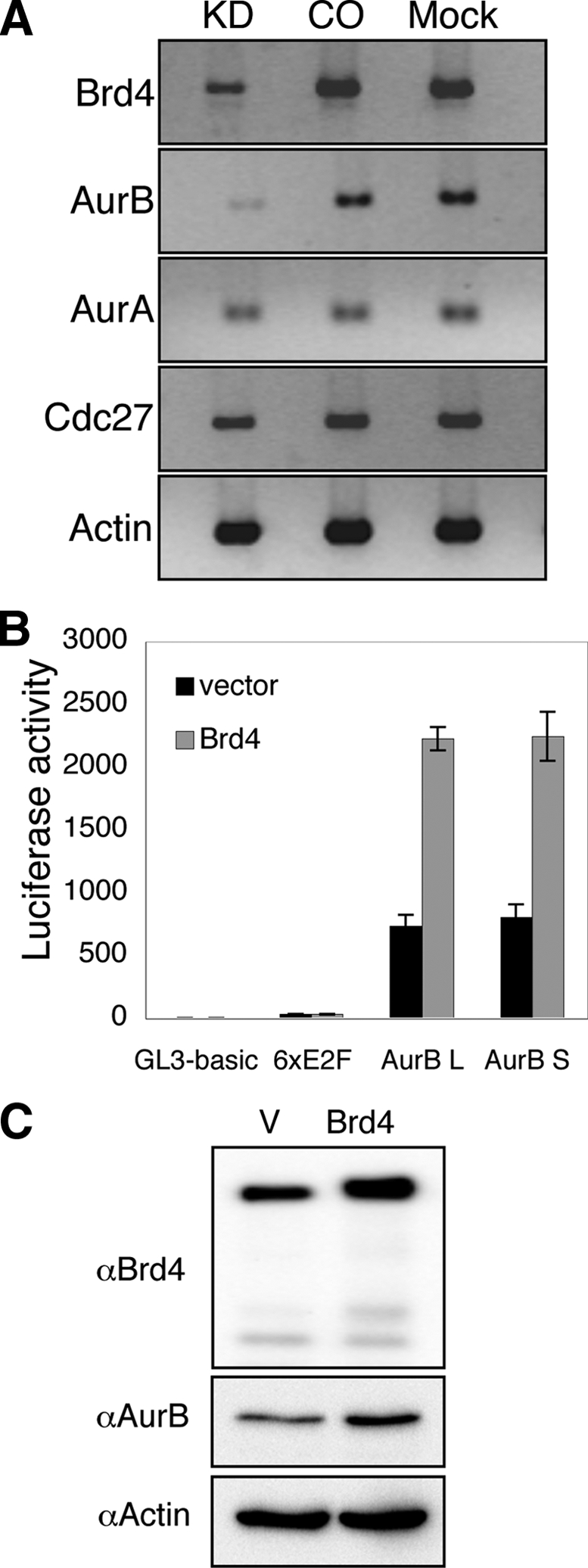
Brd4 regulates Aurora B gene transcription. (A) C33A cells were transfected with either Brd4 Si1 (KD) or a control siRNA (CO) or were mock transfected (C33A). Total RNA was harvested and subjected to RT-PCR using gene-specific primers. Actin expression is shown as a loading control. (B) Brd4 expressing construct pcDNA4C-Brd4-FL (Brd4) or an empty vector pcDNA4C (vector) was cotransfected with a luciferase reporter carrying no promoter (GL3-basic), six E2F binding sites (6xE2F), Aurora B long promoter (pGL3-1879, AurB L), or Aurora B short promoter (pGL3-185, AurB S) into 293T cells. At 24 h posttransfection, cell lysates were harvested for luciferase analysis. Results were normalized to the protein concentration in the lysates. Luciferase activity is expressed as the mean ± standard deviation of triplicate luciferase measurements. The data represents the averages from three independent experiments. (C) C33A cells were transfected with either an empty vector (V) or a Brd4-expressing construct, pCMV2-F-mBrd4 (Brd4). At 48 h posttransfection, protein lysates were analyzed by Western blotting using Brd4, Aurora B, and actin antibodies.
Aurora B is an important regulator of mitosis, and its mRNA and protein levels are tightly regulated in a cell cycle-dependent manner (21). The Aurora B promoter activity and mRNA level are upregulated during M phase, and the cell cycle-dependent element (CDE) and cell cycle gene homology region (CHR) upstream of the Aurora B transcription initiation sites regulate the cell cycle-dependent expression (21). To examine Brd4 function in regulating Aurora B promoter activity, the wild-type Aurora B promoter-luciferase construct pGL3-1879 (AurB L) and pGL3-185 (AurB S), which contains a fragment of from −185 to +359 with full promoter activity (21), were individually transfected into 293T cells with either a Brd4 expression construct or an empty vector. A pGL3-basic vector and a luciferase construct containing six E2F binding sites were also used in the experiment as specificity controls. Luciferase activity was measured at 24 h posttransfection. As shown in Fig. 6B, Brd4 expression significantly enhanced the expression of both the AurB L and AurB S reporter constructs (Fig. 6B). In contrast, luciferase reporter expression from pGL3-basic, the six-E2F-luciferase construct, or the Cdk2 promoter-luciferase constructs was not affected by Brd4 (Fig. 6B and data not shown). These data showed that Brd4 specifically stimulated Aurora B promoter activity.
To further examine the effect of Brd4 overexpression, the C33A/GFP-H2B stable cells were transfected with either an empty vector or a Brd4 expression construct. Interestingly, overexpression of Brd4 in C33A cells led to an increased number of cells containing abnormal nuclei and defective chromosomal structure (data not shown). Compared to the 1.5% mitotic index observed in the cells expressing GFP, GFP-Brd4 expression in cells reduced the mitotic index to 0%. Furthermore, overexpression of Brd4 also increased the Aurora B RNA level without affecting the Aurora A RNA level (data not shown). We also examined the effect of Brd4 overexpression on the Aurora B protein level. C33A cells were transfected with either an empty vector or a Brd4 expression construct. The increased Brd4 protein level was validated by anti-Brd4 Western blotting. The Aurora B protein level was also significantly increased in the cells with ectopic Brd4 expression (Fig. 6C). These results demonstrated that ectopic expression of Brd4 results in increased endogenous Aurora B levels and abnormal chromosome segregation. Together, the study identified the Aurora B gene as a gene whose transcription is regulated by Brd4.
Brd4 contributes to the mitotic activation of Aurora B in synchronized cells.
In view of the fact that Aurora B expression is strictly regulated in a cell cycle-dependent manner, we next examined how Brd4 affects the mitotic activation of Aurora B expression. T98G cells were synchronized to enrich for cells in mitosis using serum starvation as described previously (7). Cells were released from starvation into growth medium containing 10% FCS and analyzed hourly by DAPI staining for mitotic cells. The peak was 31 h after release, when greater than 50% of the cells were in mitosis as determined by DAPI staining. Western blot analysis revealed that the Aurora B protein level was increased more than threefold in the mitotic cells compared to nonsynchronized cells (Fig. 7A). To examine whether Brd4 contributed to this increase in Aurora B protein levels during mitosis, the T98G cells were transfected with control siRNA or Brd4 siRNA and the cells were synchronized by serum starvation, released, and analyzed during mitosis at 31 h. As shown in Fig. 7B, compared to the control siRNA-treated cells, the Aurora B protein level was dramatically reduced in the Brd4 siRNA-treated cells in mitosis. This result demonstrates that Brd4 directly contributes to the activation of Aurora B expression during mitosis.
FIG. 7.
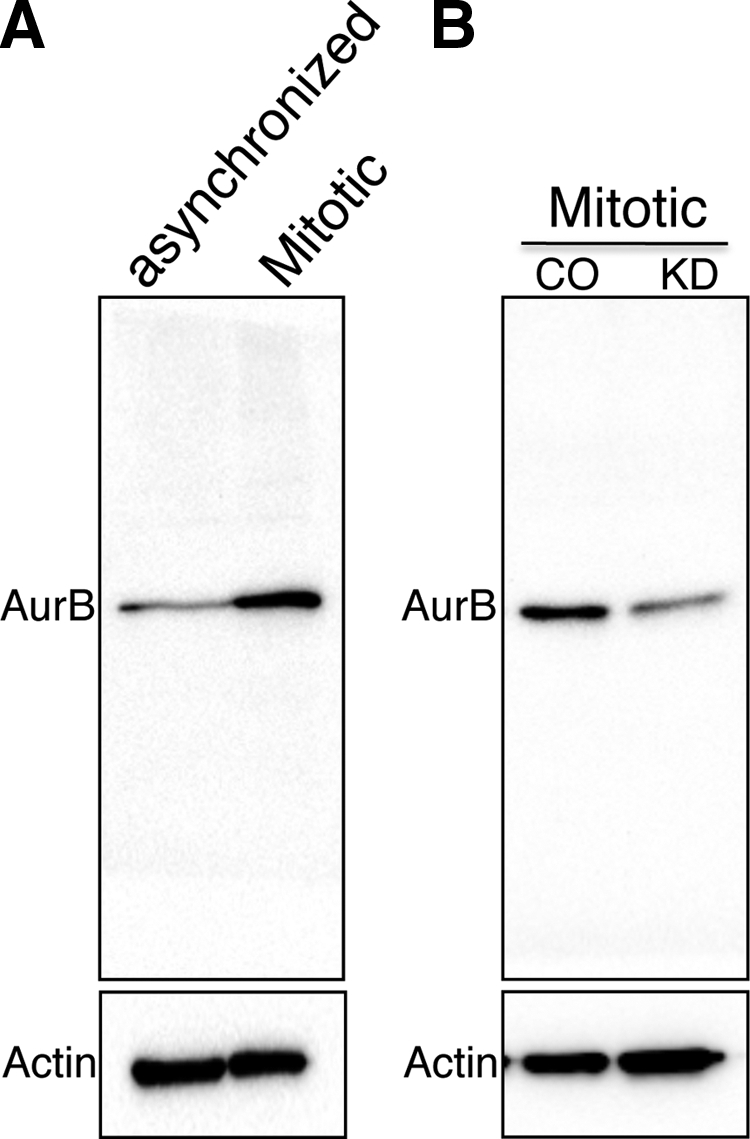
Brd4 regulates Aurora B activation during mitosis. (A) T98G cells were cultured in DMEM and 10% FCS. At 60 to 80% confluence, the cells were incubated with starvation medium (DMEM with 4.5 g/liter glucose) for 72 to 96 h. Cells were then released into release medium (DMEM with 4.5 g/liter glucose and 10% FCS). At 31 h after release, the peak for mitotic cells (>50%), protein lysates were prepared and analyzed by Western blotting using anti-Aurora B antibodies. Actin is shown as a loading control. (B) T98G cells were transfected with either Brd4 Si1 (KD) or the control siRNA (CO). At 24 h posttransfection, cells were synchronized as described above. At the peak of mitosis (31 h after release from starvation medium), protein lysates were prepared and analyzed by anti-Aurora B Western blotting. Actin is shown as a loading control.
DISCUSSION
Our previous studies have identified Brd4 as the host mitotic chromosome receptor for the BPV E2 protein (25, 51). Brd4 also functions in E2-mediated viral transcriptional regulation and is a mitotic chromosome-associated binding protein for the KSHV latency-associated nuclear antigen protein (33, 53). In cells, Brd4 plays a crucial role in cellular growth control, cell cycle progression, and tumor suppression. However, the molecular mechanisms underlying the Brd4 cellular functions are complex. In the present study, we showed that this mitotic chromosome-associated protein is essential for maintaining a properly condensed chromatin structure to ensure accurate chromosomal segregation. Knockdown of Brd4 inhibited cellular proliferation and led to abnormal centrosome accumulation, aberrant chromosome segregation and failures in cytokinesis in both cancer cells and primary cells. Interestingly, Brd4 overexpression also introduced similar chromosome segregation defects. We demonstrated that suppression of Brd4 expression by RNA interference downregulates Aurora B transcription while overexpression of Brd4 enhances the endogenous Aurora B expression in transfected cells. The impaired regulation of Aurora B expression in human cells by Brd4 knockdown or overexpression coincided with mitotic catastrophe and multinucleation that are typically observed when Aurora B is inactivated or overexpressed in cancer cells. Overall, our data suggest that Brd4 is essential for the maintenance of the cell cycle progression and the transcription of cell cycle regulatory kinase Aurora B. The phenotype of Brd4 knockdown that we observed is consistent with the work published by Ozato's group showing that, after nocodazole-induced mitotic arrest, Brd4± cells show an increased frequency of abnormal chromosomal segregation (31). Together, these studies demonstrate that properly maintained Brd4 expression is critical to ensure precise mitotic progression and that dysregulation of Brd4 leads to catastrophic mitotic defects that are frequently observed in cancer cells.
Mochizuki et al. have recently reported the results of a microarray analysis with NIH 3T3 cells following infection with a Brd4 shRNA lentivirus (30). Their study demonstrated that Brd4 stimulated G1 gene expression by binding to multiple G1 gene promoters in a cell cycle-dependent manner. Aurora B was not listed as one of the genes deregulated by Brd4 knockdown in that study. We have performed a similar microarray analysis with unsynchronized human cells transfected with Brd4 siRNA, in which we detected a 1.4-fold reduction in Aurora B expression upon Brd4 knockdown (unpublished results). Such a moderate reduction might have been below the cutoff level of the study published by Mochizuki et al. (30). The modest reduction in Aurora B expression is likely due to the fact that the Aurora B transcript level is relatively low in the asynchronized cells. By examining the cells synchronized at mitosis (Fig. 7), we were able to demonstrate a more dramatic inhibition of Aurora B expression by Brd4 knockdown.
Aurora B kinase is a chromosomal passenger protein and plays a crucial role in chromosome condensation, alignment, spindle checkpoints, chromosome segregation, and cytokinesis (28). Impaired regulation of Aurora B expression in human cells causes chromosomal abnormality and instability. Both the expression level and the kinase activity of Aurora B have been found to be upregulated in a variety of human cancers, indicating that this kinase might serve as a useful target for the development of anticancer drugs (4, 20, 43). Overexpression of Aurora B results in centrosome amplification and multinucleation in human cells, causing unequal distribution of genetic information and creating aneuploidy cells, one of the hallmarks of cancer (2, 14, 29, 32, 45). Interestingly, downregulation or overexpression of an inactive form of Aurora B yields the same polyploidization phenomenon in human cells as Aurora B overexpression. Aurora B knocked down by RNA-mediated interference prevents the formation of the midbody and leads to abnormal chromosome segregation and multinucleated cells as a consequence of cytokinesis failure (13). Depletion of Aurora B or overexpression of a kinase-inactive form in cells would also compromise the spindle checkpoint because the activity of Aurora B is required for checkpoint protein recruitment. The Brd4 knockdown-induced chromosome lagging in metaphase, chromosome segregation error, and errors in cytokinesis as presented in our study match precisely the chromosome segregation defects triggered by Aurora B knockdown or inactivation. These data were consistent with the fact that Aurora B levels were significantly reduced in Brd4 knockdown cells (Fig. 4). Due to the severe mitotic defect introduced by Brd4 knockdown, it was impossible to establish a stable Brd4 knockdown cell line. This is in agreement with the fact that deletion of the Brd4 gene is embryonically lethal in mice (15). In line with the fact that either increased or decreased Aurora B leads to phenotypically identical mitotic defects, the overexpression of Brd4, which enhanced Aurora B transcription, leads to the same mitotic abnormality as Brd4 knockdown. These observations support the idea that properly maintained Aurora B levels are essential for successful completion of chromosome segregation. Due to the Brd4 overexpression phenotype, our attempts to rescue the Brd4 siRNA defect were not successful. The rescue construct introduced severe cellular toxicity in the transfected cells, thus preventing further analysis of the Brd4 siRNA specificity. To overcome this problem, we used a set of two different Brd4 shRNAs and four different siRNAs that target different regions of the Brd4 mRNA to demonstrate the same mitotic defects, essentially ruling out the possibility of potential off-target effects being responsible for the observed phenotype. Besides the morphological analysis of the Brd4 knockdown cells, we have also examined how the Aurora B transcription, promoter activity and protein levels are modulated by Brd4 knockdown or overexpression. Despite the important function of Aurora B kinase in mitotic progression and cancer development, the mechanisms regulating Aurora B expression through the cell cycle remain largely unknown. Our functional studies provide evidence supporting a role for Brd4 in regulating the Aurora B pathway, thus contributing to the mitotic progression.
Both Aurora B promoter activity and mRNA transcription are upregulated during M phase. The cell cycle-dependent Aurora B expression is tightly regulated by the CDE and CHR upstream of the transcription initiation sites, which regulate the promoters by binding proteins (22, 35, 42, 47, 55). Highly homologous CDE-CHR elements have been identified in the promoter regions of genes such as those for Aurora A, Plk, Cdc25C, Cdc2, B-myb, cyclin A, and cyclin B2 and have been implicated in regulating gene expression in a cell cycle-dependent manner (22, 35, 42, 47, 55). The Aurora B mRNA level is regulated by a subset of E2F/DP family proteins that bind to the CDE (21). It will be interesting to investigate if Brd4 is a component of protein complexes that bind to the CDE or CHR sites in vivo. Notably, Brd4 knockdown by siRNA could not completely abrogate the transcription of Aurora B promoter luciferase reporters (data not shown), suggesting that the mechanism of Aurora B expression may be complex and that additional cellular factors besides Brd4 may contribute to regulation of Aurora B transcription. Nonetheless, the data presented here establish that Brd4 is involved in the regulation of the important cell cycle regulator Aurora B kinase and as such contributes to mitotic progression.
Most tumor cells are characterized by increased genomic instability and chromosome segregation defects, often associated with hyperamplification of centrosomes and formation of multipolar spindles. We observed a large number of macronuclei and micronuclei in cancer cells in which Brd4 had been knocked down. However, Brd4 knockdown in primary HFKs resulted primarily in binucleated or polynucleated cells. The centrosome hyperamplification in primary cells when Brd4 is knocked down did not lead to multipolarity in the primary cells. This is consistent with the theory that a process called centrosomal clustering prevented the formation of multipolar spindles in noncancer cells with accumulated centrosomes (36). However, the lack of a centrosome-clustering mechanism in cancer cells leads to a loss of centrosome coalescence, allowing multipolar spindle formation at a high frequency (36). This difference in the cancer cells might eventually result in the multipolar phenotype that we observed in the Brd4 knockdown cancer cells. Thus, our study further supports the notion that centrosome clustering may be an important mechanism for preserving genomic stability in noncancer cells.
Brd4 has emerged as an important host cell cycle regulator, tumor suppressor, and DNA tumor virus target. In order to establish Brd4 as an efficient antiviral and antitumor target, more studies are needed to understand the mechanisms that regulate the Brd4 cellular functions and its multiple roles in the life cycles of the various viruses with which it has now been associated. The specificity of Brd4 in the regulation of its downstream targets remains an interesting question for future studies. The identification of mechanisms controlling Brd4 functions in the cell cycle regulation and tumor suppressor pathways may facilitate the development of novel antitumor as well as antiviral therapeutic approaches.
Acknowledgments
We thank Randall King (Harvard Medical School) for the GFP-H2B retrovirus, Yukio Okano (Gifu University School of Medicine, Japan) for the Aurora B promoter-luciferase constructs, and the members of our laboratories for helpful discussions.
This work was supported by grants from the National Cancer Institute to P.M.H. (P01CA050661 and R01CA116720) and a McCABE Fund Pilot Award (University of Pennsylvania) to J.Y. J.Y. was supported by a fellowship from Charles A. King Trust-Bank of America, cotrustees.
Footnotes
Published ahead of print on 13 July 2009.
REFERENCES
- 1.Abbate, E. A., C. Voitenleitner, and M. R. Botchan. 2006. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 24877-889. [DOI] [PubMed] [Google Scholar]
- 2.Araki, K., K. Nozaki, T. Ueba, M. Tatsuka, and N. Hashimoto. 2004. High expression of Aurora-B/Aurora and Ipll-like midbody-associated protein (AIM-1) in astrocytomas. J. Neurooncol. 6753-64. [DOI] [PubMed] [Google Scholar]
- 3.Baxter, M. K., M. G. McPhillips, K. Ozato, and A. A. McBride. 2005. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 794806-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischoff, J. R., L. Anderson, Y. Zhu, K. Mossie, L. Ng, B. Souza, B. Schryver, P. Flanagan, F. Clairvoyant, C. Ginther, C. S. Chan, M. Novotny, D. J. Slamon, and G. D. Plowman. 1998. A homologue of Drosophila Aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 173052-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisgrove, D. A., T. Mahmoudi, P. Henklein, and E. Verdin. 2007. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 10413690-13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brannon, A. R., J. A. Maresca, J. D. Boeke, M. A. Basrai, and A. A. McBride. 2005. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein. Proc. Natl. Acad. Sci. USA 1022998-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canhoto, A. J., A. Chestukhin, L. Litovchick, and J. A. DeCaprio. 2000. Phosphorylation of the retinoblastoma-related protein p130 in growth-arrested cells. Oncogene 195116-5122. [DOI] [PubMed] [Google Scholar]
- 8.Cho, W. K., M. Zhou, M. K. Jang, K. Huang, S. J. Jeong, K. Ozato, and J. N. Brady. 2007. Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 Tax protein. J. Virol. 8111179-11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford, N. P., J. Alsarraj, L. Lukes, R. C. Walker, J. S. Officewala, H. H. Yang, M. P. Lee, K. Ozato, and K. W. Hunter. 2008. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. USA 1056380-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dey, A., F. Chitsaz, A. Abbasi, T. Misteli, and K. Ozato. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 1008758-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dey, A., J. Ellenberg, A. Farina, A. E. Coleman, T. Maruyama, S. Sciortino, J. Lippincott-Schwartz, and K. Ozato. 2000. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol. Cell. Biol. 206537-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.French, C. A., I. Miyoshi, J. C. Aster, I. Kubonishi, T. G. Kroll, P. Dal Cin, S. O. Vargas, A. R. Perez-Atayde, and J. A. Fletcher. 2001. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am. J. Pathol. 1591987-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu, J., M. Bian, Q. Jiang, and C. Zhang. 2007. Roles of Aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 51-10. [DOI] [PubMed] [Google Scholar]
- 14.Hontz, A. E., S. A. Li, W. L. Lingle, V. Negron, A. Bruzek, J. L. Salisbury, and J. J. Li. 2007. Aurora A and B overexpression and centrosome amplification in early estrogen-induced tumor foci in the Syrian hamster kidney: implications for chromosomal instability, aneuploidy, and neoplasia. Cancer Res. 672957-2963. [DOI] [PubMed] [Google Scholar]
- 15.Houzelstein, D., S. L. Bullock, D. E. Lynch, E. F. Grigorieva, V. A. Wilson, and R. S. Beddington. 2002. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 223794-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ilves, I., K. Maemets, T. Silla, K. Janikson, and M. Ustav. 2006. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 803660-3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang, M. K., K. Mochizuki, M. Zhou, H. S. Jeong, J. N. Brady, and K. Ozato. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19523-534. [DOI] [PubMed] [Google Scholar]
- 18.Kallio, M. J., M. L. McCleland, P. T. Stukenberg, and G. J. Gorbsky. 2002. Inhibition of Aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr. Biol. 12900-905. [DOI] [PubMed] [Google Scholar]
- 19.Kapasi, A. J., and D. H. Spector. 2008. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J. Virol. 82394-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katayama, H., T. Ota, F. Jisaki, Y. Ueda, T. Tanaka, S. Odashima, F. Suzuki, Y. Terada, and M. Tatsuka. 1999. Mitotic kinase expression and colorectal cancer progression. J. Natl. Cancer Inst. 911160-1162. [DOI] [PubMed] [Google Scholar]
- 21.Kimura, M., C. Uchida, Y. Takano, M. Kitagawa, and Y. Okano. 2004. Cell cycle-dependent regulation of the human Aurora B promoter. Biochem. Biophys. Res. Commun. 316930-936. [DOI] [PubMed] [Google Scholar]
- 22.Lange-zu Dohna, C., M. Brandeis, F. Berr, J. Mossner, and K. Engeland. 2000. A CDE/CHR tandem element regulates cell cycle-dependent repression of cyclin B2 transcription. FEBS Lett. 48477-81. [DOI] [PubMed] [Google Scholar]
- 23.Lin, A., S. Wang, T. Nguyen, K. Shire, and L. Frappier. 2008. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 8212009-12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruyama, T., A. Farina, A. Dey, J. Cheong, V. P. Bermudez, T. Tamura, S. Sciortino, J. Shuman, J. Hurwitz, and K. Ozato. 2002. A mammalian bromodomain protein, Brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell. Biol. 226509-6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McBride, A. A., M. G. McPhillips, and J. G. Oliveira. 2004. Brd4: tethering, segregation and beyond. Trends Microbiol. 12527-529. [DOI] [PubMed] [Google Scholar]
- 26.McPhillips, M. G., J. G. Oliveira, J. E. Spindler, R. Mitra, and A. A. McBride. 2006. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 809530-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McPhillips, M. G., K. Ozato, and A. A. McBride. 2005. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 798920-8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meraldi, P., R. Honda, and E. A. Nigg. 2004. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr. Opin. Genet. Dev. 1429-36. [DOI] [PubMed] [Google Scholar]
- 29.Meraldi, P., R. Honda, and E. A. Nigg. 2002. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 21483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mochizuki, K., A. Nishiyama, M. K. Jang, A. Dey, A. Ghosh, T. Tamura, H. Natsume, H. Yao, and K. Ozato. 2008. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2839040-9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishiyama, A., A. Dey, J. Miyazaki, and K. Ozato. 2006. Brd4 is required for recovery from antimicrotubule drug-induced mitotic arrest: preservation of acetylated chromatin. Mol. Biol. Cell 17814-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ota, T., S. Suto, H. Katayama, Z. B. Han, F. Suzuki, M. Maeda, M. Tanino, Y. Terada, and M. Tatsuka. 2002. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 625168-5177. [PubMed] [Google Scholar]
- 33.Ottinger, M., T. Christalla, K. Nathan, M. M. Brinkmann, A. Viejo-Borbolla, and T. F. Schulz. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 8010772-10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ottinger, M., D. Pliquet, T. Christalla, R. Frank, J. P. Stewart, and T. F. Schulz. 2009. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J. Virol. 834423-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philips, A., S. Chambeyron, N. Lamb, A. Vie, and J. M. Blanchard. 1999. CHF: a novel factor binding to cyclin A CHR corepressor element. Oncogene 186222-6232. [DOI] [PubMed] [Google Scholar]
- 36.Quintyne, N. J., J. E. Reing, D. R. Hoffelder, S. M. Gollin, and W. S. Saunders. 2005. Spindle multipolarity is prevented by centrosomal clustering. Science 307127-129. [DOI] [PubMed] [Google Scholar]
- 37.Rheinwald, J. G., and H. Green. 1975. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6331-343. [DOI] [PubMed] [Google Scholar]
- 38.Schreiber, E., P. Matthias, M. M. Muller, and W. Schaffner. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 176419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schweiger, M. R., J. You, and P. M. Howley. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 804276-4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senechal, H., G. G. Poirier, B. Coulombe, L. A. Laimins, and J. Archambault. 2007. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 35810-17. [DOI] [PubMed] [Google Scholar]
- 41.Shi, Q., and R. W. King. 2005. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 4371038-1042. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka, M., A. Ueda, H. Kanamori, H. Ideguchi, J. Yang, S. Kitajima, and Y. Ishigatsubo. 2002. Cell-cycle-dependent regulation of human Aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem. 27710719-10726. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka, T., M. Kimura, K. Matsunaga, D. Fukada, H. Mori, and Y. Okano. 1999. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 592041-2044. [PubMed] [Google Scholar]
- 44.Tao, Y., P. Zhang, F. Girdler, V. Frascogna, M. Castedo, J. Bourhis, G. Kroemer, and E. Deutsch. 2008. Enhancement of radiation response in p53-deficient cancer cells by the Aurora-B kinase inhibitor AZD1152. Oncogene 273244-3255. [DOI] [PubMed] [Google Scholar]
- 45.Tatsuka, M., H. Katayama, T. Ota, T. Tanaka, S. Odashima, F. Suzuki, and Y. Terada. 1998. Multinuclearity and increased ploidy caused by overexpression of the Aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 584811-4816. [PubMed] [Google Scholar]
- 46.Terada, Y., M. Tatsuka, F. Suzuki, Y. Yasuda, S. Fujita, and M. Otsu. 1998. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 17667-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchiumi, T., D. L. Longo, and D. K. Ferris. 1997. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J. Biol. Chem. 2729166-9174. [DOI] [PubMed] [Google Scholar]
- 48.Wu, S. Y., A. Y. Lee, S. Y. Hou, J. K. Kemper, H. Erdjument-Bromage, P. Tempst, and C. M. Chiang. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 202383-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, Z., N. He, and Q. Zhou. 2008. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 28967-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang, Z., J. H. Yik, R. Chen, N. He, M. K. Jang, K. Ozato, and Q. Zhou. 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19535-545. [DOI] [PubMed] [Google Scholar]
- 51.You, J., J. L. Croyle, A. Nishimura, K. Ozato, and P. M. Howley. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117349-360. [DOI] [PubMed] [Google Scholar]
- 52.You, J., M. R. Schweiger, and P. M. Howley. 2005. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J. Virol. 7914956-14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.You, J., V. Srinivasan, G. V. Denis, W. J. Harrington, Jr., M. E. Ballestas, K. M. Kaye, and P. M. Howley. 2006. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 808909-8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou, Q., and P. A. Sharp. 1995. Novel mechanism and factor for regulation by HIV-1 Tat. EMBO J. 14321-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zwicker, J., F. C. Lucibello, L. A. Wolfraim, C. Gross, M. Truss, K. Engeland, and R. Muller. 1995. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J. 144514-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]