

Abstract
Mrc1 plays a role in mediating the DNA replication checkpoint. We surveyed replication elongation proteins that interact directly with Mrc1 and identified a replicative helicase, Mcm6, as a specific Mrc1-binding protein. The central portion of Mrc1, containing a conserved coiled-coil region, was found to be essential for interaction with the 168-amino-acid C-terminal region of Mcm6, and introduction of two amino acid substitutions in this C-terminal region abolished the interaction with Mrc1 in vivo. An mcm6 mutant bearing these substitutions showed a severe defect in DNA replication checkpoint activation in response to stress caused by methyl methanesulfonate. Interestingly, the mutant did not show any defect in DNA replication checkpoint activation in response to hydroxyurea treatment. The phenotype of the mcm6 mutant was suppressed when the mutant protein was physically fused with Mrc1. These results strongly suggest for the first time that an Mcm helicase acts as a checkpoint sensor for methyl methanesulfonate-induced DNA damage through direct binding to the replication checkpoint mediator Mrc1.
Progression of the DNA replication machinery along chromosomes is a complex process. Replication forks pause occasionally when they encounter genomic regions that are difficult to replicate, such as highly transcribed regions, tRNA genes, and regions with specialized chromatin structure, like centromeric and heterochromatic regions (17). Replication forks also stall when treated with chemicals like methyl methanesulfonate (MMS), which causes DNA damage, or hydroxyurea (HU), which limits the cellular concentration of the deoxynucleoside triphosphate pool (17). Because de novo assembly and programming of the replisome do not occur after the onset of S phase (18), DNA replication forks must be protected from replicative stresses. The DNA replication checkpoint constitutes a surveillance mechanism for S-phase progression that safeguards replication forks from various replicative stresses (22, 38, 40), and malfunction of this checkpoint leads to chromosome instability and cancer development in higher organisms (4, 9).
The Saccharomyces cerevisiae DNA replication checkpoint mediator Mrc1 is functionally conserved and is involved directly in DNA replication as a component of the replisome (1, 8, 16, 19, 29, 30). Mrc1, together with Tof1 and Csm3, is required for forming a replication pausing complex when the fork is exposed to replicative stress by HU (16). The pausing complex subsequently triggers events leading to DNA replication checkpoint activation and hence stable replicative arrest. A sensor kinase complex, Mec1-Ddc2 (ATR-ATRIP homolog of higher eukaryotes), is then recruited to the complex (14, 16). Mec1-Ddc2-mediated phosphorylation of Mrc1 activates the pausing complex, and phosphorylated Mrc1 likely recruits Rad53 (a putative homolog of CHK2 of higher eukaryotes), which is then activated via phosphorylation by Mec1-Ddc2 (1, 16, 20, 30). Activated Rad53 subsequently elicits a stress responses, i.e., stabilization of replication forks, induction of repair genes, and suppression of late-firing origins (24). It remains unclear, however, whether DNA replication checkpoint activation is induced in response to DNA damage by MMS, a reagent commonly used to study the DNA replication stress response. Several lines of evidence have suggested that MMS-induced damage is also sensed directly by the replication machinery (38, 40).
Although biochemical and genetic interaction data have placed Mrc1 at the center of the replication checkpoint signal transduction cascade, its molecular function remains largely unknown. The proteins Mrc1, Tof1, and Csm3 associate with the Mcm complex (8, 27), a heterohexameric DNA helicase consisting of Mcm2 to Mcm7 proteins which unwinds the parental DNA duplex to allow replisome progression (3, 12, 18, 31, 32, 35). The Mcm complex associates with a specific set of regulatory proteins at forks to form replisome progression complexes (8). In addition to Mcm, Tof1, Csm3, and Mrc1, replisome progression complexes include factors such as Cdc45 and the GINS complex that are also required for fork progression (13, 26, 31, 32, 39). Claspin, a putative Xenopus laevis homolog of Mrc1, is also reported to associate with Cdc45, DNA polymerase ɛ (Polɛ), replication protein A, and two of the replication factor C complexes in aphidicolin-treated Xenopus egg extracts (19). Recently, Mrc1 was reported to interact directly with Polɛ (23).
The aim of this study was to provide mechanistic insight into Mrc1 function in the DNA replication checkpoint. For this purpose, it was essential to identify, among all the essential proteins in the replication machinery, a specific protein that interacts with Mrc1 and to examine the role of this interaction in the DNA replication checkpoint. We found that Mrc1 interacts with Mcm6 directly and specifically. When the interaction between Mrc1 and Mcm6 was impaired, cells no longer activated the DNA replication checkpoint in response to MMS-induced replicative stress. Interestingly and unexpectedly, this interaction was not required for DNA replication checkpoint activation in response to HU-induced replicative stress. Our results provide the first mechanistic evidence that cells use separate mechanisms to transmit replicative stresses caused by MMS and HU for DNA replication checkpoint activation.
MATERIALS AND METHODS
Saccharomyces cerevisiae strains and plasmids.
Strains and primers used for cloning, tagging, and mutagenesis in this study are listed in Tables 1 and 2, respectively. All strains constructed in this study were derived from strain BY4741. Construction of C-terminally tagged strains and mutants with deletions of Mrc1, Tof1, and Csm3 was performed as described previously (16). All tags were integrated at the 3′ end of the gene by homologous recombination at their original chromosomal loci (21). Strains expressing tagged proteins were checked for doubling times and HU and MMS sensitivities to ensure that the tagged genes behaved like their untagged counterparts. Mrc1 deletion mutants were constructed as described previously (23).
TABLE 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| BY4741 | MATahis3Δ1 leu2Δ0 met5Δ0 ura3Δ0 | Invitrogen |
| MKY0027 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mrc1Δ::LEU2 | 16 |
| MKY0029 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::LEU2 | 16 |
| MKY0120 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MRC1-6His-3FLAG-loxP-KanMX-loxP | 16 |
| MKY0122 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-mrc1(140-1096) KanMX::MRC1-FLAG | 23 |
| MKY0124 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-mrc1(312-1096) KanMX::MRC1-FLAG | 23 |
| MKY0126 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-mrc1(567-1096) KanMX::MRC1-FLAG | 23 |
| MKY0128 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-mrc1(752-1096) KanMX::MRC1-FLAG | 23 |
| MKY0188 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TOF1-6His-3FLAG-loxP-KanMX-loxP | This study |
| MKY0192 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP-PGAL1-MRC1-6His-3FLAG-loxP-KanMX-loxP | This study |
| MKY0196 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 csm3-6His-3FLAG-loxP-KanMX-loxP | This study |
| MKY0239 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 CDC45-3HA::TRP1 | 16 |
| MKY0688 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-MRC1 KanMX::mrc1(1-843)-FLAG | 23 |
| MKY0670 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-MRC1 KanMX::mrc1(1-655)-FLAG | 23 |
| MKY0672 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-MRC1 KanMX::mrc1(1-418)-FLAG | 23 |
| MKY0674 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TRP::PGAL1-MRC1 KanMX::mrc1(1-219)-FLAG | 23 |
| MKY1063 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::ura3 mcm6 IL973AA::LEU2 | This study |
| MKY1074 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MRC1-6His-3FLAG-loxP-KanMX-loxP mcm6 IL973AA::LEU2 | This study |
| MKY1076 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TOF1-6His-3FLAG-loxP-KanMX-loxP mcm6 IL973AA::LEU2 | This study |
| MKY1078 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 csm3-6His-3FLAG-loxP-KanMX-loxP mcm6 IL973AA::LEU2 | This study |
| MKY1098 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 CDC45-3HA::TRP1 mcm6 IL973AA::LEU2 | This study |
| MKY1225 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::ura3 mcm6 IL973AA::LEU2 | This study |
| MKY1227 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::LEU2 MCM6-MRC1::URA3 | This study |
| MKY1229 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::LEU2 mcm6 IL973AA-MRC1::URA3 | This study |
| MKY1231 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MCM6WT::URA3 | This study |
| MKY1233 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad9Δ::LEU2 MCM6WT::URA3 | This study |
TABLE 2.
Primers used
| Primer | Application | Sequence |
|---|---|---|
| MRC1 FLAG F | Tagging | TATAATATTTGTCTTATAAAATCCTATCATAACATGACTATGGCTTGGCCTAGACTCGGGTGCCATCTTTTTTAATGCGACTACTTCAAGACACTTCTGACTATAGGGAGACCGGCAGATC |
| MRC1 FLAG R | Tagging | CTATGGCTTGGCCTAGACTCGGGTGCCATCTTTTTTAATGCGACTACTTCAAGACAGCTTCTGGAGTTCAATCAACTTCTTCGGAAAAGATAAAAAACCAACTATAGGGAGACCGGCAGATC |
| GAL MRC1 F4 | GAL promoter | TAGCATTTCAAACACATTATGTTGGAAAAAAACCAAGAACAGACAAACAACTAAGGAAGTTCGTTATTCGCTTTTGAACTTATCACCAAATATTTTAGTGGAATTCGAGCTCGTTTAAAC |
| GAL MRC1 R2 | GAL promoter | TATTATCGTTCTCATCTAGTATGGGAACAGCAACTTTTTTGTAGGTGGTAGTTCTCTTCTTTGCAGTCAACGAGGACAAAGCATGCAAGGCATCATCCATCATTTTGAGATCCGGGTTTT |
| MCM6 F | pEG(KT)-MCM6 | GGTGGTGGTGGTTCTAGACATGTCATCCCCTTTTCCAGCTGACACACCAAGC |
| MCM6(1-969) R | pEG(KT)-MCM6(1-969) | GGTGGTGGTGGTGTCGACTTAGACCAACCTTTTTATGACTTTGAACGCTAATCTTCTC |
| MCM6(1-917) R | pEG(KT)-MCM6(1-917) | GGTGGTGGTGGTGTCGACTTAAACGATCATGTTCATCATGGACACATATTTATCATACG |
| MCM6(1-813) R | pEG(KT)-MCM6(1-813) | GGTGGTGGTGGTGTCGACTTAAGCCCTCGCAATAGCTTCTGATAATCTAATCATAC |
| MCM6(1-704) R | pEG(KT)-MCM6(1-704) | GGTGGTGGTGGTGTCGACTTACGGTGCGGTCATATTTAAATTACCCCTCAGTGATAG |
| MCM6(1-1010) R | pEG(KT)-MCM6(1-1010) | GGTGGTGGTGGTGTCGACTTATTGATCCAAAACCTCACAGTTTGGATG |
| MCM6(1-980) R | pEG(KT)-MCM6(1-980) | GGTGGTGGTGGTGTCGACTTAGGTACCATGAATCTCCATTAAAATCC |
| MCM6(969-1017) F | pEG(KT)-MCM6(969-1017) | GGTGGTGGTGGTTCTAGACATGGTCAAAGATAGGATTTTAATGGAGATTC |
| MCM6(850-1017) F | pEG(KT)-MCM6(850-1017) | GGTGGTGGTGGTTCTAGACATGGATAACATAGAGAGTCAAAGTCACGCC |
| MCM6(836-1017) F | pEG(KT)-MCM6(836-1017) | GGTGGTGGTGGTTCTAGACATGCGTGGTGATGTGGATGATGTGGAAATG |
| MCM6 R | pEG(KT)-MCM6 | GGTGGTGGTGGTGTCGACTTAGCTGGAATCCTGTGGTTCCAATTG |
| MCM6IL F | Amino acid substitution | AAAGGTTGGTCAAAGATAGGGCTGCAATGGAGATTCATGGTACCAGAC |
| MCM6IL R | Amino acid substitution | GTCTGGTACCATGAATCTCCATTGCAGCCCTATCTTTGACCAACCTTT |
| MCM6-MRC1 fusion F | YIplac fusion | GGTGGTGGTGGTGGATCCGATAACATAGAGAGTCAAAGTCACGCCGC |
| MCM6-MRC1 fusion R | YIplac fusion | GGTGGTGGTGGTGTCGACGCTGGAATCCTGTGGTTCCAATTGATCC |
| mrc1 d1 | Deletion | TAGCATTTCAAACACATTATGTTGGAAAAAAACCAAGAACAGACAAACAACTAAGGAAGTTCGTTATTCGCTTTTGAACTTATCACCAAATATTTTAGTGCCAACACCCGCTGACGCGCC |
| mrc1 d2 | Deletion | CTATGGCTTGGCCTAGACTCGGGTGCCATCTTTTTTAATGCGACTACTTCAAGACAGCTTCTGGAGTTCAATCAACTTCTTCGGAAAAGATAAAAAACCAACATTTCCCCGAAAAGTGCC |
| rad9 d1 | Deletion | TTTGTTCGTGGATATTTGCAACGATGAGCAATGTGAAGTGAGCAAGATAGAGAAACGCCATAGAAAAGAGCATAGTGAGAAAATCTTCAACATCAGGGCTCCAACACCCGCTGACGCGCC |
| rad9 d2 | Deletion | CGTGTGGGAGGATGTTCTTAGACTTAATTAAGAATCTCTAAATTTTTTTTTATTTAATCGTCCCTTTCTATCAATTATGAGTTTATATATTTTTATAATTACATTTCCCCGAAAAGTGCC |
Manipulations and growth of Saccharomyces cerevisiae were based on standard procedures. Strains carrying combinations of multiple mutant alleles were generated by genetic crosses. Cells were grown in YP (yeast extract-peptone) medium supplemented with either 2% dextrose, 2% raffinose, or 2% galactose.
Cell synchronization.
Strains were arrested in G1 phase by supplementing the media of exponential cultures with 10 μg/ml α-factor. Synchronized cells were released from G1 into fresh YPD (YP plus dextrose) to grow for 40 min when all cells showed S-phase DNA content by flow cytometry. Flow cytometry was carried out as described previously (11).
Antibodies.
Antibodies against Mcm2 and Rad53 were purchased from Abcam (Cambridge, United Kingdom). The Mcm6 antibody was a custom antibody raised against two peptides of yeast Mcm6 (GLDSQIGSRLHFPS and EIHGTRHNLRDLENE).
The antibody against Dpb2 was raised against full-length yeast Dpb2. The DNA fragment spanning the entire DPB2 open reading frame (ORF) was cloned into pET20b(+) and used for expression in Escherichia coli. Dpb2, which was insoluble after cell disruption, was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), eluted from the gel, and used to immunize rabbits.
IP assays.
Coimmunoprecipitation (co-IP) analysis of wild-type or mutant Mcm6IL was performed using strains expressing FLAG-tagged versions of Mrc1, Tof1, or Csm3. Samples (2.0 × 109 cells) were collected by centrifugation, and extract was prepared as described previously (16). The cells were crushed by glass beads in 400 μl lysis buffer (50 mM HEPES-KOH, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.25% [wt/vol] NP-40) containing protease inhibitors (1% protease inhibitor cocktail [Sigma] and 1 mM phenylmethylsulfonyl fluoride) and cleared by centrifugation to yield whole-cell extract. Whole-cell extract total protein (6 mg) was incubated with 6 μg of antibody, as indicated in the figures, for 3 h at 4°C. The anti-FLAG antibody was from Sigma-Aldrich. Precipitation was carried out with prewashed protein A beads (GE Healthcare) for 3 h at 4°C. To degrade chromosomal DNA in cell extracts, DNase I (Takara) was added to a final concentration of 200 U/ml and incubated for 1 h at 4°C before IP. DNA degradation was confirmed by agarose gel electrophoresis and ethidium bromide staining.
Antibody-bound fractions were suspended in a minimal amount of SDS loading buffer (93.75 mM Tris-HCl, pH 6.8, 3% sodium lauryl sulfate, 15% glycerol, 5% 2-mercaptoethanol), and half the sample was analyzed by SDS-PAGE using an enhanced chemiluminescence detection reagent (Amersham). Whole-cell extracts corresponding to approximately 100 μg total protein were also analyzed.
RESULTS
Specific interaction between Mrc1 and Mcm6.
Mrc1 is a component of the biochemically defined replisome progression complex that functions during S phase (8), and Mrc1 also reportedly forms a complex with individual Mcm proteins (27). We therefore searched for interacting partners of Mrc1 among the six Mcm proteins. To facilitate detection, we overexpressed a glutathione S-transferase (GST)-tagged version of each component of Mcm together with FLAG epitope-tagged Mrc1 under the control of the GAL1 promoter (21) and examined their ability to interact with each other by co-IP (Fig. 1A). Mrc1 interacted with Mcm6 when Mrc1 was used as either bait or target reciprocally (Fig. 1A, lanes 11 and 17), but Mrc1 did not interact specifically with any other component of the Mcm complex. Although we detected an interaction between Mcm7 and Mrc1 only when Mrc1 was used as the co-IP bait (Fig. 1A, lane 18), Mcm7 was also immunoprecipitated by an antibody against FLAG (anti-FLAG) from the control extract of an untagged Mrc1 strain (Fig. 1C, lane 14), suggesting that Mcm7 IP was not the result of specific interaction with Mrc1. These findings suggested that Mrc1 associates with the Mcm complex through direct interaction with Mcm6.
FIG. 1.

Interaction of Mrc1 with subunits of the Mcm complex. (A) Interaction of the full-length Mrc1 with various Mcm subunits. Cells were prepared that overexpressed Mrc1-3×FLAG along with an individual GST-tagged Mcm subunit simultaneously. For this purpose, the promoter of chromosomal MRC1 was replaced by the GAL1 promoter, and each subunit of the Mcm helicase complex was expressed using a single-copy plasmid (25) and a GAL1-promoter plasmid, pEG (10). Whole-cell extracts (WCE) were prepared from asynchronous cells as described in Materials and Methods. IP using anti-FLAG and anti-GST was carried out, respectively, and proteins in the WCE and the IPs were identified by SDS-PAGE and Western blotting. Lanes 1 to 6, overexpression of each protein was confirmed by SDS-PAGE of WCE; lanes 7 to 12, IP by anti-GST; lanes 13 to 18, IP by anti-FLAG. Blots of IPs were probed with anti-GST in the upper blot and with anti-FLAG in the lower blot on a separate gel. (B) Interaction of a central region of Mrc1 (residues 312 to 655, including the first coiled-coil region) with Mcm. N-terminal and C-terminal regions of chromosomal GAL1-MRC1 used in panel A were deleted as described in Materials and Methods. Each subunit of the Mcm helicase complex was overexpressed from a single-copy plasmid as for panel A. Lanes 1 to 6, overexpression of each protein was confirmed by Western blotting of WCE; lanes 7 to 12, IP by anti-GST; lanes 13 to 18, IP by anti-FLAG. IP was carried out as described for panel A. (C) Control experiments to show nonspecific recognition of GST-tagged MCM proteins by anti-FLAG. WCE were prepared from asynchronous cells expressing an individual GST-tagged Mcm subunit as described in Materials and Methods. IP of WCE was carried out with anti-FLAG or anti-GST, and proteins in WCE and IPs were identified by Western blotting. Lanes 1 to 7, confirmation of overexpression of each Mcm protein; lanes 8 to 14, IP by anti-FLAG; lanes 15 to 21, IP by anti-GST. Blots of IPs were probed with anti-GST (upper blots) or anti-FLAG (lower blots, from a separate gel). (D) Cells co-overexpressed each FLAG-tagged wild-type (WT) and Mrc1 deletion mutant and GST-Mcm6 from GAL1 promoters. WCE were prepared from asynchronous cells as described in Materials and Methods. IP with anti-FLAG was carried out, and proteins in the WCE and IPs were identified by Western blotting. Lanes 1 to 5 and 11 to 15, confirmation of overexpression of each protein; lanes 6 to 10 and 16 to 20, IP by anti-FLAG. Blots of IPs were probed with anti-GST (upper blots) or anti-FLAG (lower blots, from a separate gel). Deletion mutants of Mrc1 are referred to in the figure and in the text by numbers corresponding to the amino acids contained in the fragments expressed in the mrc1 mutants.
We narrowed the possible interacting regions within Mrc1 by using a set of Mrc1 deletion mutants (23). Each Mrc1 deletion mutant had a FLAG tag and was overexpressed with GST-Mcm6, and potential interactions were assessed by co-IP (Fig. 1B). Mcm6 coimmunoprecipitated with a 655-amino-acid N-terminal peptide fragment of Mrc1 (Fig. 1D, lane 8) and with a 784-residue C-terminal fragment (residues 312 to 1096) of Mrc1 (Fig. 1D, lane 18), suggesting that the region required for interaction with Mcm6 resides in the central 344-residue fragment of Mrc1 (residues 312 to 655). We further tested this possibility by overexpressing this 344-residue Mrc1 peptide together with individual Mcm proteins and examining their ability to interact with each other by co-IP (Fig. 1B). Indeed, Mrc1(312-655), when used as bait or target, interacted with Mcm6 (Fig. 1B, lanes 11 and 17). These data indicated that Mrc1(312-655), which includes the first coiled-coil region, was sufficient for interaction with full-length Mcm6. We also found that Mrc1(312-655), used as target or bait, coimmunoprecipitated with Mcm4 or Mcm3, respectively (Fig. 1B, lanes 9 and 14), suggesting that Mrc1 may also interact with these subunits in a manner distinct from that of Mcm6.
A C-terminal region of Mcm6 is responsible for interaction with Mrc1.
To determine the region of Mcm6 that interacts with Mrc1, we generated a set of Mcm6 C-terminal deletion mutants (Fig. 2). Initially, we found that the C-terminal 168-residue (850 to 1017) fragment was sufficient for the interaction with Mrc1 (Fig. 2A, lanes 4 and 5), and finally the interacting region was narrowed to less than 11 residues (Fig. 2B, lanes 8 and 9). We compared amino acid sequences of Mcm2, -4, -5, -6, and -7 and found that Mcm6 has a unique C-terminal extension containing an AAA+ ATPase motif (28) after the conserved MCM motif, and this region is the one responsible for the interaction with Mrc1 (Fig. 3A). Within the 11-residue sequence, we mutated two hydrophobic residues (isoleucine 973 and leucine 974, Fig. 3B) to the smaller amino acid alanine; we called this mutant Mcm6IL. Mcm6IL did not interact with Mrc1 (Fig. 3C, lanes 4 and 6), suggesting that at a minimum these two residues are involved in the interaction with Mrc1.
FIG. 2.

Interaction of Mrc1 with C-terminal deletion mutants of Mcm6. (A) The C-terminal 168 residues of Mcm6 are sufficient for interaction with Mrc1. Anti-FLAG IP was carried out on strains co-overexpressing Mrc1-3×FLAG and C-terminal regions of Mcm6-GST, respectively. The same construct as that in Fig. 1 was used for Mrc1 overexpression. Each C-terminal region of Mcm6 was overexpressed from pEG as for Fig. 1. Lanes 1 to 3, overexpression of each protein was confirmed by Western blotting of whole-cell extracts (WCE); lanes 4 to 6, IP by anti-FLAG. Blots of IPs were probed with anti-GST in the upper blot and with anti-FLAG in the lower blot on a separate gel. WCE were prepared, IP was carried out with the antibody indicated, and proteins in the WCE and the IP were identified by Western blotting. C-terminal regions of Mcm6 are referred to in the figure and in the text by numbers corresponding to the amino acids contained in the fragments expressed in the mcm mutants. (B) The sequence 969 to 980 in Mcm6 is sufficient for interaction with Mrc1. The indicated C-terminal mcm6 deletion mutants were tagged with GST, co-overexpressed with Mrc1, and analyzed for their ability to interact with Mrc1 as described for panel A.
FIG. 3.

Interaction between Mrc1 and a mutant Mcm6. (A) Schematic representation of Mcm2, -4, -5, -6, and -7. Gray shading indicates the conserved MCM motif. Mcm6 contains a unique C-terminal region (hatched shading). a.a., amino acids. (B) The peptide sequence around two substituted amino acid residues (I973L974 to A973A974). (C) Interaction between Mrc1 and the mutant Mcm6 overexpressed in vivo. The anti-FLAG IP was carried out on strains co-overexpressing Mrc1-3×FLAG and GST-Mcm6IL. The same construct as that in Fig. 1 was used for Mrc1 overexpression. Two amino acid substitutions were introduced into Mcm6. The mutant form of Mcm6 (Mcm6IL) was overexpressed from pEG as in Fig. 1. Lanes 1 and 2, overexpression of each protein was confirmed by Western blotting of whole-cell extracts (WCE); lanes 3 and 4, IP by anti-FLAG; lanes 5 and 6, IP by anti-GST. Blots of IPs were probed with anti-GST in the upper blot and with anti-FLAG in the lower blot on a separate gel. (D) Cell cycle progression as determined by DNA content in wild-type (WT) and various mutant strains. Cells were synchronized in G1 by α-factor and then released into YPD medium at 23°C. At the indicated times, samples were taken for fluorescence-activated cell sorting analysis. Positions of 1C and 2C cells are shown. Asyn., asynchronous. (E) Interaction of Mrc1 with the Mcm complex containing the mutant Mcm6 endogenously expressed from the mutated chromosomal gene. The two amino acid substitutions (I973L974 to A973A974) were introduced into chromosomal MCM6 as described in Materials and Methods. Chromosomal MRC1 was tagged with the 3×FLAG epitope. Cells were arrested by α-factor in G1 phase and then released into S phase for 40 min at 23°C, and WCE were prepared. IP was carried out with anti-FLAG. Western blot assays of the precipitates using antibodies against FLAG, Mcm2, Mcm6, and Dpb2 are shown. WT, wild type. (F) Interaction of Tof1 and Csm3 with Mcm proteins in WT and mcm6IL mutant strains. The anti-FLAG IP was carried out with WT and mcm6IL mutant strains expressing Tof1-3×FLAG or Csm3-3×FLAG. 3×FLAG tag was fused to the 3′ end of the chromosomal gene encoding Tof1 or Csm3. Lanes 1 to 4, expression of each protein was confirmed by Western blotting of WCE; lanes 5 to 8, IP by anti-FLAG. Western blotting of the precipitates using anti-Mcm2 and anti-Mcm6.
Amino acid substitutions encoded by mutations in MCM6 abolish the Mcm6-Mrc1 interaction in vivo.
We further investigated the importance of the putative Mrc1 interaction site in Mcm6 by mutating the cellular MCM6 gene to produce the mutant protein Mcm6IL in vivo. We fused 3×FLAG to Mrc1, expressed this 3×FLAG-Mrc1 in cells expressing wild-type Mcm6 or Mcm6IL, and then assessed the interaction between 3×FLAG-Mrc1 and Mcm6 or endogenous Mcm2. The cells were synchronized at G1 phase and then released into S phase for 40 min at 23°C. We confirmed that cells were in S phase by flow cytometric analysis of the DNA content (Fig. 3D), and whole-cell extracts were prepared. Mrc1 did not interact with Mcm6IL during S phase (Fig. 3E). Interaction of Mrc1 with another subunit of the Mcm complex, Mcm2, was hardly detectable in Mcm6IL mutant extracts, suggesting that the Mrc1 interaction with the Mcm complex as a whole depends on its interaction with the Mcm6 subunit. A recent report showed that the Polɛ complex interacts with the N- and C-terminal regions of Mrc1 (23). Thus, as a positive control for 3×FLAG-Mrc1 function, we verified whether this interaction was retained in Mcm6IL mutant extracts and found that a subunit of the Polɛ complex, Dpb2, indeed bound to 3×FLAG-Mrc1 (Fig. 3D), suggesting that the interaction between Polɛ and Mrc1 remained intact.
On the other hand, interactions of the Mcm complex with other checkpoint mediator proteins, Tof1 and Csm3, were not affected in mcm6IL mutant extracts (Fig. 3F). These data suggested that Mcm6IL specifically destabilizes the Mcm helicase complex interaction with Mrc1 but not with Tof1 or Csm3. Interestingly, cells expressing Mcm6IL showed no obvious defects in cell growth under normal conditions.
The mcm6IL mutant activates the DNA replication checkpoint in response to HU.
We next investigated the role of Mrc1-Mcm6 interaction in the DNA replication checkpoint. First, we examined cell viability under replicative stress induced by HU. Cells were synchronized in G1 phase and then released into S phase in the presence of 200 mM HU. The protein Rad9 is a specific mediator of the DNA damage checkpoint, and it can partly back up the function of Mrc1 when Mrc1 is unavailable (7, 16); thus, we prepared a double mutant of the mcm6IL mutant and a rad9 deletion mutant (rad9Δ). Unexpectedly, mcm6IL cells were not sensitive to 200 mM HU after 4 h of treatment in the absence of functional Rad9 (Fig. 4A). mcm6IL cells carrying an mrc1 deletion (mrc1Δ) were no more sensitive to HU than were mrc1Δ cells alone (survival rate, 60% [Fig. 4B]). The sensitivity of the mrc1Δ mutant to HU differed from that reported previously (1) (survival rate, ∼30%). This is probably due to the rad5-G535R missense mutation in the W303 strain (6) used in the previous study (1). Rad5 is a DNA helicase proposed to function in double-strand break repair and postreplication repair (2, 41), and therefore, mutation in rad5 may affect the strain's sensitivity to genotoxic stresses.
FIG. 4.
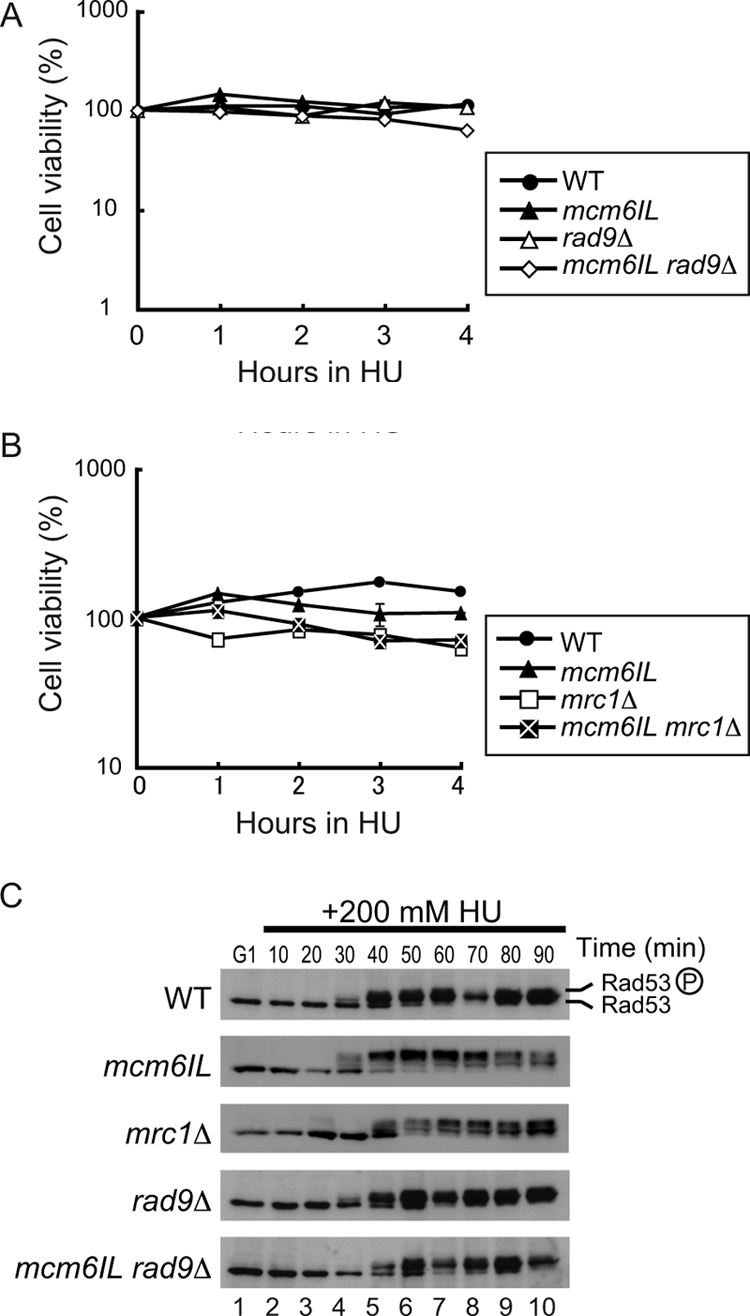
Mcm6IL can activate the DNA replication checkpoint normally in response to HU. (A) Effect of the mcm6IL, rad9Δ, and mcm6IL rad9Δ mutations on the viability of HU-treated cells. Cells were arrested by α-factor in G1 phase and then released into S phase in the presence of 200 mM HU. Cell viability was measured by plating on a YPD agar plate at the indicated times after HU addition. WT, wild type. (B) Effect of the mcm6IL, mrc1Δ, and mcm6IL mrc1Δ mutations on the viability of HU-treated cells. The procedure was the same as that in panel A. (C) Rad53 phosphorylation was induced normally in response to HU-induced replicative stress in the mcm6IL rad9Δ double mutant. Cells were arrested in G1 phase by α-factor and then released into S phase at 23°C in the presence of 200 mM HU. The samples were withdrawn at the indicated times, and cell extracts were prepared. Rad53 was detected by Western blotting using anti-Rad53. Phosphorylated Rad53 is indicated by a circled P.
Activation of Rad53, a checkpoint effector kinase that is phosphorylated in response to HU-induced replicative stress, took place even in mcm6IL and rad9 double mutant cells as effectively as in the wild-type cells (Fig. 4C). Full activation of Rad53 in mrc1Δ cells was slightly delayed compared to that in other strains, as previously described (1). These data suggested that the mcm6IL mutant responded quite normally to HU-induced replicative stress. To examine the effect of the mcm6IL mutation on Mrc1 localization at replication forks, we carried out chromatin IP on chip (ChIP-chip) analyses of Mrc1 and Cdc45. Because the mcm6IL mutant was insensitive to HU, we used HU to obtain good synchronization of the replication forks at or near replication initiation sites (Fig. 5A and B). Cells were arrested by α-factor in G1 phase and then released into S phase in the presence of 200 mM HU to slow the progression of DNA replication. ChIP using anti-FLAG (for Mrc1 to 3×FLAG) or anti-HA (for Cdc45-3×HA) was performed for the wild-type and mcm6IL mutant strains, respectively. Cdc45 binding profiles were analyzed to monitor the positions of replication forks. Replication fork pausing occurred normally in the mcm6IL mutant, and there were no differences in the binding profiles of Cdc45 between wild-type and mcm6IL mutant cells (Fig. 5A). There was almost no difference in the amounts of Mrc1 localized at replication origins between the wild type and the mcm6IL mutant, confirming that the mcm6IL mutant has specific defects in its ability to interact with Mrc1 (Fig. 5B and C). To exclude the possibility that these observations stemmed from activation of the checkpoint response by HU, we analyzed the distribution of Cdc45 and Mrc1 in the absence of HU early during DNA replication. Cells were arrested by α-factor in G1 phase and released into S phase for 45 min at 16°C to allow slow progression of S phase, as described previously (16). Both Cdc45 and Mrc1 localized to early replicating origins, and there were no differences in localization patterns of Cdc45 and Mrc1 between the wild-type and mcm6IL mutant strains (Fig. 5D and E).
FIG. 5.

Replication fork arrest and Mrc1 loading is normal in the mcm6IL mutant under HU-induced replicative stress. (A) Distribution of Cdc45 on chromosome VI in the wild-type (WT) strain and mcm6IL mutant strain in the presence of 200 mM HU. Cells were arrested in G1 phase by α-factor and then released into S phase at 23°C in the presence of 200 mM HU for 60 min. ChIP analysis was carried out as described previously (1). Each blue bar represents the binding ratio of Cdc45 at each locus and shows a significant enrichment in some of the IP fractions. The yellow line represents the average signal ratio at loci that are not enriched (gray bars) in each IP fraction. The scale of the vertical axis is log2. Positions of the centromere (CEN6) and all previously mapped autonomous replicating sequences (ARSs) are shown. The horizontal axis indicates the length of chromosome VI, in kilobases. (B) Distribution of Mrc1 on chromosome VI in the WT strain and the mcm6IL mutant strain in the presence of 200 mM HU. Cells were prepared as described for panel A. (C) Efficiency of ChIP (ratio of input versus IP) of the Mrc1-3×FLAG-chromatin complex as measured by quantitative PCR using primer pairs that specifically amplify regions indicated by arrows in panel B. Each result is the average ± standard deviation of two independent experiments. 259K, in kilobases, indicates positions of PCR primers measured from the left end of the chromosome. Mock control experiments were done following the same protocol as that for the ChIP-chip experiments described above but in the absence of the FLAG-tagged Mrc1. (D and E) Distribution of Cdc45 (D) and Mrc1 (E) on chromosome VI in WT and mcm6IL cells in early S phase without replicative stress. Cells were arrested in G1 phase by α-factor and then released into S phase at 16°C for 45 min. ChIP-chip analyses were performed as described for panel A.
The mcm6IL mutant has a specific defect in the activation of a checkpoint signal transduction cascade in response to MMS.
Because the mcm6 mutation did not affect cell sensitivity to HU stress, we performed experiments as described above using MMS, whose stress mechanism differs from that of HU. The rad9 single deletion mutant was slightly more sensitive to MMS than was the wild type (Fig. 5A). The mcm6IL rad9Δ double mutant was highly sensitive to MMS compared to the mcm6IL or rad9Δ single mutant. In contrast, the mcm6IL mrc1Δ double mutant was not more sensitive than the mrc1Δ single mutant (Fig. 6A). We could not test the mrc1Δ rad9Δ double deletion mutant because it was lethal (1).
FIG. 6.

The mcm6IL mutant cannot activate the DNA replication checkpoint in response to MMS in the absence of rad9. (A) Effect of the mrc1Δ and mcm6IL mrc1Δ mutations on the viability of MMS-treated cells. Cells were arrested by α-factor in G1 phase and then released into S phase in the presence of 0.03% MMS. Cell viability was measured by plating on YPD at the indicated times after MMS addition. WT, wild type. (B) Rad53 is not phosphorylated in response to MMS-induced replicative stress in the mcm6IL rad9Δ double mutant. Cells were arrested in G1 phase by α-factor and then released into S phase at 23°C in the presence of 0.03% MMS. Samples were withdrawn at the indicated times, and cell extracts were prepared. Rad53 was detected by Western blotting with anti-Rad53. Phosphorylated Rad53 is indicated by a circled P.
MMS slows down S-phase progression through activation of the DNA replication checkpoint cascade (33, 34), and thus we studied the effects of MMS in this regard. For all strains used in this study, more than 70% of cells completed S phase by 50 min after release from G1 arrest at 23°C (Fig. 3D). As expected, MMS blocked cell cycle progression during S phase in wild-type cells (Fig. 7B), and the progression was partially impaired in mrc1Δ or rad9Δ single mutants, as reported elsewhere (1, 33) (Fig. 7B). S-phase progression also was partially slowed by MMS in the mcm6IL mutant compared to that in the wild type. In contrast, S-phase progression was not blocked by MMS in the mcm6IL rad9Δ double mutant. In good agreement with these results, MMS did not activate Rad53 in mcm6IL rad9Δ cells (Fig. 6B and 7B).
FIG. 7.

Effect of the mcm6IL mutation on the DNA replication checkpoint in response to MMS. (A) Effect of the mcm6IL, rad9Δ, mcm6IL rad9Δ, and mcm6IL-MRC1 rad9Δ mutations on the viability of MMS-treated cells. Cells were arrested by α-factor in G1 phase and then released into S phase in the presence of 0.03% MMS. Cell viability was measured by plating on YPD at the indicated times after MMS addition. WT, wild type. (B) Effects of various mutations on cell cycle progression of MMS-treated cells. Cells were synchronized in G1 by α-factor and then released into S phase in the presence of 0.03% MMS at 23°C. At the indicated times, samples were taken for fluorescence-activated cell sorting analysis. Positions of 1C and 2C cells are shown. Asyn., asynchronous. (C) Rad53 phosphorylation in response to MMS-induced replicative stress in various mutants. Cells were arrested in G1 phase by α-factor and then released into S phase at 23°C in the presence of 0.03% MMS. Samples were withdrawn at indicated times, and cell extracts were prepared. Rad53 was detected by Western blotting using anti-Rad53. Phosphorylated Rad53 is indicated by a circled P. (D) Identification of the fusion protein in vivo. Cells carrying various mutations, as indicated, were grown to prepare cell lysates. The lysates were fractionated by SDS-PAGE, and Mcm6 was identified by Western blotting using anti-Mcm6. The asterisk indicates a nonspecific band recognized by anti-Mcm6. (E) Anaphase progression delay, DNA replication fork stabilization, and suppression of late-firing origins are defective in the mcm6IL rad9Δ double mutant in response to MMS-induced replicative stress. Suppression of spindle elongation is defective in mcm6IL rad9Δ strains in the presence of MMS. Cells were arrested by α-factor and then released into YPD containing 0.03% MMS at 23°C. Cells were collected at 90 min after release into MMS and fixed by addition of formaldehyde to the culture. Nuclear and microtubule structures were visualized with DAPI (4′,6-diamidino-2-phenylindole) and antitubulin, respectively, as described previously (3). At least 150 cells were examined for each strain. (F) Fork stabilization and suppression of late-firing origins are defective in mcm6IL rad9Δ cells in response to MMS-induced replicative stress. Distribution of Cdc45 on chromosome VI in wild-type, rad9Δ, mcm6IL rad9Δ, and mcm6IL-MRC1 rad9Δ cells in the presence of 0.03% MMS was determined by ChIP-chip experiments. Cells were arrested in G1 phase by α-factor and then released into S phase at 23°C in the presence of 0.03% MMS for 30 min. ChIP analysis was carried out as described previously (1). Each blue bar represents the binding ratio of Cdc45 at each locus and shows significant enrichment in some of the IP fractions. The yellow line represents the average signal ratio at loci that are not enriched (gray bars) in each IP fraction. The scale of the vertical axis is log2. Positions of the centromere (CEN6) and all previously mapped autonomous replicating sequences (ARSs) are shown. The horizontal axis indicates the length of chromosome VI in kilobases.
To confirm that the mcm6IL rad9Δ double mutant is defective for the DNA replication checkpoint in response to MMS, we counted the number of cells proceeding through anaphase in the presence of MMS (36). Cells were synchronized with α-factor and released into medium containing 0.03% MMS for 90 min at 23°C. Most of the wild-type, mcm6IL mutant, and mrc1Δ mutant cells were arrested as large budded cells with short spindles (Fig. 7E). In contrast, 12% and 38% of rad9Δ and mcm6IL rad9Δ cells, respectively, exhibited elongated spindles, suggesting that mcm6IL rad9Δ cells are defective for the DNA replication checkpoint in response to MMS. Moreover, to examine the effect of the mcm6IL mutation on progression of replication forks in the presence of MMS, we carried out ChIP-chip analyses of Cdc45-3×HA. Cells were arrested by α-factor in G1 phase and then released into S phase in the presence of 0.03% MMS for 30 min at 23°C. ChIP-chip analyses were performed for the wild-type, rad9Δ single mutant, and mcm6IL rad9Δ double mutant strains (Fig. 7F). In the wild-type and rad9Δ cells, Cdc45 localized around early replicating regions. In contrast, very little Cdc45 localized to forks in the mcm6IL rad9Δ strain, suggesting that replication forks were unstable in this double mutant. Furthermore, Cdc45 localized to the late-firing origin ARS603 (43) in the mcm6IL rad9Δ strain. Because the activity of the late-firing origin is suppressed in response to DNA replication checkpoint activation by MMS (37), these data suggested that the mcm6IL rad9Δ double mutant is defective for activation of the DNA replication checkpoint in response to MMS and could neither stabilize replication forks nor suppress activity of a late-firing origin. We thus concluded that the mcm6IL mutation causes a defect in the function of the Mcm/Mrc1 complex specifically in the activation of the DNA replication checkpoint in response to MMS.
Expression of an Mcm6ILP-Mrc1P fusion protein completely suppresses the checkpoint defects caused by the mcm6IL mutation.
To confirm that the observed mcm6IL defects on the DNA replication checkpoint were caused by the loss of interaction between Mcm6 and Mrc1, we fused the mcm6IL ORF with the Mrc1 ORF and examined the effects of the expressed fusion protein on cell cycle progression, cell viability, and Rad53 phosphorylation under MMS-induced replicative stress. We replaced the termination codon of mcm6IL with the initiation codon of MRC1 to yield a continuous fusion of the entire ORFs. The fusion protein was expressed in mcm6IL rad9Δ double mutant cells as the sole source of Mcm6. The expression of the fusion construct was confirmed by Western blotting (Fig. 7D). Expression of the fusion protein in mcm6IL rad9Δ cells restored cell viability, cell cycle delay, Rad53 phosphorylation, fork stabilization, suppression of late-firing origins, and suppression of spindle elongation in response to MMS treatment, to the same level as in rad9Δ cells (Fig. 7A, B, C, and E), suggesting that the physical interaction between Mcm6 and Mrc1 is essential for the activation of the DNA replication checkpoint in response to MMS.
DISCUSSION
Here we screened for binding partners of Mrc1 among Mcm helicase subunits and found that Mrc1 bound specifically to Mcm6. We narrowed the interaction site to within the 11-residue C-terminal region and the 344-residue middle region of Mcm6 and Mrc1, respectively. Mutation of two relatively large hydrophobic residues (isoleucine and leucine) within the 11-residue region of Mcm6 to alanine completely abolished the interaction between Mcm6 and Mrc1. These results strongly suggest that association between these two proteins involves a direct hydrophobic residue-mediated interaction between defined domains. We generated a new mutant allele of MCM6, mcm6IL, in the yeast chromosome, which harbored the isoleucine/alanine and leucine/alanine substitutions. Although binding between Mrc1 and the Mcm complex, as represented by Mcm6 and Mcm2, was weakened in mcm6IL cells, the interaction between the Mcm complex and Tof1 or Csm3 was not affected, suggesting that Tof1 and Csm3 interact with subunits or sites other than Mcm6 in the Mcm complex. The mcm6IL mutant alone was insensitive to HU or MMS, but the mcm6IL rad9Δ double mutant showed additive sensitivity to MMS but not HU. From these data, we hypothesized that binding between Mcm6 and Mrc1 is specifically required for activation of the replication checkpoint in response to MMS-induced replicative stress. This hypothesis was strongly supported by our finding that all MMS-related checkpoint defects were suppressed upon expression of an Mcm6IL-Mrc1 fusion protein. In the absence of a replication checkpoint, DNA damage is likely to accrue. The presence of the DNA damage then leads to the engagement of the Rad9-dependent DNA damage checkpoint, resulting in only modest sensitivity to the presence of alkylating DNA damage in S phase despite the absence of the replication checkpoint. Upon removal of both checkpoint pathways, sensitivity is greatly increased, and all of our data are consistent with this interpretation.
Our results provide new insight on the mechanism by which Mrc1 and Mcm6 sense DNA damage during S phase. The loss of interaction between these proteins led to a complete and selective loss of DNA replication checkpoint activation in response to MMS-induced DNA damage, suggesting that the Mrc1-Mcm6 interaction serves as a specific sensor of MMS-induced DNA aberrations in S phase as well as an important mediator between Mec1 signaling and Rad53 activation. Recently, Mrc1 was reported to interact with Polɛ, the catalytic subunit of DNA Polɛ that is essential for leading-strand DNA replication and for checkpoint activation (23). Mrc1 was reported to interact independently with both the N- and C-terminal halves of Polɛ. Interestingly, the region of Mrc1 that we mapped for interaction with Mcm6 is located between the sites of Polɛ interaction. In the mcm6IL mutant, we could still detect interaction between Mrc1 and Dpb2, a subunit of Polɛ, at the same level as that in wild-type cells, suggesting that the Mrc1-Polɛ interaction may be involved mainly in sensing HU-induced replicative stress. Thus, the interaction of Mrc1 with both a helicase and a polymerase is important for proper Mrc1 function to sense different types of replicative stress for the activation of the DNA replication checkpoint and protection of the replisome. Mcm was reported to physically interact with ATRIP and is phosphorylated by ATM and ATR in HeLa cell extracts (5). The Mcm complex may provide a platform for multiple regulatory inputs through interaction with Mrc1 and ATR to regulate DNA replication in higher eukaryotes.
Biochemical studies in vitro suggest that during DNA unwinding Mcm binds to only one strand of a duplex corresponding to the leading strand of a replication fork, and Mcm progression stops at a nick in either strand (15). As a DNA nick is the major intermediate during repair of alkylated DNA (42), the Mrc1-Mcm interaction may function to sense helicases that are arrested in front of nicked DNA. Claspin, a putative homolog of Mrc1 in Xenopus laevis, was reported to participate in the detection of chromosomal double-stranded DNA breaks (44), which may be relevant to our findings. As such, it would be interesting to examine changes in the biochemistry of the Mcm helicase upon binding to Mrc1.
Acknowledgments
We thank Yasuo Kawasaki and Bik Tye for the gift of Mcm-GST plasmids. We thank Takashi Sutani for his suggestions on the construction of the mcm6IL mutant.
K.S. is supported in part by grants-in-aid on priority areas from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). M.K. is a research associate of the Global Center of Excellence Program (GCOE) from MEXT.
Footnotes
Published ahead of print on 20 July 2009.
REFERENCES
- 1.Alcasabas, A. A., A. J. Osborn, J. Bachant, F. Hu, P. J. Werler, K. Bousset, K. Furuya, J. F. Diffley, A. M. Carr, and S. J. Elledge. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3958-965. [DOI] [PubMed] [Google Scholar]
- 2.Blastyák, A., L. Pintér, I. Unk, L. Prakash, S. Prakash, and L. Haracska. 2007. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 28167-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bochman, M. L., and A. Schwacha. 2008. The Mcm2-7 complex has in vitro helicase activity. Mol. Cell 31287-293. [DOI] [PubMed] [Google Scholar]
- 4.Branzei, D., and M. Foiani. 2005. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 17568-575. [DOI] [PubMed] [Google Scholar]
- 5.Cortez, D. 2005. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 191007-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan, H. Y., K. K. Cheng, and H. L. Klein. 1996. Mutations in the RNA polymerase II transcription machinery suppress the hyperrecombination mutant hpr1 delta of Saccharomyces cerevisiae. Genetics 142749-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foss, E. J. 2001. Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 157567-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gambus, A., R. C. Jones, A. Sanchez-Diaz, M. Kanemaki, F. van Deursen, R. D. Edmondson, and K. Labib. 2006. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 8358-366. [DOI] [PubMed] [Google Scholar]
- 9.Harper, J. W., and S. J. Elledge. 2007. The DNA damage response: ten years after. Mol. Cell 28739-745. [DOI] [PubMed] [Google Scholar]
- 10.Homesley, L., M. Lei, Y. Kawasaki, S. Sawyer, T. Christensen, and B. K. Tye. 2000. Mcm10 and the MCM2-7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes Dev. 14913-926. [PMC free article] [PubMed] [Google Scholar]
- 11.Hori, Y., K. Shirahige, C. Obuse, T. Tsurimoto, and H. Yoshikawa. 1996. Characterization of a novel CDC gene (ORC1) partly homologous to CDC6 of Saccharomyces cerevisiae. Mol. Biol. Cell 7409-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishimi, Y. 1997. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 27224508-24513. [DOI] [PubMed] [Google Scholar]
- 13.Kanemaki, M., A. Sanchez-Diaz, A. Gambus, and K. Labib. 2003. Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature 423720-724. [DOI] [PubMed] [Google Scholar]
- 14.Kanoh, Y., K. Tamai, and K. Shirahige. 2006. Different requirements for the association of ATR-ATRIP and 9-1-1 to the stalled replication forks. Gene 37788-95. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan, D. L., M. J. Davey, and M. O'Donnell. 2003. Mcm4, 6, 7 uses a “pump in ring” mechanism to unwind DNA by steric exclusion and actively translocate along a duplex. J. Biol. Chem. 27849171-49182. [DOI] [PubMed] [Google Scholar]
- 16.Katou, Y., Y. Kanoh, M. Bando, H. Noguchi, H. Tanaka, T. Ashikari, K. Sugimoto, and K. Shirahige. 2003. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 4241078-1083. [DOI] [PubMed] [Google Scholar]
- 17.Labib, K., and B. Hodgson. 2007. Replication fork barriers: pausing for a break or stalling for time? EMBO Rep. 8346-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labib, K., J. A. Tercero, and J. F. Diffley. 2000. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 2881643-1647. [DOI] [PubMed] [Google Scholar]
- 19.Lee, J., D. A. Gold, A. Shevchenko, A. Shevchenko, and W. G. Dunphy. 2005. Roles of replication fork-interacting and Chk1-activating domains from Claspin in a DNA replication checkpoint response. Mol. Biol. Cell 165269-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, S. J., J. K. Duong, and D. F. Stern. 2004. A Ddc2-Rad53 fusion protein can bypass the requirements for RAD9 and MRC1 in Rad53 activation. Mol. Biol. Cell 155443-5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle.1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14953-961. [DOI] [PubMed] [Google Scholar]
- 22.Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani, M. Muzi-Falconi, C. S. Newlon, and M. Foiani. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412557-561. [DOI] [PubMed] [Google Scholar]
- 23.Lou, H., M. Komata, Y. Katou, Z. Guan, C. C. Reis, M. Budd, K. Shirahige, and J. L. Campbell. 2008. Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol. Cell 32106-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melo, J., and D. Toczyski. 2002. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14237-245. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell, D. A., T. K. Marshall, and R. J. Deschenes. 1993. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast 9715-722. [DOI] [PubMed] [Google Scholar]
- 26.Moyer, S. E., P. W. Lewis, and M. R. Botchan. 2006. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 10310236-10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nedelcheva, M. N., A. Roguev, L. B. Dolapchiev, A. Shevchenko, H. B. Taskov, A. Shevchenko, A. F. Stewart, and S. S. Stoynov. 2005. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J. Mol. Biol. 347509-521. [DOI] [PubMed] [Google Scholar]
- 28.Neuwald, A. F., L. Aravind, J. L. Spouge, and E. V. Koonin. 1999. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 927-43. [PubMed] [Google Scholar]
- 29.Noguchi, E., C. Noguchi, W. H. McDonald, J. R. Yates III, and P. Russell. 2004. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 248342-8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborn, A. J., and S. J. Elledge. 2003. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 171755-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacek, M., A. V. Tutter, Y. Kubota, H. Takisawa, and J. C. Walter. 2006. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol. Cell 21581-587. [DOI] [PubMed] [Google Scholar]
- 32.Pacek, M., and J. C. Walter. 2004. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 233667-3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paulovich, A. G., and L. H. Hartwell. 1995. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82841-847. [DOI] [PubMed] [Google Scholar]
- 34.Paulovich, A. G., R. U. Margulies, B. M. Garvik, and L. H. Hartwell. 1997. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics 14545-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shechter, D., C. Y. Ying, and J. Gautier. 2004. DNA unwinding is an Mcm complex-dependent and ATP hydrolysis-dependent process. J. Biol. Chem. 27945586-45593. [DOI] [PubMed] [Google Scholar]
- 36.Shimomura, T., S. Ando, K. Matsumoto, and K. Sugimoto. 1998. Functional and physical interaction between Rad24 and Rfc5 in the yeast checkpoint pathways. Mol. Cell. Biol. 185485-5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi, C. Obuse, T. Tsurimoto, and H. Yoshikawa. 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature 395618-621. [DOI] [PubMed] [Google Scholar]
- 38.Tercero, J. A., and J. F. Diffley. 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412553-557. [DOI] [PubMed] [Google Scholar]
- 39.Tercero, J. A., K. Labib, and J. F. Diffley. 2000. DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J. 192082-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tercero, J. A., M. P. Longhese, and J. F. Diffley. 2003. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 111323-1336. [DOI] [PubMed] [Google Scholar]
- 41.Torres-Ramos, C. A., S. Prakash, and L. Prakash. 2002. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 222419-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyatt, M. D., and D. L. Pittman. 2006. Methylation agents and DNA repair response: methylated bases and sources of strand breaks. Chem. Res. Toxicol. 191580-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamashita, M., Y. Hori, T. Shinomiya, C. Obuse, T. Tsurimoto, H. Yoshikawa, and K. Shirahige. 1997. The efficiency and timing of initiation of replication of multiple replicons of Saccharomyces cerevisiae chromosome VI. Genes Cells 2655-665. [DOI] [PubMed] [Google Scholar]
- 44.Yoo, H. Y., S. Y. Jeong, and W. G. Dunphy. 2006. Site-specific phosphorylation of a checkpoint mediator protein controls its responses to different DNA structures. Genes Dev. 20772-783. [DOI] [PMC free article] [PubMed] [Google Scholar]