

Abstract
Mismatch repair plays an essential role in reducing the cellular mutation load. Paradoxically, proteins in this pathway produce A·T mutations during the somatic hypermutation of immunoglobulin genes. Although recent evidence implicates the translesional DNA polymerase η in producing these mutations, it is unknown how this or other translesional polymerases are recruited to immunoglobulin genes, since these enzymes are not normally utilized in conventional mismatch repair. In this report, we demonstrate that A·T mutations were closely associated with transversion mutations at a deoxycytidine. Furthermore, deficiency in uracil-N-glycolase (UNG) or mismatch repair reduced this association. These data reveal a previously unknown interaction between the base excision and mismatch repair pathways and indicate that an abasic site generated by UNG within the mismatch repair tract recruits an error-prone polymerase, which then introduces A·T mutations. Our analysis further indicates that repair tracts typically are ∼200 nucleotides long and that polymerase η makes ∼1 error per 300 T nucleotides. The concerted action of Msh2 and UNG in stimulating A·T mutations also may have implications for mutagenesis at sites of spontaneous cytidine deamination.
The affinity maturation of the antibody response depends on the somatic hypermutation (SHM) process. The enzyme activation-induced cytidine deaminase (AID) initiates SHM in germinal center B cells by deaminating C within immunoglobulin (Ig) genes, yielding a G·U lesion that is resolved by several mechanisms (29). Replication across the U generates G·C to A·T transition mutations, while the removal of the U by uracil-N-glycolase (UNG) leads to transversion and transition mutations at the original G·C base pair (33). The AID-generated G·U lesion is also a substrate for the mismatch repair (MMR) proteins Msh2, Msh6, and Exo1. Unlike their normal role in DNA repair, the processing of this lesion by these MMR proteins during SHM paradoxically leads to the production of mutations at A·T base pairs (see below).
MMR is a DNA repair process utilized by prokaryotes and eukaryotes (25). This pathway repairs DNA errors caused by the misincorporation of nucleotides during DNA synthesis. The initial mismatch is detected by MutSα, which consists of Msh2 and Msh6 in mammalian cells. The ability of MMR to discriminate between the mutated and unmutated strands of DNA is thought to be dictated by nicks or gaps on the newly synthesized lagging strand between Okazaki fragments or by strand ends on the leading strand at the replication fork (18). The MutLα endonuclease (Mlh1/Pms2) uses the DNA nick or end as a marker of the newly synthesized, and therefore mutated, strand to introduce a new nick on either side of the mismatch (15). This nicked strand is then excised by the 5′-to-3′ exonuclease Exo1, and the ensuing gap is repaired by the replicative polymerase δ. However, since AID acts primarily during G1 of the cell cycle (11, 36), it is unclear whether Msh2/6 is capable of distinguishing between the AID-mutated and unmutated strands prior to strand excision.
Consistently with their role in DNA repair, deficiency in Msh2, Msh6, or Exo1 generally leads to an increase in mutation frequencies in different tissues (40). However, in the case of SHM of Ig genes, the loss of these MMR proteins reduces the frequency of mutations at A·T base pairs (4, 5, 10, 16, 22, 30, 32, 37, 41, 42). One possible difference between conventional and mutagenic MMR is the involvement of the error-prone DNA polymerase η in the latter process. Indeed, both mice and humans lacking polymerase η resemble Msh2-deficient mice, in that mutations at A·T base pairs in the V region are less frequent (6, 7, 47). Moreover, the error spectrum of polymerase η on undamaged DNA matches the mutation spectrum of A·T mutations in the V region (35). While it is now well established that mutations at A·T base pairs are produced largely by proteins involved in the MMR pathway, it is not known how DNA polymerase η is recruited during SHM.
One possible explanation for the use of error-prone polymerases is the occurrence of replication-blocking lesions, such as an abasic site or a modified nucleotide, in the V region of Ig genes. Evidence that a replication block leads to mutagenic MMR comes from recent studies showing the requirement of ubiquitinated PCNA for mutagenic MMR (1, 19, 34). Monoubiquitination at the K164 residue of PCNA in response to DNA damage leads to translesional synthesis (1), and SHM at A·T base pairs is reduced in PCNAK164R/K164R mice to levels observed in MMR-deficient mice (19, 34). In addition, the finding that translesional DNA polymerases are involved in SHM (9, 31, 45-47) suggests that replication-blocking lesions are common at the Ig locus during SHM. Taken together, these observations suggest a model in which replication-blocking lesions recruit error-prone polymerases, which then generate mutations at nearby A·T base pairs. As reported here, we have tested this model by examining the correlated mutations in V region sequences from hypermutating Ramos cells and in murine centroblasts.
MATERIALS AND METHODS
Cell culture, subcloning, and flow cytometry.
Ramos 67 cells were maintained as previously described (48), and Abelson pre-B cell lines 15-63 (Msh2+/−) and 8-58 (Msh2−/−) were maintained in RPMI medium (Invitrogen) with 10% bovine calf serum (HyClone), penicillin (100 U/ml), and streptomycin (0.1 mg/ml; Sigma). For subcloning, Ramos 67 cells were plated at 0.1 cell per well into 96-well plates. After ∼15 cell divisions (∼32,000 cells), cells were harvested and prepared for flow cytometry cell sorting or enzyme-linked immunospot (ELISPOT) assays. To isolate IgM-positive and IgM-negative clones, Ramos cells were stained with fluorescein isothiocyanate- or biotin-conjugated anti-IgM Fab fragment antibody (Jackson ImmunoResearch Laboratories), with the latter followed by staining with allophycocyanin-conjugated-streptavidin (eBioscience), and single IgM-positive and IgM-negative cells were sorted (FacsAria; BD) directly into 96-well plates. Based on the previously measured mutation rate in these cells (10−5 mutation/bp/generation) (48), we estimate that under this protocol, 15% of IgM-reverted cells had undergone multiple independent mutational events in the 1,000-bp V region (10−5 mutation/bp/generation × 1,000 bp × 15 generations = 0.15 mutation per V region).
Measurement of MMR activity.
To quantify MMR activity, pCA-OF-expressing clones were harvested and washed in phosphate-buffered saline (PBS) (Gibco, Invitrogen), and the green fluorescent protein (GFP) reversion frequency was determined using a flow cytometer (FACSCalibur; BD) as previously described (39). Flow cytometry data were analyzed using FlowJo software.
ELISPOT assays.
The ELISPOT assay for IgM secretion was performed as previously described (21).
Plasmids and transfections.
To measure MMR activity, the microsatellite-like plasmid pCA-OF was used. To inhibit UNG activity, pEF (control) and pEF-UGI plasmids were used. Plasmids were linearized with MluI (pEF and pEF-UGI) and BglII (pCA-OF). For cell transfections, ∼4 × 106 log-phase cells were mixed with 10 μg of linearized plasmid DNA in 4-mm cuvettes and electroporated (Gene Pulser Xcell) at 250 V and 950 μF for Ramos cells and at 450 V, 950 μF, and 150 Ω for the pre-B cells. Cells were diluted in appropriate media and plated in 96-well plates. After incubation at 37°C for 24 h, stable clones were selected with puromycin (pEF and pEF-UGI; 0.8 μg/ml) or blasticidin (pCA-OF; 2.5 μg/ml for Ramos and 25 μg/ml for pre-B cells).
DNA extraction, PCR, and sequencing in Ramos.
Genomic DNA was extracted as previously described (21). For V region amplification from IgM-positive and IgM-negative clones, Taq polymerase was used with the following cycling parameters: 95°C for 2 min for 1 cycle, and then 35 cycles of 95°C for 45 s, 58°C for 30 s, and 72°C for 90 s. The forward and reverse primers were 5′RamV5316 (5′ ACAGCCAGCATACACCTCCC) and 3′RamV6209 (5′ CAACCTGAG-TCCCATTTTCC), respectively. PCR products were purified using the Wizard PCR Preps purification system (Promega) according to the manufacturer's specifications and sequenced using 5′ RamV_Inner (5′ CACCAACTACAACCCGTCCC) and 3′ RamV_Inner (5′GTGGCCATTCTTACCTGAGG). To measure V region mutation rates by PCR, amplifications were performed on DNA from unselected Ramos clones using PFU Ultra II (Stratagene) as previously described (21).
Amplification and analysis of murine Ig sequences.
All murine V region sequence data were generated from sorted PNAhi B220+ germinal center B cells isolated from Peyer's patches or spleen as described previously (22). Mutations were analyzed in the intronic JH2-JH4 region or the intronic VHJ558-JH4 rearrangement flanking region (wild-type and UNG−/− mice). Wild-type mouse data were obtained from previously published works (3, 22, 33). UNG−/− sequence data were generously provided by J. Di Noia, C. Rada, and M. Neuberger. Additional UNG−/− sequence data were generated from genomic DNA kindly provided by H. Ming and U. Storb (37). The VHJ558-JH4 flanking region was amplified from genomic DNA as previously described (33). To compensate for the unequal distribution of nucleotides in the sequenced region (the C:G ratio was ∼0.7:1, and the A:T ratio was ∼0.8:1), the data shown in Fig. 4C were normalized for nucleotide content according to the following formula: percent bottom strand C mutations = 100(bottom strand C mutations/number of bottom strand C's)/[(bottom strand C mutations/number of bottom strand C's) + (top strand C mutations/number of top strand C's)]. The same formula was used to normalize for A·T mutations.
FIG. 4.

Transversion mutations at C on the bottom strand are associated with mutations at A on the top strand in wild-type mice. (A) Percentages of sequences with associated A·T and G·C mutations from nine wild-type mice and five UNG−/− mice. All analyzed sequences contained up to five mutations per sequence with a minimum of one mutation at an A·T base pair. (B) Comparison of G·C transition and G·C transversion mutations in sequences with and without A·T mutations in wild-type (WT) and UNG−/− mice (for the wild-type mice, n = 31 without A·T mutations and n = 24 with A·T mutations; for UNG−/− mice, n = 20 without A·T mutations and n = 41 with A·T mutations). Sequences with G·C transversion mutations that also contained G·C transition mutations were scored as sequences with G·C transversion mutations. Statistical analyses were performed using Fisher's exact test (**, P = 0.0023 for sequences with and without A mutations from wild-type mice for G·C transversion mutations). (C) Percentages of mutations located on the bottom strand were determined for wild-type and UNG−/− mice in unrestricted (UnRes; columns 1 to 3 and 7 to 9) and restricted (Res; columns 4 to 6 and 10 to 12) data sets. Sequences that did not contain A·T mutations were excluded from this analysis. Mutations were normalized for nucleotide content in the sequenced region (see Materials and Methods). Statistical analyses were conducted using the t test (*, P = 0.02 for the comparison of C transversion mutations in the restricted data set between wild-type and UNG−/− mice). (D) Location of C and A mutations in the JH2-JH4 and VHJ558-JH4 intronic regions of wild-type mice. Data shown are from sequences containing A mutations in the top strand (top) or the bottom strand (middle) or containing no A mutations (bottom). Top strand mutations and bottom strand mutations are depicted above and below the midline, respectively. Statistical analyses were performed using the t test (*, P = 0.0249 for bottom strand transition mutations [top panel]; ***, P < 0.0001 for bottom strand transversion mutations [top panel]). (E) Strand bias of A mutations in sequences containing a single C mutation in the top strand (top) or bottom strand (bottom). A mutations that are linked to a C transition mutation are represented by a square, and those linked to a C transversion mutation are represented by a circle. Statistical analyses for the strand bias of A mutations were performed using the t test (***, P < 0.0001), and those for the location of A mutations relative to C transition and transversion mutations were performed using Fisher's exact test (P = 0.0015).
In vitro UDG assay.
The inhibition of UNG by uracil-DNA glycosylase inhibitor (UGI) was confirmed using the uracil DNA glycosylase assay as previously described (8), with minor modifications. Briefly, a double-stranded oligonucleotide containing a single U:G mismatch was 5′ labeled with [γ-32P]dATP. The labeled substrate was incubated with 1 U of uracil DNA glycosylase (NEB) or with serially diluted Ramos nuclear extracts (1 to 10 μg) in uracil glycosylase buffer (NEB) for 3 h at 37°C, followed by incubation with sodium hydroxide (100 mM) for 10 min at 98°C. Samples were electrophoresed on a 20% denaturing acrylamide gel with a running buffer of 1× TBE (Tris-borate-EDTA) at 300 V for 3 h and visualized using a PhosphorImager (Molecular Dynamics). Quantitation was performed using ImageQuant software, version 5.0 (Molecular Dynamics).
Statistical analysis.
Ramos and murine data were graphed using GraphPad software (Prism), and statistical analyses were performed using the unpaired t test, Fisher's exact test, and Mann-Whitney test.
RESULTS
Transversion mutations at C are linked to A·T mutations.
As noted above, SHM of both G·C and A·T base pairs depends wholly on AID, while A·T mutations depend additionally on mismatch repair and potentially on replication-blocking lesions. These observations suggest the specific model illustrated in Fig. 1A. As in other models, the G-U mismatch generated by AID is detected by the MMR system. Since translesional synthesis is the final stage of MutSα-mediated repair during SHM, we investigated the mutational outcomes of MutSα when it targeted either the AID-mutated or unmutated DNA strand. Assuming that MutSα targets both strands equally for repair, 50% of the time the mutated (U-containing) strand is degraded by Exo1. The resynthesis of the ensuing gap restores the original G·C base pair. If, however, the unmutated (G-containing) strand is excised, as illustrated in Fig. 1A, the degradation of the unmutated strand by Msh2/Exo1 exposes U in the opposite strand. The excision of the U by UNG, which is threefold more active on single-stranded DNA (ssDNA) than on double-stranded DNA (17), generates an abasic site. The resynthesis of the degradation tract begins faithfully but stalls at the abasic site, thus inducing the ubiquitination of PCNA and the assembly of translesional polymerases that can then bypass this lesion. Random nucleotide insertion opposite the abasic site leads to a G·C transversion mutation 50% of the time; further extension 3′ by the translesional polymerase η then introduces A·T mutations into the V region.
FIG. 1.

Model for mutagenic repair by MutSα. (A) A cytidine in the representative DNA sequence is deaminated by AID, thus generating a G·U lesion, which stimulates the MMR-directed Exo1 excision of the unmutated DNA strand. This yields a U in the opposite strand within an ssDNA region. The removal of the U by UNG generates an abasic site. The resynthesis of the excised strand begins with an error-free replicative polymerase until the abasic site is encountered, causing the polymerase to stall. This leads to translesional synthesis, with the recruitment of error-prone polymerases to bypass the abasic site as well as polymerase η to generate mutations at A·T base pairs. (B) On the top is a schematic of the IgH locus of Ramos 67 (R67) cells with the position of the TAA nonsense codon represented by an arrow. On the bottom is a diagram describing the enrichment for Ramos clones containing A·T mutations, including subcloning, sorting based on IgM expression, and the sequencing of the V region. FITC, fluorescein isothiocyanate.
The scenario described above predicts that sequences with mutations at A·T base pairs will be enriched for mutations at G·C base pairs and that the G·C mutations will be predominantly transversions. To test these predictions, we examined sequences from hypermutating Burkitt's lymphoma Ramos cells and from hypermutating centroblasts in mice. For the Ramos cell analysis, we used an IgM-negative variant Ramos 67 that harbors a TAA nonsense codon in the endogenous Ig heavy chain (IgH) locus, truncating translation in the variable domain. Consequently, sorting for IgM-positive Ramos 67 cells was expected to select for cells that had undergone a mutation at an A·T base pair. We therefore isolated IgM-positive cells and examined the associated mutations in an ∼1-kb V region-containing segment of the IgH locus.
In these experiments, it was important to minimize the occurrence of unrelated mutations. The mutation rate of the V region in Ramos 67 is known to be ∼10−5 mutations/bp/generation (48). Therefore, Ramos 67 was subcloned and allowed to expand only ∼15 cell divisions before sorting. This protocol was expected to yield an IgM-positive cell population in which only ∼15% of cells had undergone multiple independent mutations within the V region being analyzed (see Materials and Methods for the calculations). Subclones of Ramos 67 were stained with fluorescein isothiocyanate-labeled anti-IgM, and IgM-positive and -negative clones were sorted directly into individual wells on a 96-well plate. Sorted cells then were expanded, genomic DNA was extracted, and an ∼1-kb region encompassing the V region was amplified by PCR (Fig. 1B). To minimize PCR errors, we sequenced the PCR products directly.
To assess the likelihood that G·C mutations were associated with A·T mutations, we measured the excess of G·C mutations in the IgM-positive and IgM-negative clones. Under conditions in which we limited proliferation to ∼15 cell divisions, most unselected IgM-negative Ramos clones did not harbor a mutation. Indeed, there was an ∼5-fold increase in mutation frequency (not including the reverted nonsense codon) in selected IgM-positive clones compared to that of the unselected IgM-negative clones (Fig. 2A), indicating that selecting for mutations at A·T base pairs enriched for other types of mutations.
FIG. 2.

Sequences with A·T mutations are enriched for transversion mutations at C on the bottom strand in an UNG-dependent manner. (A) Mutation frequency at the V region in R67 clones is shown for sorted IgM+ and IgM− clones. Only unique mutations from independent clones were scored. The reverting mutation at the TAA nonsense codon was excluded. The data represent 51 IgM+ and 68 IgM− R67 clones and 18 IgM+ and 52 IgM− UGI-expressing R67 clones. Statistical analyses were conducted using the Fisher's exact test (***, P < 0.0001; *, P = 0.0284). (B) The percentage of sequences containing associated A·T and G·C mutations was determined for reverted IgM+ untransfected (left) and UGI-expressing (right) R67 clones. (C) Mutation characteristics in the V region of IgM+ and IgM− clones are shown. Individual mutations are represented by white, gray, or black circles. The location of C transition (TRN) and C transversion (TRV) mutations and A mutations (MUT) relative to the reverted mutation at the TAA nonsense codon (vertical line at x = 0) also is depicted. Mutations on the top strand (above the midline) and bottom strand (below the midline) are shown for R67 IgM+ (top), R67 IgM− (middle), and R67 IgM+ UGI-expressing clones (bottom). Statistical analyses to compare top and bottom strand mutations were conducted using the t test (***, P < 0.0001 for transversion mutations at C [top panel]; **, P = 0.0052 for mutations at A [top panel]; *, P = 0.0486 for transition mutations at C [bottom panel]).
Of 51 independent IgM-positive clones, 21 clones (41%) contained mutations at G·C base pairs, whereas of 68 unselected clones only 8 clones (12%) had a mutated G·C (Fig. 2B; also see Table S1 in the supplemental material), supporting the model shown in Fig. 1A that mutation at A·T enriches for mutations at G·C. Sequences derived from Ramos 67 IgM-positive clones with associated G·C mutations are shown in Fig. S1 in the supplemental material. Of note, the TAA nonsense codon in all IgM-positive clones was reverted exclusively to either TAC or TAT (see Fig. S1 in the supplemental material), which encodes tyrosine. Two mechanisms that cause this limited set of revertants might be due to the mutagenic preference of polymerase η (24, 35) or the assembly of IgM. That is, the TAA codon lies opposite the sequence 5′-TTA-3′, and polymerase η preferentially misincorporates nucleotides opposite a T in the motif TT or TA (the underlined nucleotide is mutated). The reversion pattern most probably reflects the fact that the nonsense codon is located within the conserved Tyr-Tyr-Cys motif in the FR3 region and indicates that other amino acids in this motif do not allow the assembly of membrane-bound IgM.
We compared the types of mutations in the IgM-positive and IgM-negative clones (Fig. 2C; also see Table S1 in the supplemental material). The mutations in the unselected IgM-negative clones predominantly were transition mutations at G·C base pairs, similarly to that reported earlier (48). However, the mutation spectrum of the IgM-positive pool differed significantly from that of the IgM-negative pool, in that there were more A·T mutations (P = 0.0204) and more G·C transversion mutations (P < 0.0001). Seventy-five to 80% of G·C mutations in both populations occurred in AID hot spot motifs (i.e., WRC), suggesting that these mutations arose from a processed AID deamination event. Moreover, 65% of mutations at A occurred at polymerase η WA motifs (40% of A in the Ramos V region are in WA motifs), as expected for mutations due to the SHM process.
We also examined the location of these mutations with respect to the TAA nonsense codon (Fig. 2C). Mutations at G·C base pairs are initiated by altering the C. If the C is in the top (or bottom) strand, mutations at G·C base pairs correspondingly are deemed to be mutations in the top (or bottom) strand. In the case of the unselected IgM-negative clones, 30% of mutations in these clones were found within a distance of 100 bp either upstream or downstream of the TAA codon. In contrast, in the IgM-positive clones, 71% of all mutations were localized within 100 bp on either side of the TAA codon. These data further corroborate the notion that most mutations in the IgM-positive clones were associated with mutations at the TAA codon. Moreover, these results suggest that the typical repair tract is ∼200 nucleotides.
Strikingly, C transversion mutations clustered on the bottom strand in IgM-positive cells (P < 0.0001), while no bias was seen in the unselected IgM-negative clones (P = 0.6874). These transversion mutations in the bottom strand were found equally upstream and downstream of the A·T mutation. This pattern of clustering is not predicted by the model in Fig. 1A. That is, this model hypothesizes that polymerase η is recruited when replication is blocked at an abasic site and then proceeds to introduce mutations at A·T base pairs. This hypothesis therefore predicts that A·T mutations will be associated with an upstream C in the bottom strand and a downstream C in the top strand. The implications of this observation will be considered in the Discussion.
The reversion of the TAA codon also was associated with the occurrence of other A·T mutations. As considered in the Discussion, the frequency of these A·T mutations can be used to estimate the error rate of the translesional polymerase. Eight out of these 10 A·T mutations occurred with A on the top strand (P = 0.005). This is consistent with observations in wild-type mice, which also have been interpreted to reflect the preferential excision of the top strand by Msh2/Exo1, followed by preferential misincorporation opposite T (24, 43) (see Fig. 4C). In keeping with the strong preference for misincorporation opposite T, in this paper we refer to mutations at A·T base pairs as A mutations.
AID does not show a strong strand bias preference.
We next tested whether the strand bias of the C transversion mutations reflects a preferential specificity for AID mutating the bottom strand in Ramos cells. For this analysis, we assembled previously obtained sequence data from Ramos clones that were carried in culture but not subjected to any form of selection (e.g., IgM expression). Only unique mutations at G·C base pairs were included in this analysis, unless genealogies indicated that the mutation was unique or that the same mutation occurred in different clones. As suggested by the data in Fig. S2 in the supplemental material, after correcting for base composition, AID marginally prefers to mutate the top strand by a ratio of 0.48:0.44 (63 mutations/131 C on the top strand, 67 mutations/151 C on the bottom strand), supporting previous findings (23, 43). In addition, using this data set, we found that mutations at AID hot spot motifs (i.e., WRC) occur on both strands at approximately similar frequencies (data not shown), which is consistent with the notion that AID mutates both strands approximately equally. This suggests that the bias for C transversion mutations on the bottom strand in IgM-positive clones is not due to AID directly but likely reflects a strand preference of the repair process (see below).
Inhibition of UNG abrogates linked C transversion and A mutations.
An abasic site produced by a DNA glycosylase is likely an intermediate for C transversion mutations within AID hot spot motifs. To test whether these transversions depend on UNG, we inhibited UNG by stably transfecting Ramos cells with a UGI-expressing plasmid (8) and examined whether the inhibition of UNG altered the frequency or spectrum of mutations. We screened transfected clones and measured residual UNG activity in nuclear extracts using a previously described assay (8). As shown in Fig. 3A, nuclear extracts from UGI-expressing Ramos 67 had no detectable residual glycosylase activity relative to that of empty vector controls, indicating that UNG is the major glycosylase in Ramos cells. By sequencing the V region in unselected Ramos clones, we found that transversion mutations at G·C base pairs were reduced in UGI-expressing clones by at least sixfold (58.7 × 10−6 versus <9.01 × 10−6; P = 0.015) (Fig. 3B), similarly to that reported for UNG−/− mice (33). To determine the effect of UGI expression on the mutation frequency at A·T base pairs, we measured the reversion frequency of the TAA codon using an ELISPOT assay for IgM secretion (Fig. 3C). The IgM reversion frequency in UGI-transfected clones was reduced ∼3-fold relative to that of the empty vector control (P = 0.007), indicating that at least ∼2/3 of the mutations at A·T base pairs depended on UNG, possibly in concert with the Msh2/Exo1 pathway (see below). As expected, UNG inhibition did not perturb MMR activity, as measured by a microsatellite instability assay (Fig. 3D) (39).
FIG. 3.

Expression of UNG inhibitor UGI in R67 cells reduces both A·T mutations and C transversion mutations. (A) The level of UNG activity in control and UGI-expressing Ramos clones was determined using a substrate cleavage assay. A substrate containing a single U:G mismatch was incubated alone (lane 1), with uracil DNA glycosylase (lane 2), or with 3× serial dilutions of nuclear extracts from two control (lanes 3 to 8) and two UGI-expressing (lanes 9 to 14) R67 clones. TRN, transition; TRV, transversion; MUT, mutation. (B) Mutation characteristics in the V region of control (left) and UGI-expressing (right) R67 clones are shown. Statistical analyses comparing mutation types were performed using the t test (the asterisk indicates P = 0.0150 for G·C transition mutations and P = 0.0492 for G·C transversion mutations). TRN, transition; TRV, transversion; MUT, mutation. (C) The IgM reversion frequency was determined using the ELISPOT assay for IgM secretion. Individual subclones are represented as light gray circles; black circles represent clones in which no IgM reversion events were observed among ∼5 × 10−5 cells. Statistical analyses comparing median reversion frequencies were performed using the Mann-Whitney test (***, P = 0.0007). (D) MMR activity for Msh2−/− and Msh2+/− pre-B cell lines and R67 clones was quantified using a microsatellite instability assay based on the GFP reversion frequency. The GFP open reading frame is interrupted with an out-of-frame dinucleotide microsatellite-like sequence (39), and microsatellite instability can lead to GFP expression. Statistical analyses were performed using the Mann-Whitney test (**, P = 0.003 for results with Msh2−/− pre-B cells compared to those with Msh2+/− pre-B cells).
To determine whether UNG activity is required for linked C transversion and A mutations, we measured whether UGI expression reduced the fraction of IgM-positive clones in which the reversion mutation was associated with a C transversion. As shown in Fig. 2B and C, ∼31% (16/51) of IgM-positive Ramos 67 clones had a linked transversion, compared to ∼17% (3/18) for the UGI-expressing cells. Combined with the reduced mutation frequency at A·T base pairs in UGI-expressing Ramos cells (Fig. 3B), these data indicate that ∼2/3 of A·T mutations in Ramos cells are produced by replication across an UNG-generated abasic site on the bottom strand. Although A·T mutations occurred when UNG was inhibited (Fig. 2C and 3C), the residual A·T mutations were not associated with C transversions. These data suggest that a mechanism that is independent of an abasic site also generates A·T mutations during SHM.
Murine Ig sequences with A·T mutations are enriched for C transversion mutations.
Our analysis showed that IgM-positive Ramos clones that have an A·T mutation in their V region are enriched for a nearby C transversion mutation on the bottom strand. We tested whether a similar relationship holds in the V regions of murine B cells that were hypermutating in vivo. For this analysis, we examined the JH2-JH4 or VHJ558-JH4 intronic Ig sequence from PNAhi Peyer's patch B cells from nine wild-type mice and five UNG−/− mice (see Fig. S3 in the supplemental material). A limitation in examining linked A·T and G·C mutations in Ig sequences derived from mice is that they often are heavily mutated, which likely is due to multiple rounds of mutational events during the life cycle of the centroblast. Therefore, we restricted our analysis to sequences containing a maximum of five mutations per sequence (see Table S2 in the supplemental material), thereby minimizing the possibility that mutations arose independently. As shown in Fig. 4A, ∼57 and ∼85% of Ig sequences that contained at least one A·T mutation and had up to five total mutations also harbored a G·C mutation in wild-type and UNG−/− mice, respectively. To examine the relationship between C transversion mutations and A·T mutations in the wild-type-restricted data set, we measured the number of sequences with C transition or C transversion mutations with respect to the presence or absence of A·T mutations. For wild-type mice, in sequences with no A·T mutations, only 21% (5/24) of those sequences harbored a C transversion mutation (Fig. 4B). However, among sequences that contain A·T mutations, 65% (20/31) also contained C transversion mutations (P = 0.0023) (Fig. 4B). Thus, sequences with A·T mutations are enriched for C transversion mutations in murine B cells, thereby corroborating the Ramos cell data. However, Ig sequences from UNG−/− mice showed no difference in C transversion mutations whether or not a sequence harbored an A·T mutation (Fig. 4B), highlighting, just as with the UGI-Ramos data, that a mechanism independent of abasic sites produces A·T mutations during SHM.
We next examined whether specific mutations displayed a strand bias exclusively in sequences with A·T mutations. To eliminate bias due to the unequal distribution of nucleotides in the sequenced region, all values were normalized for nucleotide content (see Materials and Methods). As shown in Fig. 4C, only 28% of A·T mutations had the A on the bottom strand, indicating that A mutations occurred preferentially in the top strand (P < 0.0001), consistently with previous data for mice (24) and for Ramos cells (Fig. 2C). Importantly, 72% of C transversion mutations occurred on the bottom strand (P = 0.00392) (Fig. 4C) in wild-type mice, displaying the same strand preference as that in Ramos cells (Fig. 2D), and most of these mutations depended on UNG (Fig. 4C). Moreover, in wild-type mice, in sequences containing A mutations in the top strand, 100% of transversion mutations and 65% of transition mutations at C were located on the bottom strand (P < 0.0001 and P = 0.0249, respectively) (Fig. 4D, top). In contrast, sequences containing A mutations on the bottom strand showed no strand preference for C mutations (P = 0.6261; P = 0.2195 for C transversion and C transition mutations, respectively) (Fig. 4D, middle). There also was no strand preference in sequences with no mutations at A·T base pairs (P = 0.5796; P = 0.5734 for C transversion and C transition mutations, respectively) (Fig. 4D, bottom).
To extend this analysis, we determined whether A mutations display a strand bias in sequences harboring a single C mutation. Sequences with a top strand C mutation do not display a significant strand bias with A mutations (P = 0.1114) (Fig. 4E, top). In contrast, sequences with a bottom strand C mutation display a bias of A mutations to the top strand (P < 0.0001) (Fig. 4E, bottom), which is consistent with data shown in Fig. 4D and with Ramos data (Fig. 2C). Notably, bottom strand C transversion mutations are preferentially located upstream of A mutations (P = 0.0015) (Fig. 4E, bottom), which is consistent with the model shown in Fig. 1A. However, as noted above, this relationship was not observed in Ramos cells (Fig. 2C). Collectively, these data show that A mutations on the top strand are associated with C transversion mutations on the bottom strand in murine B cells, supporting the notion that the excision of the top strand by Exo1 and the removal of U on the bottom strand by UNG generates A mutations on the top strand.
Deficiency of MMR leads to decreased C transversion mutations.
The model (Fig. 1A) envisages that the AID-generated G-U mismatch recruits the MMR system, which sometimes excises the G-containing strand exposing the U to UNG, leading to frequent transversion mutations. Because UNG displays greater activity on U in ssDNA than in double-stranded DNA (17), deficiency in MMR should protect U from UNG and lead to an increase in transition mutations at C on the bottom strand. As such, we examined Ig sequences from wild-type and MMR-compromised (i.e., Msh2−/−, Msh2G674A, Msh6−/−, and Exo1−/−) mice from previously published data (4, 20, 22) (see Fig. S4 in the supplemental material).
Figure 5 shows the percentage of C mutations on the top or bottom strand in wild-type and MMR-deficient mice. While transition and transversion mutations at C were similar between wild-type and MMR-deficient mice on the top strand, we observed a twofold decrease in the fraction of C mutations that were C-to-G transversion mutations (P = 0.014) on the bottom strand in MMR-deficient mice (P = 0.003) and a corresponding ∼1.3-fold increase in C-to-T transition mutations in MMR-deficient mice (P = 0.003). These data indicate that ∼50% of C transversion mutations on the bottom strand are produced indirectly by MMR proteins and suggest that in the absence of MMR, a larger proportion of AID-generated U on the bottom strand is instead being replicated, leading to an increase in C transition mutations. Since AID is slightly more active on the top than the bottom strand and the UNG pathway has not been shown to display a strand bias, this result indicates that the MMR and base excision repair pathways cooperate to produce C transversion mutations on the bottom strand and A mutations on the top strand, as inferred from the analysis of Ramos cells.
FIG. 5.
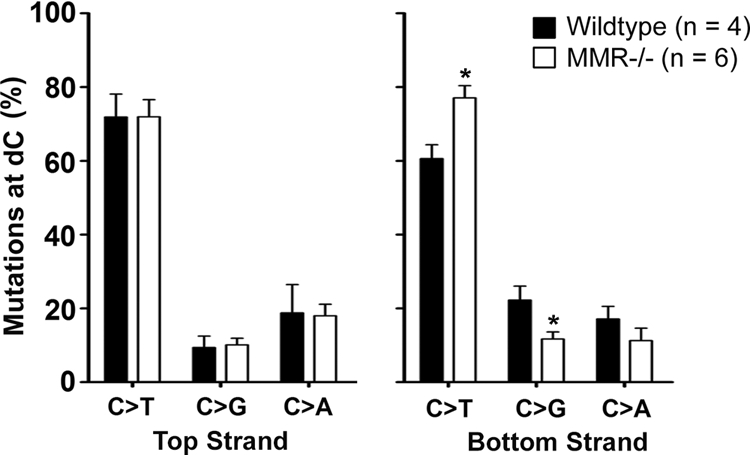
Bottom, but not top, strand C transversion mutations are reduced in MMR-deficient mice. The percentages of C-to-T, C-to-G, and C-to-A mutations on the top strand (left) and on the bottom strand (right) in wild-type and MMR-deficient mice is displayed. Data were collected from the JH2-JH4 region of wild-type (two Msh2G674A+/+, one Msh2G674A+/M, and one Exo1+/−) and MMR-deficient (one Msh2−/−, one Exo1−/−, one Msh6−/−, and three Msh2G674AM/M) mice from previous publications (see Fig. S4 in the supplemental material). Statistical analyses were performed using the t test (the asterisk indicates P = 0.0250 for C to T mutations on the bottom strand and P = 0.0121 for C to G mutations on the bottom strand).
DISCUSSION
SHM is initiated by the AID-mediated deamination of cytidines. Proteins involved in the MMR pathway have been coopted by the SHM process to extend mutagenesis from this initial G·C base pair to A·T base pairs. To gain insight into this process, we used a hypermutating B cell line, Ramos, in which we selected for mutation at a specific A·T base pair and then analyzed the other, correlated mutations. We also tested for similar correlations in Ig sequences from mice. We have interpreted our results based on the model in Fig. 1A. This model postulates that the creation of an abasic site, the result of cytidine deamination followed by uridine excision, recruits an error-prone polymerase that introduces mutations at A·T base pairs. As predicted by the model, we observed that C transversions, a proxy measurement for the replication of an abasic site, were strongly associated with A·T mutations in Ramos cells and in mice (Fig. 2 and 4).
It is evident from our data and from data for UNG−/− mice (33) that not all mutations at A·T base pairs arise from this pathway, as A·T mutations occurred even when UNG was deficient (Fig. 2A; also see Fig. S3 in the supplemental material), and the residual A·T mutations were not strongly correlated with the occurrence of C transversions (Fig. 2B and 4B). A possible explanation is that these mutations result from an MMR-dependent but UNG-independent mechanism. These data argue that in the absence of UNG, A·T mutations are made by a qualitatively different mechanism, perhaps still MMR dependent but not involving an abasic site.
Our analysis of C transition mutations suggested that both top and bottom strands were mutated to a similar extent (see Fig. S2 in the supplemental material), indicating that AID targets both strands equally. However, C transversions occurred predominantly in the bottom strand (Fig. 2 and 4), suggesting that the MMR system preferentially excises the top strand, leading to DNA polymerase η-induced A mutations in the top strand. One potential explanation for the preferential excision of the top strand by the MMR pathway is that the lagging strand is synthesized off the bottom strand during DNA replication. This would place Okazaki fragments on the top strand in the new daughter cell. Since the MMR system repairs mismatches more efficiently on the lagging strand than on the leading strand (28), this would lead to the preferential excision of the top strand (28). The disproportionate position of Okazaki fragments in the top strand could occur if the Ig genes in B cells were replicated by an origin of replication that is located 3′ of the V region, such as those found in the 3′ enhancer region (49) and the intronic μ enhancer (2).
The model illustrated in Fig. 1A, in which translesional synthesis begins at the abasic site, predicts that all C transversion mutations on the bottom strand are upstream of the mutated A·T base pair. However, we found that bottom strand C transversions occurred both upstream and downstream of the mutated A·T base pair in Ramos cells (Fig. 2 and 4E). One explanation has been suggested by Ohm-Laursen and Barington, who observed an inverse correlation between the mutation rate at A·T base pairs and the distance to the nearest 3′ WRC motif (27). The authors proposed that this correlation was due to a 3′-to-5′ exonuclease or endonuclease, such as MRE11, which has been shown to increase SHM when ectopically overexpressed in Ramos cells (44). The 3′-to-5′ exo/endonuclease would be recruited after replication stalls at the abasic site, thus extending the gap (∼100 nucleotides) to include an upstream A·T, followed by error-prone gap filling. As a result, A mutations are induced both upstream and downstream of the abasic site, resulting in the accumulation of C transversion mutations on the bottom strand located 3′ and 5′ of the A mutation.
Our data reveal that in sequences containing A·T mutations with associated C transversion mutations, a C-to-G mutation predominates. This indicates that a deoxycytidine is frequently inserted opposite the abasic site. We propose that Rev1, a known deoxycytidil transferase, is involved at this step. Studies of yeast show that Rev1 preferentially inserts C opposite abasic sites in a gapped duplex substrate (12), which mimics an MMR-induced excision tract containing an abasic site in the template strand. Moreover, C-to-G transversion mutations were significantly reduced in mutated Ig genes in Rev1-deficient mice (14). Although Rev1 efficiently inserts C opposite abasic sites and other lesions, it does not readily extend from them (13). Following the initial insertion event, Rev1 is replaced by a second translesional polymerase that can extend from a C-abasic site mispair, and in hypermutating B cells this is likely polymerase η. The reason for the utilization of polymerase η is not fully understood but may be related to the finding that Msh2/6 proteins associate physically with, and stimulate the catalytic activity of, polymerase η (42).
Figure 2 shows that most mutations at G·C base pairs were located within 100 nucleotides on either side of the mutated A·T base pair. These data suggest that the Exo1 degradation tract is ∼200 nucleotides in length. Similar results were found in a study that examined the length of MMR-dependent ssDNA in yeast and mammalian cells (26). Using electron microscopy, the authors demonstrated that the length of Exo1 tracts peaked at ∼200 nucleotides. Furthermore, Unniraman and Schatz (38) showed, using a transgenic mouse model, that mutations at A·T base pairs accumulated up to ∼30 nucleotides away from a G·C-rich tract, suggesting that the Exo1 tract is ∼60 nucleotides in length. Based on data generated in our study as well as by Mojas et al. and Unniraman and Schatz, we conclude that the excision tract produced by Exo1 during MMR is ∼200 nucleotides (26, 38).
Our analysis also can be used to estimate the error rate of the translesional polymerase η in vivo. Ten of the 51 V genes that we sequenced from the IgM-positive Ramos revertants contained A·T mutations in addition to the A·T mutation that restored IgM production; 8 of these 10 mutations had A in the top strand. Considering that transversions at G·C occurred mostly with C on the bottom strand, these eight A·T mutations probably represent misincorporations opposite T in the bottom strand. These eight A mutations were generated in the course of repairing 51 excision tracts (i.e., 51 revertant clones analyzed) that we estimate to be ∼200 nucleotides long in a sequence of 100 bp on either side of the TAA nonsense codon, which contains 24% T's. This calculation thus implies that the error rate of polymerase η opposite template T is ∼3.3 × 10−3 (i.e., 8/[51 × 200 × 0.24]). This error rate is ∼10-fold lower than the in vitro error rate of human polymerase η (3.5 × 10−2) (35), which may be due to the repair of polymerase η-generated errors in our in vivo system.
Like other seeming paradoxes, the discovery that somatic hypermutation coopts machinery that otherwise prevents mutations has proved very instructive. The lesson from the immune system is that a G-U mismatch, which AID creates frequently by deaminating cytidine, entrains further mutations. This process of mutagenesis has implications for non-B cells, because all cells must cope with the spontaneous deamination of cytidine. Although the spontaneous rate of deamination is low, the target size (all of the C's in the genome) is large, so the mutational load potentially is high. If MMR and UNG were to act on these spontaneous G-U mismatches in the same manner as that in Ig genes, the system would extend mutagenesis beyond the G·U lesion, clearly a deleterious situation for non-Ig cells. We can anticipate from this apparent paradox that some method of avoiding the hypermutation problem has evolved. The solution to this puzzle will prove very interesting to elucidate.
Supplementary Material
Acknowledgments
We are grateful to M. Ratcliffe, S. Lewis, G. Wu, and the Martin laboratory for helpful discussions and to Maribel Berru for technical help. We thank J. Di Noia for the UGI plasmid, N. Rosenberg for the Abelson murine pre-B cell lines, C. Her for the pCA-OF plasmid, and H. Ming, U. Storb, C. Rada, and M. Neuberger for sequence data and reagents.
This research was supported by a grant from the Canadian Institute of Health Research (165697) to A.M., who also is supported by a Canada Research Chair award. M.L. is supported by a Terry Fox fellowship through the National Cancer Institute of Canada.
The authors have no conflicting financial interests, and all agreed to the submission of the manuscript.
Footnotes
Published ahead of print on 13 July 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Arakawa, H., G. L. Moldovan, H. Saribasak, N. N. Saribasak, S. Jentsch, and J. M. Buerstedde. 2006. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 4e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ariizumi, K., Z. Wang, and P. W. Tucker. 1993. Immunoglobulin heavy chain enhancer is located near or in an initiation zone of chromosomal DNA replication. Proc. Natl. Acad. Sci. USA 903695-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell, P. D., A. Martin, E. Wong, Z. Li, W. Edelmann, and M. D. Scharff. 2003. Cutting edge: the G-U mismatch glycosylase methyl-CpG binding domain 4 is dispensable for somatic hypermutation and class switch recombination. J. Immunol. 1701620-1624. [DOI] [PubMed] [Google Scholar]
- 4.Bardwell, P. D., C. J. Woo, K. Wei, Z. Li, A. Martin, S. Z. Sack, T. Parris, W. Edelmann, and M. D. Scharff. 2004. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 5224-229. [DOI] [PubMed] [Google Scholar]
- 5.Cascalho, M., J. Wong, C. Steinberg, and M. Wabl. 1998. Mismatch repair co-opted by hypermutation. Science 2791207-1210. [DOI] [PubMed] [Google Scholar]
- 6.Delbos, F., S. Aoufouchi, A. Faili, J. C. Weill, and C. A. Reynaud. 2007. DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 20417-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delbos, F., A. De Smet, A. Faili, S. Aoufouchi, J. C. Weill, and C. A. Reynaud. 2005. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2011191-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Noia, J., and M. S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 41943-48. [DOI] [PubMed] [Google Scholar]
- 9.Diaz, M., L. K. Verkoczy, M. F. Flajnik, and N. R. Klinman. 2001. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase zeta. J. Immunol. 167327-335. [DOI] [PubMed] [Google Scholar]
- 10.Ehrenstein, M. R., and M. S. Neuberger. 1999. Deficiency in msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 183484-3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faili, A., S. Aoufouchi, Q. Gueranger, C. Zober, A. Leon, B. Bertocci, J. C. Weill, and C. A. Reynaud. 2002. AID-dependent somatic hypermutation occurs as a DNA single-strand event in the BL2 cell line. Nat. Immunol. 3815-821. [DOI] [PubMed] [Google Scholar]
- 12.Gibbs, P. E., and C. W. Lawrence. 1995. Novel mutagenic properties of abasic sites in Saccharomyces cerevisiae. J. Mol. Biol. 251229-236. [DOI] [PubMed] [Google Scholar]
- 13.Haracska, L., I. Unk, R. E. Johnson, E. Johansson, P. M. Burgers, S. Prakash, and L. Prakash. 2001. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 15945-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen, J. G., P. Langerak, A. Tsaalbi-Shtylik, P. van den Berk, H. Jacobs, and N. de Wind. 2006. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 203319-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadyrov, F. A., L. Dzantiev, N. Constantin, and P. Modrich. 2006. Endonucleolytic function of MutLα in human mismatch repair. Cell 126297-308. [DOI] [PubMed] [Google Scholar]
- 16.Kim, N., G. Bozek, J. C. Lo, and U. Storb. 1999. Different mismatch repair deficiencies all have the same effects on somatic hypermutation: intact primary mechanism accompanied by secondary modifications. J. Exp. Med. 19021-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krokan, H., and C. U. Wittwer. 1981. Uracil DNA-glycosylase from HeLa cells: general properties, substrate specificity and effect of uracil analogs. Nucleic Acids Res. 92599-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunkel, T. A., and D. A. Erie. 2005. DNA mismatch repair. Annu. Rev. Biochem. 74681-710. [DOI] [PubMed] [Google Scholar]
- 19.Langerak, P., A. O. Nygren, P. H. Krijger, P. C. van den Berk, and H. Jacobs. 2007. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2041989-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, Z., C. Zhao, M. D. Iglesias-Ussel, Z. Polonskaya, M. Zhuang, G. Yang, Z. Luo, W. Edelmann, and M. D. Scharff. 2006. The mismatch repair protein Msh6 influences the in vivo AID targeting to the Ig locus. Immunity 24393-403. [DOI] [PubMed] [Google Scholar]
- 21.Martin, A., P. D. Bardwell, C. J. Woo, M. Fan, M. J. Shulman, and M. D. Scharff. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature 415802-806. [DOI] [PubMed] [Google Scholar]
- 22.Martin, A., Z. Li, D. P. Lin, P. D. Bardwell, M. D. Iglesias-Ussel, W. Edelmann, and M. D. Scharff. 2003. Msh2 ATPase activity is essential for somatic hypermutation at A-T basepairs and for efficient class switch recombination. J. Exp. Med. 1981171-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martomo, S. A., D. Fu, W. W. Yang, N. S. Joshi, and P. J. Gearhart. 2005. Deoxyuridine is generated preferentially in the nontranscribed strand of DNA from cells expressing activation-induced cytidine deaminase. J. Immunol. 1747787-7791. [DOI] [PubMed] [Google Scholar]
- 24.Mayorov, V. I., I. B. Rogozin, L. R. Adkison, and P. J. Gearhart. 2005. DNA polymerase eta contributes to strand bias of mutations of A versus T in immunoglobulin genes. J. Immunol. 1747781-7786. [DOI] [PubMed] [Google Scholar]
- 25.Modrich, P., and R. Lahue. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65101-133. [DOI] [PubMed] [Google Scholar]
- 26.Mojas, N., M. Lopes, and J. Jiricny. 2007. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 213342-3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohm-Laursen, L., and T. Barington. 2007. Analysis of 6912 unselected somatic hypermutations in human VDJ rearrangements reveals lack of strand specificity and correlation between phase II substitution rates and distance to the nearest 3′ activation-induced cytidine deaminase target. J. Immunol. 1784322-4334. [DOI] [PubMed] [Google Scholar]
- 28.Pavlov, Y. I., I. M. Mian, and T. A. Kunkel. 2003. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr. Biol. 13744-748. [DOI] [PubMed] [Google Scholar]
- 29.Peled, J. U., F. L. Kuang, M. D. Iglesias-Ussel, S. Roa, S. L. Kalis, M. F. Goodman, and M. D. Scharff. 2008. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 26481-511. [DOI] [PubMed] [Google Scholar]
- 30.Phung, Q. H., D. B. Winter, A. Cranston, R. E. Tarone, V. A. Bohr, R. Fishel, and P. J. Gearhart. 1998. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. J. Exp. Med. 1871745-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poltoratsky, V., M. F. Goodman, and M. D. Scharff. 2000. Error-prone candidates vie for somatic mutation. J. Exp. Med. 192F27-F30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rada, C., J. M. Di Noia, and M. S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell 16163-171. [DOI] [PubMed] [Google Scholar]
- 33.Rada, C., G. T. Williams, H. Nilsen, D. E. Barnes, T. Lindahl, and M. S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 121748-1755. [DOI] [PubMed] [Google Scholar]
- 34.Roa, S., E. Avdievich, J. U. Peled, T. Maccarthy, U. Werling, F. L. Kuang, R. Kan, C. Zhao, A. Bergman, P. E. Cohen, W. Edelmann, and M. D. Scharff. 2008. Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc. Natl. Acad. Sci. USA 10516248-16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogozin, I. B., Y. I. Pavlov, K. Bebenek, T. Matsuda, and T. A. Kunkel. 2001. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2530-536. [DOI] [PubMed] [Google Scholar]
- 36.Schrader, C. E., J. E. Guikema, E. K. Linehan, E. Selsing, and J. Stavnezer. 2007. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J. Immunol. 1796064-6071. [DOI] [PubMed] [Google Scholar]
- 37.Shen, H. M., A. Tanaka, G. Bozek, D. Nicolae, and U. Storb. 2006. Somatic hypermutation and class switch recombination in Msh6−/− Ung−/− double-knockout mice. J. Immunol. 1775386-5392. [DOI] [PubMed] [Google Scholar]
- 38.Unniraman, S., and D. G. Schatz. 2007. Strand-biased spreading of mutations during somatic hypermutation. Science 3171227-1230. [DOI] [PubMed] [Google Scholar]
- 39.Vo, A. T., F. Zhu, X. Wu, F. Yuan, Y. Gao, L. Gu, G. M. Li, T. H. Lee, and C. Her. 2005. hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair. EMBO Rep. 6438-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei, K., A. B. Clark, E. Wong, M. F. Kane, D. J. Mazur, T. Parris, N. K. Kolas, R. Russell, H. Hou, B. Kneitz, G. Yang, T. A. Kunkel, R. D. Kolodner, P. E. Cohen, and W. Edelmann. 2003. Inactivation of exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 17603-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiesendanger, M., B. Kneitz, W. Edelmann, and M. D. Scharff. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191579-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson, T. M., A. Vaisman, S. A. Martomo, P. Sullivan, L. Lan, F. Hanaoka, A. Yasui, R. Woodgate, and P. J. Gearhart. 2005. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J. Exp. Med. 201637-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao, Z., M. Ray, C. Jiang, A. B. Clark, I. B. Rogozin, and M. Diaz. 2007. Known components of the immunoglobulin A:T mutational machinery are intact in Burkitt lymphoma cell lines with G:C bias. Mol. Immunol. 442659-2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yabuki, M., M. M. Fujii, and N. Maizels. 2005. The MRE11-RAD50-NBS1 complex accelerates somatic hypermutation and gene conversion of immunoglobulin variable regions. Nat. Immunol. 6730-736. [DOI] [PubMed] [Google Scholar]
- 45.Zan, H., A. Komori, Z. Li, A. Cerutti, A. Schaffer, M. F. Flajnik, M. Diaz, and P. Casali. 2001. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity 14643-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng, X., G. A. Negrete, C. Kasmer, W. W. Yang, and P. J. Gearhart. 2004. Absence of DNA polymerase eta reveals targeting of C mutations on the nontranscribed strand in immunoglobulin switch regions. J. Exp. Med. 199917-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng, X., D. B. Winter, C. Kasmer, K. H. Kraemer, A. R. Lehmann, and P. J. Gearhart. 2001. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2537-541. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, W., P. D. Bardwell, C. J. Woo, V. Poltoratsky, M. D. Scharff, and A. Martin. 2001. Clonal instability of V region hypermutation in the Ramos Burkitt's lymphoma cell line. Int. Immunol. 131175-1184. [DOI] [PubMed] [Google Scholar]
- 49.Zhou, J., N. Ashouian, M. Delepine, F. Matsuda, C. Chevillard, R. Riblet, C. L. Schildkraut, and B. K. Birshtein. 2002. The origin of a developmentally regulated Igh replicon is located near the border of regulatory domains for Igh replication and expression. Proc. Natl. Acad. Sci. USA 9913693-13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.