

Abstract
The circadian clock is encoded by a transcription-translation feedback loop that synchronizes behavior and metabolism with the light-dark cycle. Here we report that both the rate-limiting enzyme in mammalian NAD+ biosynthesis, nicotinamide phosphoribosyltransferase (NAMPT), and levels of NAD+, display circadian oscillations which are regulated by the core clock machinery in mice. Inhibition of NAMPT promotes oscillation of the clock gene Per2 by releasing CLOCK:BMAL1 from suppression by SIRT1. In turn, the circadian transcription factor CLOCK binds to and up-regulates Nampt, thus completing a feedback loop involving NAMPT/NAD+ and SIRT1/CLOCK:BMAL1.
Many aspects of mammalian behavior and physiology are coordinated through interconnected networks of 24 hr central and peripheral oscillators that synchronize cycles of fuel storage and utilization to maintain organismal homeostasis. In mice, circadian disruption has been tied to metabolic disturbance (1, 2), while conversely, high-fat diet alters both behavioral and molecular rhythms (3, 4). The underlying mechanism of the mammalian clock consists of a transcription-translation feedback loop in which CLOCK and BMAL1 activate transcription of Cryptochrome (Cry1 and 2) and Period (Per1, 2, and 3), leading to subsequent repression of CLOCK:BMAL1 by CRY and PER proteins (5). An additional feedback loop involves the transcriptional regulation of Bmal1 by RORα and REV-ERBα (6, 7). Previous studies have also implicated a role for cellular NAD+ in the regulation of CLOCK and NPAS2 activity (8), an observation consistent with the recent finding that the NAD+-dependent protein deacetylase SIRT1 modulates activity of the clock complex (9, 10).
NAD+ is a classic co-enzyme that is synthesized from three major precursors – tryptophan, nicotinic acid, and nicotinamide (Fig. S1). In yeast, it has been reported that enzymes involved in the NAD+ salvage pathway play an important role in the regulation of Sir2 activity (11, 12). On the other hand, in mammals, the predominant NAD+ biosynthetic pathway involves the conversion of nicotinamide and 5′-phosphoribosyl-pyrophosphate to nicotinamide mononucleotide (NMN) by the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT) (Fig. S1) (13-15). NAMPT-mediated NAD+ biosynthesis has been demonstrated to play a critical role in a number of biological processes through the NAD+-dependent deacetylase SIRT1 (Fig. S1) (13, 16-18). These findings led us to hypothesize that circadian regulation of NAD+ may provide a mechanism for regulation of the core clock. Expression levels of Nampt RNA displayed a robust diurnal pattern in both wild-type mouse liver and white adipose tissue (WAT), peaking at the beginning of the dark period (zeitgeber time (ZT) 14) (Fig. 1A). Expression levels of NAMPT protein also showed a diurnal pattern of oscillation across the 24 hr light-dark cycle in liver (Fig. 1B), with a reduction in NAMPT protein levels prior to the onset of the dark period (Fig. 1B; Supporting Description 1). Nampt RNA oscillation is circadian in nature, as we found a robust oscillation of Nampt RNA for 48 hr in liver isolated from wild-type mice that had been maintained in constant darkness (p<0.01, one-way ANOVA) (Fig. 1C). Furthermore, the levels of Nampt RNA and protein were lower across the entire light-dark cycle in liver and WAT of ClockΔ19 mutant mice, and the diurnal oscillation of Nampt RNA and protein was abolished in ClockΔ19 mutant mice (Fig. 1A-B). The robust Nampt RNA oscillation during constant darkness was also abolished completely in ClockΔ19 mutant mouse liver (Fig. 1C), indicating that the core clock machinery is required for the circadian control of NAMPT expression.
Fig. 1.
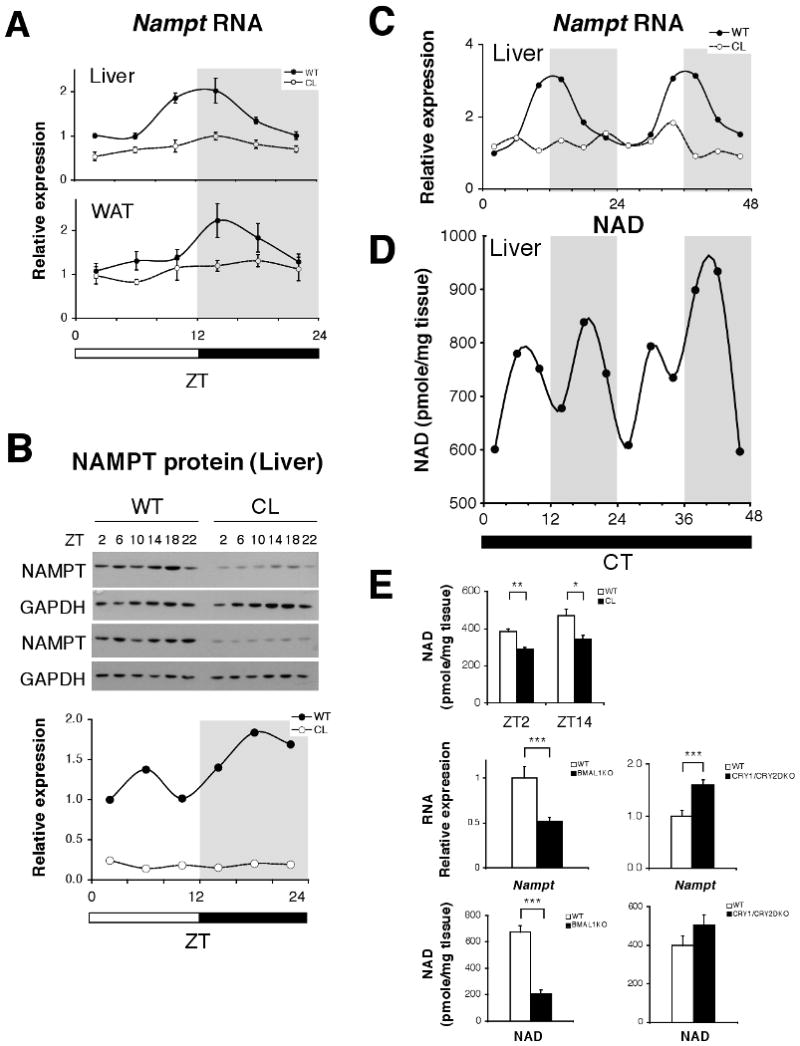
Oscillation of NAMPT-mediated NAD+ biosynthesis. (A) Relative expression levels of Nampt RNA in liver and white adipose tissue (WAT) across the 24 hr light:dark cycle. Gray shading indicates dark period (n=4-6 mice/genotype/time point). (B) Western blots showing NAMPT and GAPDH at indicated ZTs. Quantitation of NAMPT normalized to GAPDH. (C) Relative expression levels of Nampt RNA across 48 hrs in liver of WT and ClockΔ19 mice in constant darkness. Shading indicates where light and dark periods would normally occur under 12:12 LD conditions. (n=2 WT, n=1 ClockΔ19/time point); (p<0.01, one-way ANOVA for WT oscillation). (D) NAD+ levels across 48 hrs in liver of WT mice in constant darkness. (n=2 WT/time point). (E) NAD+ levels in WT and ClockΔ19 mutant liver at ZT2 and 14 (n=3) (upper panel). Relative expression levels of Nampt RNA (middle panels) and NAD+ (lower panels) in Bmal1-/-, Cry1-/-/Cry2-/-, and WT liver at ZT7 (n=4-13). *p<0.05; **p<0.01; ***p<0.001. All data is presented as mean ± SEM.
Because NAMPT is the rate-limiting enzyme in the predominant NAD+ biosynthetic pathway in mammals, we tested whether tissue NAD+ levels also display a circadian oscillation pattern in wild-type liver across 48 hrs from mice maintained in constant darkness. Liver NAD+ levels showed a very similar bimodal circadian oscillation pattern to that of NAMPT protein, and the lowest levels of NAD+ occurred with a rhythmicity of 24 hrs (Fig. 1D). A decrease in NAD+ levels was also observed around CT10-14 and again 24 hrs later (CT34), generating the observed bimodal oscillation pattern (Fig. 1D; Supporting Description 1). The degree to which NAD+ levels increased from baseline (40-53%) during these daily cycles, and the concentrations of NAD+ that we measured, fall within the physiological range of reported alterations in NAD+ levels (Supporting Description 2) (19, 20).
NAD+ levels were significantly reduced in ClockΔ19 mutant liver during both the light (ZT2) and dark (ZT14) periods (Fig. 1E). Furthermore, we observed that mice deficient in Bmal1 (Bmal1-/-) (21), the heterodimeric binding partner of CLOCK, also exhibited a significant reduction in both Nampt RNA and NAD+ levels in liver (Fig. 1E). Finally, mice deficient for both CRY1 and CRY2 (22), displayed a significant increase in Nampt RNA levels and a corresponding increase in NAD+ levels in liver, which is consistent with Nampt being a target gene of CLOCK:BMAL1 (Fig. 1E). The rhythmic oscillation in RNA and protein levels of NAMPT thus leads to a circadian oscillation of NAD+ levels in the living animal.
The peak in NAMPT and NAD+ in liver of wild-type mice (Fig. 1B, D-E) is consistent with a previous report that endogenous SIRT1, an NAD+-dependent and nutrient-responsive deacetylase, displays a diurnal oscillation in activity with a peak around ZT15 (9). We therefore examined whether SIRT1 activity is affected by altering NAMPT-mediated NAD+ biosynthesis in primary hepatocytes using the specific chemical inhibitor of NAMPT, FK866 (23). FK866 significantly inhibited NAD+ biosynthesis (Fig. 2A), resulting in ∼30% reduction in mRNA expression levels of two major SIRT1 target genes in the liver, Pepck (phosphoenolpyruvate carboxykinase) and G6Pase (glucose-6-phosphatase) (19), but not an unrelated gene, Igf-1 (insulin-like growth factor-1) (Fig. 2B). Using Per2:luciferase transcriptional reporter assays in HEK293 cells (Fig. 2C-E; S2), we show that inhibition of NAMPT by FK866 led to a significant increase in the CLOCK:BMAL1-driven transcription of the Per2:luciferase reporter (Fig. 2C), indicating that reduced NAMPT-mediated NAD+ biosynthesis released CLOCK:BMAL1 from the SIRT1-dependent suppression. Consistent with this notion, SIRT1 suppressed transcription of Per2:luciferase in a dose-dependent manner (Fig. 2D). Additionally, resveratrol, a putative SIRT1 activator, also inhibited Per2:luciferase transcription (Fig. S2, S3A). In contrast, inhibition of SIRT1 by high doses of nicotinamide activated Per2:luciferase transcription, and the selective SIRT1 inhibitor EX-527 showed a similar trend (Fig. S2, S3B-C). Furthermore, FK866 abrogated the SIRT1-dependent suppression of CLOCK:BMAL1-mediated transcription (Fig. 2E).
Fig. 2.

NAMPT, NAD+, and SIRT1 regulate CLOCK:BMAL1 activity. (A-B) NAD+ levels (A) and relative expression levels of Pepck, G6Pase, and Igf-1 RNA (B) in primary hepatocytes incubated with 200nM FK866 for 24 hours (n=3/group). (Pepck, phosphoenolpyruvate carboxykinase; G6Pase, glucose-6-phosphatase; Igf-1, insulin-like growth factor-1). (C-E) Relative luciferase activity of Per2-luciferase in the presence of: (C) 10nM FK866 for 24 or 48 hours, (D) increasing doses of Sirt1 (200, 400, 800 ng), and (E) Sirt1 (800 ng) alone or Sirt1 (800 ng) and 10nM FK866 for 48 hours. *p<0.05; **p<0.01; ***p<0.001; NS not significant. All data is presented as mean ± SEM.
To investigate the effect of this regulatory pathway on clock gene expression, we first confirmed that SIRT1 binds to BMAL1 (Fig. S4) (9, 10), and then determined that SIRT1 localizes to the E-box of the Per2 promoter (Fig. 3A), suggesting that Per2 is a target of the NAMPT/NAD+-driven pathway. We next examined the effect of FK866 on Per2 rhythms in primary hepatocytes. Serum shock induced a Per2 oscillation in vehicle-treated hepatocytes that damped rapidly after one cycle (2.2 day damping rate), whereas FK866 treatment prolonged the oscillation of Per2 (5.3 day damping rate) (Fig. 3B). Reduction in NAMPT-mediated NAD+ biosynthesis may promote a more robust oscillation of clock target gene expression by releasing CLOCK:BMAL1 from SIRT1-mediated suppression. Consistent with this notion, adenoviral Sirt1 transduction of primary hepatocytes suppressed the expression and the oscillation of Per2, although adenoviral infection itself altered the Per2 oscillation pattern (Fig. 3C). Therefore, the NAMPT/NAD+-driven pathway modulates circadian transcription patterns in mammals.
Fig. 3.
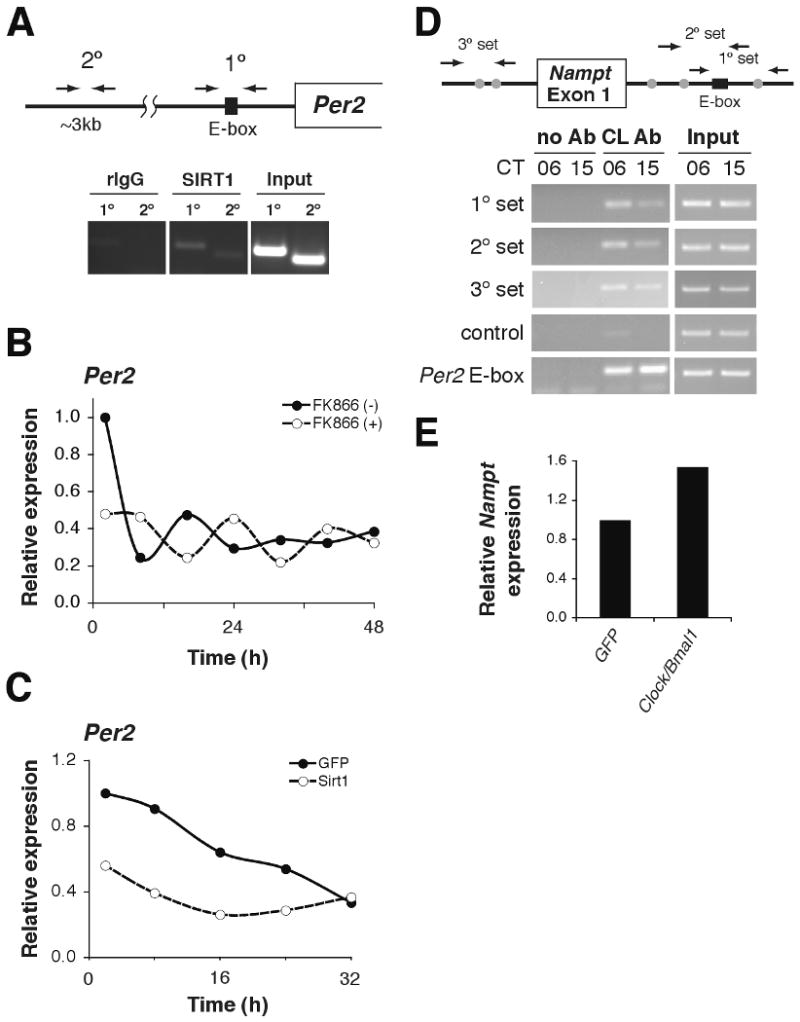
NAMPT/NAD+-driven feedback loop through SIRT1/CLOCK:BMAL1. (A) ChIP assays in primary hepatocytes for Per2 with rIgG control (left), SIRT1 antibody (middle), and input (right). Primer locations are schematically shown above. (B-C) Relative expression levels of Per2 RNA in primary hepatocytes following serum shock. Hepatocytes were either (B) incubated with 200nM FK866 or (C) infected with SIRT1- and GFP (control)-expressing adenovirus (average of 2 independent experiments). (D) ChIP assays in liver isolated from mice at CT6 and CT15 for Nampt with no Ab control (left), CLOCK antibody (middle), and input (right). Primer locations are shown schematically above. The black box indicates a canonical E-box, while gray circles represent non-canonical E-boxes. Control primers are located ∼5kb upstream of the Nampt transcriptional start site. Per2 E-box primers are included as a positive control. (E) Relative Nampt expression levels in adenovirally GFP- and Clock/Bmal1-infected mouse embryonic fibroblasts (average of 2 independent experiments).
To determine if this pathway comprises a feedback loop involving regulation of the Nampt gene by CLOCK:BMAL1, we performed chromatin immunoprecipitation (ChIP) to test whether the CLOCK:BMAL1 complex binds to canonical or non-canonical E-box motifs in the promoter and first intron of the Nampt gene (Fig. S5). ChIP detected CLOCK in these regions (Fig. 3D, first, second, and third panels), but not in a non-specific upstream region (Fig. 3D, fourth panel). We observed a stronger signal for CLOCK at CT6 compared to CT15 (Fig. 3D), suggesting rhythmic binding of CLOCK:BMAL1 in a promoter context-dependent manner (24, 25). Transduction of Clock/Bmal1 into mouse embryonic fibroblasts appeared to up-regulate Nampt expression ∼1.6-fold (Fig. 3E). Together, these data demonstrate that NAMPT-mediated NAD+ biosynthesis comprises a feedback loop in which NAD+ functions as a metabolic oscillator and regulates the core clock machinery through SIRT1 (Fig. S6; Supporting Description 3). Given the central role of NAD+ as a biological cofactor, the rhythmic production of NAD+ may have a cascade of effects on downstream pathways, including chromatin regulation, metabolism, and aging. We thus propose that the circadian feedback loop through NAMPT-mediated NAD+ biosynthesis may function to fine-tune the daily cycles of energy storage and utilization and to coordinate these processes with the rest-activity cycle.
Supplementary Material
Acknowledgments
We thank Seung-Hee Yoo and members of the Bass, Imai, and Takahashi laboratories for discussions. Work was supported by grants from NIDDK (T32 DK007169) to K.M.R.; NIA (AG02150), Ellison Medical Foundation, Longer Life Foundation to S.I.; NIH (PO1 AG011412), Chicago Biomedical Consortium Searle Funds, and JDRF to J.B; and (P50 MH074924) to J.S.T. J.Y. is supported by the Japan Research Foundation for Clinical Pharmacology and Keio University Medical Science Fund. J.S.T. is an Investigator in the Howard Hughes Medical Institute and a cofounder of ReSet Therapeutics Inc., and J.S.T. and J.B. are members of its scientific advisory board. J.B. is also an advisor and receives support from Amylin Pharmaceuticals. S.I. holds a patent related to the work reported in this paper, specifically NAMPT and NMN.
References
- 1.Turek FW, et al. Science. 2005;308:1043. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudic RD, et al. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohsaka A, et al. Cell Metab. 2007;6:414. doi: 10.1016/j.cmet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Barnea M, Madar Z, Froy O. Endocrinology. 2009;150:161. doi: 10.1210/en.2008-0944. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi JS, Hong HK, Ko CH, McDearmon EL. Nat Rev Genet. 2008;9:764. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preitner N, et al. Cell. 2002;110:251. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 7.Sato TK, et al. Neuron. 2004;43:527. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 8.Rutter J, Reick M, Wu LC, McKnight SL. Science. 2001;293:510. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- 9.Nakahata Y, et al. Cell. 2008;134:329. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asher G, et al. Cell. 2008;134:317. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 11.Anderson RM, et al. J Biol Chem. 2002;277:18881. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 12.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nature. 2003;423:181. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Revollo JR, Grimm AA, Imai S. J Biol Chem. 2004;279:50754. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 14.Revollo JR, et al. Cell Metab. 2007;6:363. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai S. Curr Pharm Des. 2009;15:20. doi: 10.2174/138161209787185814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Veer E, et al. J Biol Chem. 2007;282:10841. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 17.Fulco M, et al. Dev Cell. 2008;14:661. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H, et al. Cell. 2007;130:1095. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodgers JT, et al. Nature. 2005;434:113. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 20.Chen D, et al. Genes Dev. 2008;22:1753. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bunger MK, et al. Cell. 2000;103:1009. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitaterna MH, et al. Proc Natl Acad Sci U S A. 1999;96:12114. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasmann M, Schemainda I. Cancer Res. 2003;63:7436. [PubMed] [Google Scholar]
- 24.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Cell. 2001;107:855. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 25.Ripperger JA, Schibler U. Nat Genet. 2006;38:369. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.