

Abstract
The special blood group antigen Mi.III exhibits a characteristic hybrid structure of glycophorin A (GPA) and glycophorin B, termed Gp.Mur. This phenotype has exceptionally high occurrence rates in several indigenous tribes in Taiwan (∼21.2%-88.4%). Because glycophorin/Miltenberger begins interaction with anion exchanger-1 (AE1) in the endoplasmic reticulum, we hypothesized that the AE1-based macrocomplexes on erythrocyte membranes obtained from Mi.III+ people could be differentiated from those obtained from non-Miltenberger people. Quantitative mass spectrometric comparison of the AE1-based complexes by iTRAQ™ (Applied Biosystems) revealed 25% to 67% higher expression of AE1 in Mi.III+ erythrocytes. In accordance with the higher AE1 level, the Mi.III+ erythrocytes exhibited superior HCO3− capacities, pH homeostasis, and osmotic resistance. Cotransfection experiments in HEK293 cells showed that Gp.Mur, like GPA, enhanced trafficking of AE1 to the plasma membrane. In summary, the increased surface expression of AE1 in Mi.III+ erythrocytes could be attributed to the additive effect of GPA and Gp.Mur coexpression.
Introduction
Miltenberger antigens belong to the complex MNS blood group system.1 They most likely evolved from specific gene mutation or crossover events of homologous glycophorin A (GYPA), glycophorin B (GYPB), and glycophorin E (GYPE). The occurrence frequencies of the Miltenberger blood group antigens such as type III (Mi.III) are extremely low in white2 (0.0098%) and Japanese (0.006%) people. However, the frequencies are exceptionally high in several Taiwanese aboriginal tribes (up to 90%), compared with 2% to 3% in Han Taiwanese (Minnan).3 Mi.III exhibits a unique glycophorin hybrid peptide (Gp.Mur), the likely product of specific gene insertion of GYPA into GYPB (denoted BAB as in Figure 1A).4 Because transfusion with incompatible Miltenberger blood could result in severe hemolytic diseases,5–8 blood bank screening of the Miltenberger phenotypes before transfusion is required in Taiwan.
Figure 1.

The expression levels of GPB and Gp.Mur in Mi.III+ RBCs were complementary. (A) Mi.III-specific Gp.Mur probably evolved from homologous gene recombination between GYPA and GYPB, and shows a unique glycophorin B-A-B structure and the antigenic Mur (marked as checkered). The protein sequences of full-length Gp.Mur and GPB, and GPA lacking a cytoplasmic domain, were aligned by the CLUSTALW program. (B) Glycophorin immunoblot of ghost lysates from 6 Mi.III+ and 6 non-Mi.III (control) samples (30 mg/lane). Solubilized ghosts were resolved on 10% SDS–polyacrylamide gel electrophoresis. (Left) GPA immunoblot by E4 antibody. (Right) Immunoblot against GAB by E3 antibody. Homozygous Mi.III samples are marked +/+.
Previous work has shown that the glycophorin A (GPA) interacts with glycophorin B (GPB) and band 3 (also known as anion exchanger-1 [AE1]), as well as Rh polypeptides, aquaporin, glucose transporter type I, 4.1R, and spectrin, among other proteins in large macromolecular complexes.9–11 Bruce et al have suggested that these individual protein components or complexes function interactively as a “gas-exchange metabolon,”11 in which AE1 acts as the central pillar for the entire assembly. The “macrocomplex” concept implies that variations among individual components or complexes are interconnected.
GPA exerts a stabilizing effect on AE1 expression, specifically by assisting the folding and maturation, and by enhancing the anion exchange activity of AE1.12–14 GPA and AE1 begin coassembly in the endoplasmic reticulum and Golgi, and their protein expression is tightly coupled based on the studies of AE1 knockout mice and human GPA transgenic mice.10,15 In contrast, GPB that is highly homologous to GPA, but lacks a cytoplasmic domain,16 does not seem to influence AE1 activities when expressed in Xenopus oocytes.12 The function of GPB remains unclear.17
In this study, we sought to identify the structural and functional impact of the Mi.III blood type commonly observed among Taiwanese. We reasoned that the hybrid structure of Gp.Mur might engender compositional or structural differences in the AE1-based complexes, which, in turn, might manifest differences in erythrocyte membrane functions. By comparing the protein compositions of AE1-based complexes in erythrocyte ghosts obtained from Mi.III+ and non-Miltenberger (control) people, we found a significant increase of AE1 on Mi.III+ membrane. Their higher AE1 level was correlated with functional changes, including superior HCO3−-transporting capacities, acid-base homeostasis, and osmotic resistance, which contrast with the phenotype of certain kinds of hereditary spherocytosis characterized by a marked reduction of AE1 expression. By unveiling the functional relevance of the Miltenberger antigen, our work suggests that its evolutional emergence is beneficial.
Methods
Red blood cell samples
The Mackay Memorial Hospital Institutional Review Board has approved the collection of human blood from consented donors free of infectious diseases. All donors provided informed consent in accordance with the Declaration of Helsinki. The Mi.III phenotype was verified serologically using anti-Mia, anti-Mur, anti-Hil, and anti-Anek antisera (Table 1). Mi.III homozygosity (Mi.III++) was identified by the presence of Gp.Mur and the absence of GPB.
Table 1.
Electrolyte and RBC evaluation for Mi.III+ and control red cells
| RBC count (106/μL) | MCV (fL) | Plasma Cl− (mM) | Plasma K+ (mM) | Plasma Na+ (mM) | Cholesterol (mg/dL) | HDL (mg/dL) | LDL (mg/dL) | Triglyceride (mg/dL) | |
|---|---|---|---|---|---|---|---|---|---|
| Control | 4.48 ± 0.34 (5) | 84.65 ± 9.6 (5) | 106.0 ± 3.4 (8) | 4.05 ± 0.1 (8) | 138 ± 3.3 (8) | 196.3 ± 45.0 (8) | 57.3 ± 20.0 (8) | 122.5 ± 30.3 (8) | 114.5 ± 94.8 (8) |
| Mi.III | 4.40 ± 0.21 (6) | 88.95 ± 6.57 (6) | 104.3 ± 3.7 (10) | 4.13 ± 0.4 (10) | 139.7 ± 3.6 (10) | 195.3 ± 35.4 (10) | 57.3 ± 8.1 (10) | 118.6 ± 41.6 (10) | 82.0 ± 48.9 (10) |
Data are expressed as mean ± SD. The number of tested samples is noted within parentheses.
MCV indicates mean corpuscular volume; HDL, high-density lipoproteins; and LDL, low-density lipoproteins.
Immunoprecipitation
Anti-AE1 monoclonal antibodies used include AE12-M (Alpha Diagnostic), BRIC170, and BRIC71 (Bristol Institute for Transfustion Sciences [BITS]). Anti-GPA and GPB monoclonal antibodies include E318 (Sigma-Aldrich) and R1.319 (BITS). E3 recognizes a nonglycosylated, homologous region close to the transmembrane segment (residues 61-64 or 64-67 [GPA]; residues 32-35 [GPB]),18 whereas R1.3 recognizes the N-terminal nonsialylated residues common to GPA and GPB.19 Erythrocyte ghosts were solubilized with an equal volume of the doubly concentrated lysis buffer containing 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 2% Nonidet P-40, 0.05% sodium dodecyl sulfate (SDS), phosphate-buffered saline, and complete protease inhibitor mixtures (Calbiochem). Anti-AE1 was dimethyl pimelimidate dihydrochloride cross-linked to Dynabeads; equal quantities of the ghost lysates (usually 1 mg per sample) were mixed with the beads for immunoprecipitation at 4°C for 12 to 16 hours, as described previously.20
Quantitative mass spectrometry by iTRAQ™
For iTRAQ™, 4 mg of the ghost lysates per sample was used as the starting materials for immunoprecipitation (IP). To facilitate mass spectrometry–based protein identification, coimmunoprecipitated carbohydrates were removed by chemical and enzymatic deglycosylation (supplemental Figure 1A-B, available on the Blood website; see the Supplemental Materials link at the top of the online article). The samples were subsequently trichloracetic acid precipitated, individually resolubilized, reduced, alkylated, and digested with trypsin, followed by iTRAQ™ labeling (Applied Biosystems; see supplemental Figure 1). Proteins from the Mi.III samples (tagged with 116- and 117-Da reporter ions) whose ratios relative to the control samples (tagged with 114- and 115-Da reporters) consistently exceeded 1.2 or were less than 0.8 were deemed targets of interest. Further details are in the supplemental Methods.
The DIDS labeling of intact red blood cell surface
Equal numbers of intact erythrocytes were labeled with 5 μM DIDS (4,4′-di-isothiocyanato-2,2′-disulfostilbene) at room temperature for 20 minutes, followed by 2 washes. The amount of DIDS bound to cell surface was measured by a microplate spectrofluorometer (SpectraMAX Gemini XS; Molecular Devices) at 450 nm emission.
Measurement of HCO3−/Cl− transport capacities
HCO3−/Cl− transport across red blood cell (RBC) membrane was assessed by the concentration changes of intracellular Cl− ([Cl−]in) with respect to that of extracellular Cl− ([Cl−]out). Fresh erythrocytes were labeled with 5 mM Cl−-sensitive dye 6-methoxy-N-(3-sulfopropyl) quinolinium (SPQ; Invitrogen), as previously described.21 SPQ fluorescence from wet erythrocytes was excited at 350 nm, and its emission collected at 430 nm. [Cl−]in was calculated based on individual calibration equations.21 Further details are provided in the supplemental Methods.
Intracellular pH measurement by flow cytometry
Fresh erythrocytes were loaded with 1 μM fluorescent pH indicator carboxy SNARF-1 (Invitrogen) for 10 minutes, followed by Hanks balanced salt solution wash. For intracellular pH (pHi) calibration, SNARF-1–loaded cells were incubated with nigericin-containing, high K+ buffer. SNARF-1 fluorescence was excited at 488 nm, and its emission at yellow and red fluorescence channels was collected by FACSCalibur. Because SNARF-1 exhibits a pH-dependent spectral shift, pHi was calculated from the ratios of fluorescence intensities.22 Further details are provided in the supplemental Methods.
Osmotic fragility test
A modified osmotic fragility test was performed to determine the range of tolerable osmotic stresses on erythrocytes. Equal quantities of fresh RBCs were incubated in 0.2% to 1% NaCl for 30 minutes at room temperature. Percentage of hemolysis was recorded after 5 minutes of 100g centrifugation.
Assessment of AE1 expression by FACS
The coding region of human AE1 (GenBank accession M27819) was subcloned into expression vector pCIG (pIRES2-EGFP) to generate pCIG-AE1. Gp.Mur (EU338225) and GPA (NM002099) were individually subcloned into pcDNA3.1. HEK293 cells were transiently transfected with pCIG-AE1 alone, or together with pcDNA-GPA or pcDNA-Gp.Mur, at equimolar concentrations. To assess AE1 surface expression, intact cells were stained with anti-AE1 antibody BRIC71 at 36 to 72 hours posttransfection, followed by secondary staining with Alexa Fluor 660–conjugated anti–mouse antibody (Invitrogen). For total AE1 production, intact cells were fixed and permeabilized first, followed by BRIC71 labeling. Transfected cells were selected for green fluorescent protein expression, and AE1 levels were assessed using FACSCalibur.
Results
Comparison of the AE1-based complexes from Mi.III+ and the control erythrocytes
To characterize the Mi.III blood group proteome, 6 Mi.III (including 2 homozygous Mi.III) and 6 control samples were collected. Their glycophorin profiles were assessed by immunoblot. The E3 monoclonal antibody, which recognizes both GPA and GPB (anti-GAB), showed monomeric and oligomeric GPA, GPB, and Gp.Mur in the ghost lysates (Figure 1B). Most oligomers, that is, GPB-GPB, GPB-Gp.Mur, Gp.Mur-Gp.Mur, GPA-GPA, and GPA-GPB, could be deduced from aligning the GAB and GPA immunoblots. GPB expression in heterozygous Mi.III+ was reduced relative to the control. Homozygous Mi.III+/+ cells were devoid of GPB, but expressed more Gp.Mur than heterozygous Mi.III. This complementarity between the levels of GPB and Gp.Mur suggests that the combined level of GPB and Gp.Mur in Mi.III (or the Gp.Mur level in Mi.III+/+) is probably equivalent to that of GPB in the control. Their GPA levels also appeared consistent.
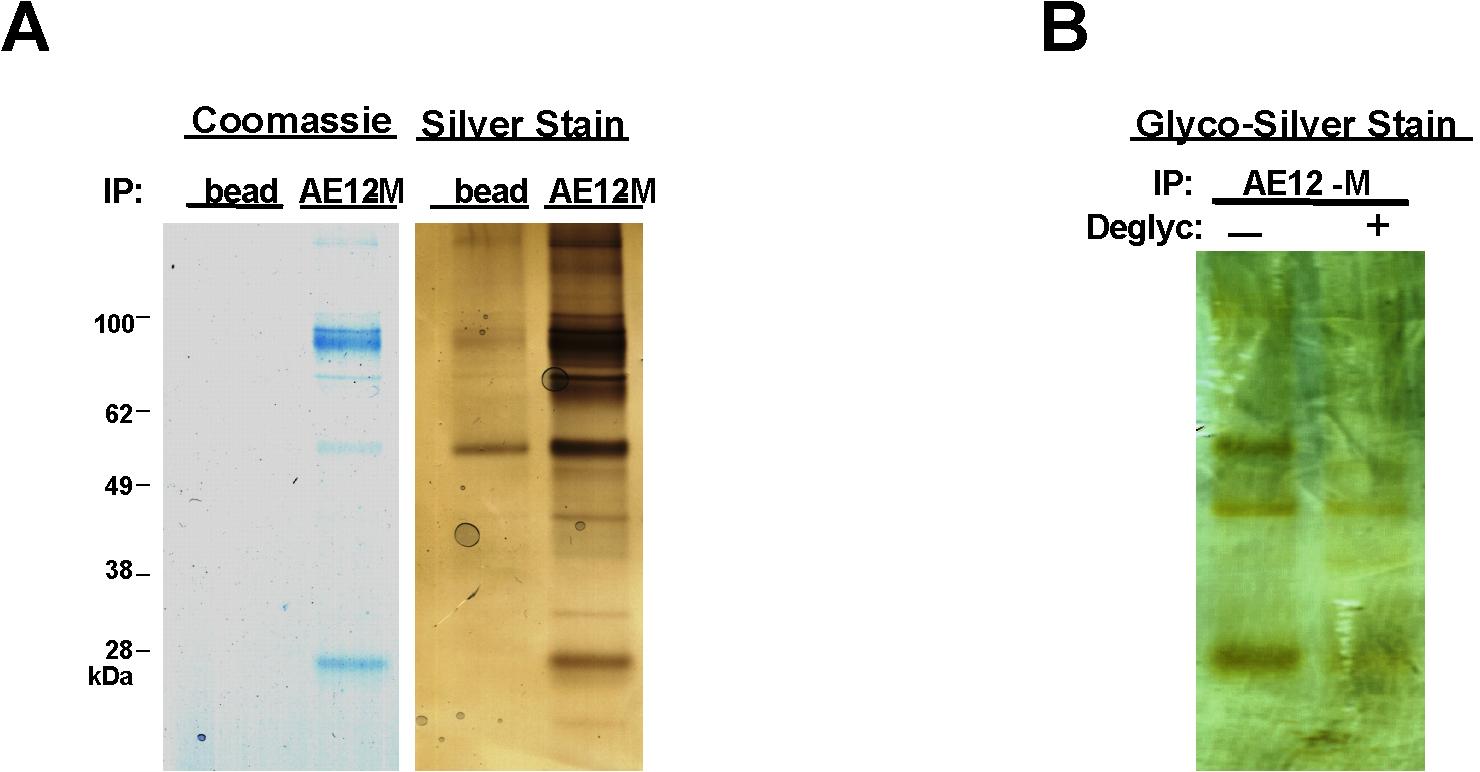
To study the Mi.III blood group proteome comparatively, we used 2 different anti-AE1 antibodies (AE12-M and BRIC170), opting against using glycophorin/Miltenberger antisera in the pulldown experiments to avoid possible selection biases toward glycophorin variants. AE1 immunoprecipitates were deglycosylated and then pooled for iTRAQ™-based quantitative mass spectrometry. The proteins identified in our immunoprecipitates are presented in Table 2. Our results confirm the components of the band 3–based macromolecular complexes identified previously, including Rh polypeptides, α- and β-spectrins, β-hemoglobin, ankyrin-1, glucose transporter type I, 4.1R, and EPB42.11 Among them, α-tropomyosin, band 4.9, aquaporin-1 (AQP1), flotillin-1, flotillin-2, and stomatin were previously unreported (Table 2 and supplemental Table 1). The first 2 components, α-tropomyosin and band 4.9, are established members of the submembranous cytostructural junctional complexes, which additionally include spectrin tetramers, short actin filaments, 4.1R, myosin, and adducin.23 The other newly identified components, flotillins and stomatin, belong to distinct erythrocyte lipid rafts.24,25 Some of these cytoskeleton-associated proteins might be interconnected rather than directly binding to AE1. The mass spectrometry (MS)–identified proteins were validated by immunoblot analysis (Figure 2). We note that certain proteins, like Rh-associated glycoprotein and glycophorins, were present in our experiments (Figures 2–3), but proved recalcitrant to MS/MS identification. Despite our best efforts to deglycosylate the proteins (as shown by glycosilver stain in supplemental Figure 1), residual glycosylation might yet have been a confounder.
Table 2.
Components of glycophorin/band 3 macrocomplexes identified and quantified by iTRAQ™
| No. | % Sequence Coverage | Accession | Name | 115:114 |
116:114 |
117:114 |
|||
|---|---|---|---|---|---|---|---|---|---|
| Ratio | P | Ratio | P | Ratio | P | ||||
| 1 | 33.83 | P02549 SPTA1_HUMAN | Spectrin α chain, erythrocyte | 1.09 | < .005 | 1.06 | .003 | 1.11 | < .005 |
| 2 | 30.93 | P11277 SPTB1_HUMAN | Spectrin β chain, erythrocyte | 1.15 | < .005 | 1.08 | < .005 | 1.12 | < .005 |
| 3 | 25.57 | P16157 ANK1_HUMAN | Ankyrin-1 (erythrocyte ankyrin) | 1.08 | .006 | 1.15 | < .005 | 1.22 | < .005 |
| 4 | 29.42 | P02730 B3AT_HUMAN | Band 3 anion transport protein (anion exchange protein 1) | 0.97 | .361 | 1.67 | < .005 | 1.25 | < .005 |
| 5 | 31.40 | P16452 EPB42_HUMAN | Erythrocyte membrane protein band 4.2 | 1.02 | .797 | 1.14 | .009 | 1.32 | < .005 |
| 6 | 32.00 | P60709 ACTB_HUMAN | Actin, cytoplasmic* | 1.21 | .000 | 1.08 | .101 | 1.26 | < .005 |
| 7 | 27.16 | Q08495 DEMA_HUMAN | Dematin (erythrocyte membrane protein band 4.9) | 1.20 | .158 | 1.11 | .211 | 1.00 | > .999 |
| 8 | 24.07 | P11171 41_HUMAN | Protein 4.1 (band 4.1) | 0.87 | .002 | 1.08 | .018 | 1.13 | .016 |
| 9 | 29.51 | P27105 STOM_HUMAN | Stomatin (protein 7.2b) | 0.98 | .689 | 0.94 | .141 | 1.10 | .014 |
| 10 | 34.30 | Q14254 FLOT2_HUMAN | Flotillin-2 | 1.27 | .026 | 1.02 | .730 | 1.08 | .372 |
| 11 | 9.35 | P11166 GTR1_HUMAN | Solute carrier family 2, facilitated glucose transporter member 1 | 0.83 | .009 | 1.07 | .060 | 1.26 | .000 |
| 12 | 47.57 | P62805 H4_HUMAN | Histone H4† | 0.96 | .520 | 0.51 | .067 | 0.65 | .021 |
| 13 | 13.73 | P04406 G3P_HUMAN | Glyceraldehyde-3-phosphate dehydrogenase | 0.90 | .420 | 0.94 | .261 | 1.07 | .158 |
| 14 | 26.46 | O75955 FLOT1_HUMAN | Flotillin-1 | 0.98 | .794 | 0.89 | .313 | 1.02 | .689 |
| 15 | 22.45 | P68871 HBB_HUMAN | Hemoglobin‡ | 1.06 | .859 | 1.06 | .448 | 0.89 | .289 |
| 16 | 27.78 | Q99877 H2B1N_HUMAN | Histone H2B† | 1.17 | .302 | 0.33 | .011 | 0.57 | .020 |
| 17 | 56.58 | P62988 UBIQ_HUMAN | Ubiquitin | 1.27 | .151 | 1.22 | .159 | 1.16 | .200 |
| 18 | 30.77 | Q6FI13 H2A2A_HUMAN | Histone H2A† | 0.25 | 0.19 | 0.40 | |||
| 19 | 6.71 | Q02161 RHD_HUMAN | Blood group Rh§ | 1.23 | .659 | 1.00 | > .999 | 1.54 | .032 |
| 20 | 50.00 | Q71DI3 H32_HUMAN | Histone H3† | 1.17 | .147 | 0.53 | .003 | 0.60 | .004 |
| 21 | 25.00 | P06753 TPM3_HUMAN | Tropomyosin α‖ | 1.00 | > .999 | 0.97 | .677 | 0.89 | .356 |
| 22 | 6.51 | P35611 ADDA_HUMAN | α-Adducin | 0.76 | .127 | 1.20 | .141 | 0.91 | .419 |
| 23 | 2.60 | P29972 AQP1_HUMAN | AQP1 | 0.78 | 0.84 | 1.41 | |||
The β and γ isoforms could not be differentiated.
Multiple isoforms could not be differentiated.
The β and δ isoforms could not be differentiated.
Rh polypeptide 1 and Rh polypeptide 2 could not be differentiated.
The α-3 and α-4 isoforms could not be differentiated.
Figure 2.

iTRAQ™ validation by immunoblot. Equal quantities of ghost lysates were pooled from 6 to 8 donors per group (control vs Mi.III) for IP, and one-tenth of the pulldown (vol/vol) was loaded onto 4% to 12% SDS-polyacrylamide gel electrophoresis for immunoblot comparison. (A) Immunoblots confirmed that glucose transporter type I, ANK1, EPB42, α spectrin, and β spectrin were not quantitatively different in AE1 immunoprecipitates from both groups. The pulldown experiments were repeated 2 to 8 times, and IP was also confirmed using another anti-AE1, BRIC170 (data not shown). (B) Both anti-4.1R and anti-Rh antibodies pulled down GPA, GPB, Gp.Mur, and AE1, indicating that 4.1R and Rh polypeptides were part of the AE1-based complexes. (C) Rh-associated glycoprotein was coimmunoprecipitated with AE1. AE1 IP was repeated and confirmed with both anti-AE1 (AE12-M and BRIC170). (D) AQP1 was more substantially associated with AE1 on Mi.III+ membrane. (Left) AQP1 immunoblot of the pooled ghost lysates showed similar expression levels for both Mi.III and the control. (Right) Both anti-AE1 antibodies coimmunoprecipitated more AQP1 from Mi.III. The coimmunoprecipitation experiment has been repeated and confirmed 7 times.
Figure 3.

AE1 expressed more in Mi.III+ RBCs. (A top panel) An outline for the iTRAQ™-based quantitative proteomic method. To compare the composition of AE1-based complexes in Mi.III versus the control, RBC samples were collected from 6 Mi.III+ and 6 control donors, and each subjected to immunoprecipitation and then deglycosylation. The deglycosylated samples were independently digested with trypsin, and then combined for labeling with 1 of the 4 iTRAQ™ reagents. Two pools of 3 control samples and 2 pools of 3 Mi.III+ samples were randomly formed from the 6 donors from each group. The iTRAQ™-labeled peptides from all 4 pools were mixed, fractionated by strong cation chromatography, and analyzed by liquid chromatography–MS/MS. (Bottom panel) A representative fragmentation spectrum on iTRAQ™-labeled AE1. A high-scoring spectrum with overlapping b- and y-series fragment ions assigned to the peptide ADFLEQPVLGFVR (99% confidence) from AE1. (Inset) Expansion of x-axis demonstrates the abundance (area under the curve) of the isobaric tags at 114, 115, 116, and 117 Da. (B) A representative immunoprecipitation experiment using AE12-M antibody. Equal protein quantities of ghost lysates (m/m) from 6 to 8 donors per group (control vs Mi.III) were pooled for immunoprecipitation. One-tenth of the immunoprecipitate (vol/vol) was loaded for immunoblot comparison. The IP experiment has been repeated 7 times and confirmed by another anti-AE1, BRIC170 (data not shown). (Left) Silver stain of the AE1 immunoprecipitates from Mi.III and the control groups. (Right) Immunoblots for AE1, GPA, and GAB (E3). More AE1 was expressed by Mi.III+ cells, whereas the levels of GPA and GPB/Gp.Mur were not significantly different between the 2 groups. (C) DIDS labeling of AE1 was significantly higher in Mi.III+ than the control erythrocytes. Fresh erythrocytes from 3 donors per group were labeled with DIDS. The background fluorescence intensities from the unlabeled erythrocytes were subtracted. Data are expressed as mean ± SE; *P < .01.
The major quantitative difference revealed by iTRAQ™ was 25% to 67% increase of AE1 in Mi.III, for which a representative spectrum is presented in Figure 3A. This observation was confirmed by immunoblot (Figure 3B). To confirm that the increased AE1 levels in these experiments represented a true change in protein expression and not confounding issues related to immunoprecipitation, we further assessed AE1 levels by comparative binding of AE1 inhibitor, DIDS, to the surface of fresh intact RBCs (Figure 3C), and by immunoblot analysis of deglycosylated ghost (supplemental Figure 2).
There was general concordance between iTRAQ™ and immunoblot analyses of the pulldown experiments. A notable exception was AQP1, for which immunoblot revealed greater amounts in pulldowns from Mi.III, which in this case is most likely more reliable given that MS/MS identified AQP1 by only one peptide (2.6% sequence coverage in Table 2). Whereas the overall AQP1 levels were similar between Mi.III and the control, AQP1 was more substantially associated with AE1 in Mi.III (Figure 2D). This suggests that the AE1-AQP1 complex in Mi.III+ RBCs is unique, and might result from their significantly higher AE1 expression.
Higher anion exchange capacity of Mi.III+ erythrocytes
Erythrocyte AE1 has 2 important physiologic roles, each corresponding to a structural region: (1) its multispan transmembrane primarily serves as a Cl−/HCO3− exchanger for blood CO2 transport; and (2) its N-terminal cytoplasmic domain binding to ankyrin, 4.1R and EPB42, provides a mechanical support for RBC membranes and shapes.
The fact that more AE1 was expressed on Mi.III+ RBCs hints that their anion exchange capacity should be larger. We evaluated the capacity of Cl−/HCO3− exchange by loading erythrocytes with a fluorescent probe for Cl−, SPQ. When challenging the SPQ-loaded RBCs with different [Cl−]out, we observed small changes of [Cl−]in with respect to changes of [Cl−]out in the milieu of 5 mM HCO3− (Figure 4A). When extracellular bicarbonate increased to 15 mM, the changes of Cl− content (Δ[Cl−]in/Δ[Cl−]out) were still small for the control, but were significant for Mi.III. The intracellular Cl− concentrations were 57.1 plus or minus 10.6 mM (control) and 84.0 plus or minus 5.5 mM (Mi.III) in the milieu of 15 mM HCO3− and 90 mM Cl−, compared with 62.6 plus or minus 13.4 mM (control) and 58.3 plus or minus 5.9 mM (Mi.III) in the milieu of 5 mM HCO3− and 90 mM Cl− (Figure 4A). Mi.III+ erythrocytes showed significantly higher Cl− permeability at high [HCO3−]out.
Figure 4.

Mi.III+ RBCs exhibited higher Cl−/HCO3− exchange capacity upon HCO3− stimulation. (A) Intracellular Cl− was labeled by fluorescent dye SPQ, and the intracellular Cl− concentration ([Cl−]out) was measured by the degree of SPQ quenching inside erythrocytes. When the extracellular bicarbonate ([HCO3−]out) was 5 mM, [Cl−]in increased little with respect to [Cl−]out. [Cl−]in was similar between the control and Mi.III+ erythrocytes. In the milieu of 15 mM HCO3−, Mi.III+ erythrocytes contained more Cl− and showed higher Cl− permeability than the control cells. Data are expressed as mean ± SE; *P < .05 at 90 mM [Cl−]out. The numbers of donors tested were indicated next to sample labels. (B) The concentrations of intracellular bicarbonate ([HCO3−]in) were predicted according to Donnan equilibrium. In the milieu of 15 mM HCO3−, Mi.III+ erythrocytes also contained more HCO3− and showed higher HCO3− permeability.
If Donnan equilibrium is assumed, [HCO3−]in could be approximated from the conserved Donnan ratio between [Cl−]in/[Cl−]out and [HCO3−]in/[HCO3−]out. [HCO3−]in are estimated 3.5 plus or minus 0.7 mM (control) and 3.2 plus or minus 0.3 mM (Mi.III) in the milieu of 5 mM HCO3− and 90 mM Cl−, and 9.5 plus or minus 1.8 mM (control) and 14.0 plus or minus 0.9 mM (Mi.III) at 15 mM [HCO3−]out (Figure 4B). Mi.III+ RBCs again showed higher HCO3− permeability than the control at high bicarbonate. The better Cl− and HCO3− permeability of Mi.III+ RBCs could be attributed to their higher AE1 expression. However, how their transport capacity would expand at high [HCO3−]out remains unclear.
Greater pHi-buffering capacity of Mi.III+ erythrocytes
In blood pH homeostasis (CO2 + H2O ⇔ H+ + HCO3−), H+ is buffered by the high concentration of Hb− inside RBC, and HCO3− is considered the major determinant for pHi.26 Because Mi.III+ erythrocytes showed higher HCO3−/Cl− exchange capacities (Figure 4), we tested whether their differences were reflected in pHi homeostasis. Erythrocytes were loaded with fluorescent pH indicator carboxy SNARF-1, and the elicited pHi responses were quantified by flow cytometry.
At pHout 7.5, the pHi values for Mi.III+ and control erythrocytes were both approximately 7.2 (7.26 ± 0.032 [Mi.III] vs 7.15 ± 0.038 [control]). Depletion of extracellular bicarbonate drove HCO3− efflux, resulting in pHi drop. Notably, pHi drop was more substantial for the control cells (6.75 ± 0.088) than Mi.III (6.95 ± 0.047) when [HCO3−]out was depleted (Figure 5). Thus, Mi.III+ erythrocytes showed greater pHi-buffering capacity upon changes of HCO3− gradients.
Figure 5.

The pHi-buffering capacities of Mi.III+ erythrocytes were superior. Fresh RBCs were loaded with fluorescent pH indicator SNARF-1, and its intracellular pH measurement at pHout 7.5 was measured by flow cytometry. In the absence of extracellular bicarbonate, the control cells became more acidified than Mi.III. Depletion of extracellular Cl− maximized HCO3− loading for both Mi.III+ and the control cells, and diminished their pHi differences. The number of donors tested was indicated next to each bar. Data are expressed as mean ± SE; *P < .05.
To verify the role of Cl−/HCO3− exchange in pHi regulation, we replaced extracellular Cl− with membrane-impermeable gluconate−, which halted the coupled transport of Cl− influx and HCO3− efflux. As the coupled transport of Cl− efflux and HCO3− influx remained possible and the chemical gradient for Cl− was substantial ([Cl−]out: 143→0 mM), bicarbonate was forced to accumulate intracellularly. Consequently, the intracellular pH increased from approximately 7.2 to 7.71 plus or minus 0.037 (control) and to 7.70 plus or minus 0.044 (Mi.III), whereas pHout remained 7.5 (Figure 5). The fact that complete depletion of extracellular Cl− resulted in the same maximal pHi for both groups of cells indicates that their HCO3−-buffering capacities could be forced to expand to the same maxima. The asymmetry of their pHi responses from Cl− depletion and HCO3− depletion experiments suggests that other erythrocyte transporters, like Na+/H+ exchanger, may also contribute to the differences in pHi buffering. Future experiments are required to elucidate these asymmetric pHi responses between Mi.III and the control RBCs.
Functional implication in membrane resistance to osmotic stress
From osmotic fragility tests, Mi.III+ erythrocytes exhibited superior resistance toward osmotic pressure. Osmotic stress was induced by decreasing NaCl concentrations in the saline. Control cells began hemolysis at 0.600 plus or minus 0.05% NaCl, whereas Mi.III+ cells began at 0.557 plus or minus 0.03% NaCl (Figure 6). We did not find significant alteration in their cytoskeletal densities, arrangements, or spectrin lengths by electron microscopy (supplemental Figure 3). Our results support the previous finding that AE1 directly interacts with cell membranes to prevent surface loss and to maintain cell morphology.27 Conceivably, higher AE1 expression in Mi.III could increase contacts within the AE1-cytoskeletal network and thereby strengthen membrane resistance to osmotic stress.
Figure 6.

Mi.III+ erythrocytes were more resistant to osmotic stress. Osmotic fragility tests showed that Mi.III+ erythrocytes began and completed hemolysis at more hypotonic concentrations than the control cells. *P < .05. Data are expressed as mean ± SE. The number of donors tested was indicated next to each bar.
Gp.Mur enhanced the biosynthesis and surface expression of AE1
To determine how the glycophorin complement of Mi.III blood group engenders increased AE1 expression, we tested the hypothesis that Gp.Mur, essentially a variant of GPB containing an element of GPA, might mimic GPA with respect to its ability to act as a chaperone for AE1.12,13 We cloned Gp.Mur (GenBank accession EU338225) and GPA, and cotransfected them individually with AE1 into HEK293 cells. We found that GPA promoted the overall production of AE1 by 1.18- plus or minus 0.045-fold, compared with the singly transfected AE1 (Figure 7). GPA enhanced the surface expression of AE1 more substantially (1.58- ± 0.10-fold). Coexpression of Gp.Mur showed similar functionality to GPA. Gp.Mur also enhanced AE1 biosynthesis by 1.17- plus or minus 0.093-fold, and surface expression by 1.43- plus or minus 0.05-fold (Figure 7). Notably, the GPA levels were similar between Mi.III and the control; the GPB levels (or that combining Gp.Mur and GPB) were similar, too (Figure 1B). Thus, the increased surface expression of AE1 in Mi.III is best explained by the additive effect of trafficking-enhancing GP.Mur that replaces GPB, while retaining levels of GPA.
Figure 7.

Gp.Mur exhibited similar functionality as GPA in promoting AE1 expression. (A) Both GPA and Gp.Mur promoted AE1 biosynthesis. AE1 was subcloned in pCIG, a bicistronic construct containing the reporter gene green fluorescent protein. pCIG-AE1 was expressed alone, or together with GPA or Gp.Mur, in HEK293 cells. The transiently transfected cells were fixed, permeabilized, and stained with anti-AE1, BRIC71. The degrees of BRIC71 staining in the fixed and permeabilized cells reflected the relative expression levels of AE1 on intracellular and plasma membranes. (B) Both GPA and Gp.Mur promoted similar degrees of AE1 surface expression. Intact HEK293 cells were directly stained with BRIC71 after scraped off from culture plates. (Top) A representative BRIC71 histogram from flow cytometry. (Bottom) The intensities of BRIC71 staining for different coexpression groups were compared with that for the singly expressed AE1 (set at 1). Data were averaged from 8 to 9 independent experiments (as indicated next to each bar), and expressed as mean ± SE. P > .05 was deemed not significant (n.s.).
Discussion
In this study, we identified a major function of blood group antigen Mi.III in promotion of AE1 expression. Quantitative proteomics by iTRAQ™ revealed 25% to 67% more AE1 protein in Mi.III+ erythrocytes (Table 2 and Figure 3). GPA, a chaperone for AE1, is expressed at similar levels in Mi.III+ and the control erythrocytes (Figure 1B). Because Mi.III additionally expresses the functionally equivalent Gp.Mur, its elevated AE1 level could be attributed to the additive effect of Gp.Mur and GPA.
The functional equivalency between Gp.Mur and GPA is, itself, intriguing in light of our current understanding of the role of glycophorin domains in AE1 trafficking (Figure 7). Previous structural-functional studies on GPA and GPB attribute the enhanced AE1 trafficking to the cytoplasmic region of GPA.12 But Gp.Mur, like GPB, also lacks a cytoplasmic domain that GPA has. Therefore, the extracellular and the transmembrane domains where Gp.Mur and GPA share homologies are important for facilitating the expression of AE1.
Could the unique interaction between Gp.Mur and AE1 modify the intrinsic kinetics of AE1 antiport? From protein and transport studies, both homozygous and heterozygous Mi.III+ erythrocytes showed similar AE1 levels (supplemental Figure 2) and similar Cl−/HCO3− exchange capacities (Figure 4). However, there was more Gp.Mur expressed in homozygous Mi.III (Figure 1B). If the direct effect of Gp.Mur on the catalytic activity of AE1 were substantial, the homozygous and heterozygous Mi.III+ erythrocytes would have differential Cl−/HCO3− exchange capacities because of their different Gp.Mur levels. Thus, greater surface expression of AE1 in Mi.III appears to be the primary factor for its superior anion exchange.
The higher level of AE1 of Mi.III+ erythrocytes has dual impacts: (a) it expands anion exchange and pH-buffering capacities (Figures 4–5); (b) it strengthens cell membranes toward osmotic stress (Figure 6). The majority of blood CO2 is transported in the form of HCO3−. CO2 is converted into HCO3− primarily by carbonic anhydrase II inside erythrocytes. Although CO2 can permeate through RBC membranes easily, HCO3− is membrane impermeable and must cross membranes via AE1. Because AE1-mediated transport is at least one order of magnitude slower than the enzymatic activity of carbonic anhydrase II, it is the rate-limiting step for CO2 transport.28 Increase of AE1 expression could lift the limit and expand the capacity of CO2 respiration. In contrast, blood bicarbonate and CO2 capacities have been shown much reduced in the AE1-deficient cattle.29 Thus, one might predict that those with the Mi.III phenotype are capable of enduring higher physical stresses (eg, during exercise; at altitudes). Indeed, as an anecdote, a disproportional number of the world-class athletes in Taiwan, including the “iron man of Asia,”30,31 are from the aborigine tribe with the highest Mi.III occurrence (80%-90%). It would be intriguing to determine whether the high degree of athleticism can be correlated with the Mi.III phenotype.
Although AE1 was elevated in Mi.III, most of its binding partners were not elevated accordingly in the pulldowns from Mi.III+ RBCs (Figure 2). One exception is AQP1, whose interaction with AE1 was more substantial in Mi.III (Figure 2D). Interestingly, Cho et al showed that AE1 and AQP1 diffuse laterally at different rates on erythrocyte surface, and concluded that the 2 belong to distinct complexes.32 It is therefore conceivable that the AE1-AQP1 complex found more substantially on Mi.III+ cells is distinctly different from the band 3 complex11 or the 4.1R complex33 previously delineated. Their unique interaction in Mi.III might further facilitate CO2 respiration, presumably through coupling between H2O transport and intracellular bicarbonate formation, which requires H2O.
The glycophorin gene family is probably the second fastest evolving gene following immunoglobin.34 Although the function of glycophorin/Miltenberger seems auxiliary, glycophorins are the major host receptors for Plasmodium.35,36 The speedy evolution of glycophorin was proposed as a decoy or an evasion mechanism against malaria infection.34,37 But it is unclear whether there was a correlation between malaria and the emergence of Mi.III in the coastal lowland aborigines in Taiwan.34,38 Conceivably, the expression of Mi.III could tilt the host-pathogen interaction with its unique glycophorin structure, and/or with a higher AE1 level. It remains an intriguing question as to whether the higher AE1 level of Mi.III could help alleviate major malarial symptoms such as acidosis.39,40 Perhaps this blood group antigen has evolved not to avoid encountering malaria,34,37,38 but to assist host survival upon infection.
Supplementary Material
Acknowledgments
We thank Mackay Memorial Hospital blood bank specialists for guidance on the identification of Miltenberger blood group antigens; S. Chen for electron microscopy services (Mackay Memorial Hospital); Y. Yang, S. Huang, Y. Deng, Z. Chen, and director Dr C.-P. Chen for providing research assistance (Mackay Memorial Hospital); Prof D. Chandanayingyong for providing anti-Mur and anti-Anek antibodies (Siriraj Hospital); Dr M. Uchikawa for providing anti-Hil antibodies (Japan Red Cross Central Blood Center); Prof D. Anstee for providing the R1.3 and BRIC71 antibodies (BITS); and J. Rucker for technical assistance (Johns Hopkins University).
This work was supported by Taiwan National Science Council (95-2320-B-195-002-MY2; 97-2314-B-195-021) and by Mackay Memorial Hospital intramural grants to K.H. Support for proteomic analysis was provided through a grant from the National Heart, Lung, and Blood Institute (contract N01-HV-28180) to J.E.V.E. and R.N.C.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.H. designed and performed the research, analyzed the data, and wrote the paper; N.C. performed the research and analyzed the data; M.G. performed the mass spectrometry research and analyzed the data; R.N.C. and J.E.V.E. designed the research and helped write the paper; M.L. helped conceive the thesis; and D.B.F. designed and performed the research, analyzed the data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kate Hsu, Mackay Memorial Hospital Transfusion Medicine Laboratory, 45 Min-Sheng Rd, Research Bldg 616, Tamsui, Taiwan 251; e-mail: khsu1@ms1.mmh.org.tw.
References
- 1.Cleghorn TE. A memorandum on the Miltenberger blood groups. Vox Sang. 1966;11:219–222. doi: 10.1111/j.1423-0410.1966.tb04226.x. [DOI] [PubMed] [Google Scholar]
- 2.Tippett P, Reid ME, Poole J, Green CA, Daniels GL, Anstee DJ. The Miltenberger subsystem: is it obsolescent? Transfus Med Rev. 1992;6:170–182. doi: 10.1016/s0887-7963(92)70167-9. [DOI] [PubMed] [Google Scholar]
- 3.Lin M, Broadberry RE. Immunohematology in Taiwan. Transfus Med Rev. 1998;12:56–72. doi: 10.1016/s0887-7963(98)80090-4. [DOI] [PubMed] [Google Scholar]
- 4.Huang CH, Blumenfeld OO. Molecular genetics of human erythrocyte MiIII and MiVI glycophorins: use of a pseudoexon in construction of two δ-α-δ hybrid genes resulting in antigenic diversification. J Biol Chem. 1991;266:7248–7255. [PubMed] [Google Scholar]
- 5.Lin CK, Mak KH, Szeto SC, et al. First case of haemolytic disease of the newborn due to anti-Mur in Hong Kong. Clin Lab Haematol. 1996;18:19–22. doi: 10.1111/j.1365-2257.1996.tb00731.x. [DOI] [PubMed] [Google Scholar]
- 6.Lin CK, Mak KH, Yuen CM, Chan NK, Liu HW, Cheng G. A case of hydrops fetalis, probably due to antibodies directed against antigenic determinants of GP: Mur (Miltenberger class III) cells. Immunohematology. 1996;12:115–118. [PubMed] [Google Scholar]
- 7.Wu KH, Chang JG, Lin M, et al. Hydrops foetalis caused by anti-Mur in first pregnancy: a case report. Transfus Med Rev. 2002;12:325–327. doi: 10.1046/j.1365-3148.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- 8.Broadberry RE, Lin M. The incidence and significance of anti-“Mia” in Taiwan. Transfusion. 1994;34:349–352. doi: 10.1046/j.1537-2995.1994.34494233585.x. [DOI] [PubMed] [Google Scholar]
- 9.Knowles DW, Chasis JA, Evans EA, Mohandas N. Cooperative action between band 3 and glycophorin A in human erythrocytes: immobilization of band 3 induced by antibodies to glycophorin A. Biophys J. 1994;66:1726–1732. doi: 10.1016/S0006-3495(94)80965-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auffray I, Marfatia S, de Jong K, et al. Glycophorin A dimerization and band 3 interaction during erythroid membrane biogenesis: in vivo studies in human glycophorin A transgenic mice. Blood. 2001;97:2872–2878. doi: 10.1182/blood.v97.9.2872. [DOI] [PubMed] [Google Scholar]
- 11.Bruce LJ, Beckmann R, Ribeiro ML, et al. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180–4188. doi: 10.1182/blood-2002-09-2824. [DOI] [PubMed] [Google Scholar]
- 12.Groves JD, Tanner MJ. Glycophorin A facilitates the expression of human band 3-mediated anion transport in Xenopus oocytes. J Biol Chem. 1992;267:22163–22170. [PubMed] [Google Scholar]
- 13.Groves JD, Tanner MJ. The effects of glycophorin A on the expression of the human red cell anion transporter (band 3) in Xenopus oocytes. J Membr Biol. 1994;140:81–88. doi: 10.1007/BF00234488. [DOI] [PubMed] [Google Scholar]
- 14.Bruce LJ, Pan RJ, Cope DL, et al. Altered structure and anion transport properties of band 3 (AE1, SLC4A1) in human red cells lacking glycophorin A. J Biol Chem. 2004;279:2414–2420. doi: 10.1074/jbc.M309826200. [DOI] [PubMed] [Google Scholar]
- 15.Hassoun H, Hanada T, Lutchman M, et al. Complete deficiency of glycophorin A in red blood cells from mice with targeted inactivation of the band 3 (AE1) gene. Blood. 1998;91:2146–2151. [PubMed] [Google Scholar]
- 16.Huang CH, Johe KK, Seifter S, Blumenfeld OO. Biochemistry and molecular biology of MNSs blood group antigens. Baillieres Clin Haematol. 1991;4:821–848. doi: 10.1016/s0950-3536(06)80032-6. [DOI] [PubMed] [Google Scholar]
- 17.Blumenfeld OO, Huang CH. Molecular genetics of the glycophorin gene family, the antigens for MNSs blood groups: multiple gene rearrangements and modulation of splice site usage result in extensive diversification. Hum Mutat. 1995;6:199–209. doi: 10.1002/humu.1380060302. [DOI] [PubMed] [Google Scholar]
- 18.Telen MJ, Scearce RM, Haynes BF. Human erythrocyte antigens III: characterization of a panel of murine monoclonal antibodies that react with human erythrocyte and erythroid precursor membranes. Vox Sang. 1987;52:236–243. doi: 10.1111/j.1423-0410.1987.tb03035.x. [DOI] [PubMed] [Google Scholar]
- 19.King MJ, Poole J, Anstee DJ. An application of immunoblotting in the classification of the Miltenberger series of blood group antigens. Transfusion. 1989;29:106–112. doi: 10.1046/j.1537-2995.1989.29289146826.x. [DOI] [PubMed] [Google Scholar]
- 20.Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell. 2004;14:259–267. doi: 10.1016/s1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]
- 21.Pilas B, Durack G. A flow cytometric method for measurement of intracellular chloride concentration in lymphocytes using the halide-specific probe 6-methoxy-N-(3-sulfopropyl) quinolinium (SPQ). Cytometry. 1997;28:316–322. [PubMed] [Google Scholar]
- 22.van Erp PE, Jansen MJ, de Jongh GJ, Boezeman JB, Schalkwijk J. Ratiometric measurement of intracellular pH in cultured human keratinocytes using carboxy-SNARF-1 and flow cytometry. Cytometry. 1991;12:127–132. doi: 10.1002/cyto.990120205. [DOI] [PubMed] [Google Scholar]
- 23.Gilligan DM, Bennett V. The junctional complex of the membrane skeleton. Semin Hematol. 1993;30:74–83. [PubMed] [Google Scholar]
- 24.Salzer U, Zhu R, Luten M, et al. Vesicles generated during storage of red cells are rich in the lipid raft marker stomatin. Transfusion. 2008;48:451–462. doi: 10.1111/j.1537-2995.2007.01549.x. [DOI] [PubMed] [Google Scholar]
- 25.Salzer U, Hinterdorfer P, Hunger U, Borken C, Prohaska R. Ca+/+-dependent vesicle release from erythrocytes involves stomatin-specific lipid rafts, synexin (annexin VII), and sorcin. Blood. 2002;99:2569–2577. doi: 10.1182/blood.v99.7.2569. [DOI] [PubMed] [Google Scholar]
- 26.Juel C, Lundby C, Sander M, Calbet JA, Hall G. Human skeletal muscle and erythrocyte proteins involved in acid-base homeostasis: adaptations to chronic hypoxia. J Physiol. 2003;548:639–648. doi: 10.1113/jphysiol.2002.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters LL, Shivdasani RA, Liu SC, et al. Anion exchanger 1 (band 3) is required to prevent erythrocyte membrane surface loss but not to form the membrane skeleton. Cell. 1996;86:917–927. doi: 10.1016/s0092-8674(00)80167-1. [DOI] [PubMed] [Google Scholar]
- 28.Reithmeier RA. A membrane metabolon linking carbonic anhydrase with chloride/bicarbonate anion exchangers. Blood Cells Mol Dis. 2001;27:85–89. doi: 10.1006/bcmd.2000.0353. [DOI] [PubMed] [Google Scholar]
- 29.Inaba M, Yawata A, Koshino I, et al. Defective anion transport and marked spherocytosis with membrane instability caused by hereditary total deficiency of red cell band 3 in cattle due to a nonsense mutation. J Clin Invest. 1996;97:1804–1817. doi: 10.1172/JCI118610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang F. Taiwan's ‘iron man’ to return home to hospital. Taipei Times. 2001 Jan 4;:1. [Google Scholar]
- 31.Maruyama H. Nihonryon jidai nokoshita taiwan no ijieisei gyouseki [Medicine and Public Health in Taiwan under Japanese Colonial Rule] Yokohama, Japan: 1957. [Google Scholar]
- 32.Cho MR, Knowles DW, Smith BL, et al. Membrane dynamics of the water transport protein aquaporin-1 in intact human red cells. Biophys J. 1999;76:1136–1144. doi: 10.1016/S0006-3495(99)77278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salomao M, Zhang X, Yang Y, et al. Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc Natl Acad Sci U S A. 2008;105:8026–8031. doi: 10.1073/pnas.0803225105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang HY, Tang H, Shen CK, Wu CI. Rapidly evolving genes in human I: the glycophorins and their possible role in evading malaria parasites. Mol Biol Evol. 2003;20:1795–1804. doi: 10.1093/molbev/msg185. [DOI] [PubMed] [Google Scholar]
- 35.Pasvol G, Wainscoat JS, Weatherall DJ. Erythrocytes deficiency in glycophorin resist invasion by the malarial parasite Plasmodium falciparum. Nature. 1982;297:64–66. doi: 10.1038/297064a0. [DOI] [PubMed] [Google Scholar]
- 36.Rayner JC, Vargas-Serrato E, Huber CS, Galinski MR, Barnwell JW. A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J Exp Med. 2001;194:1571–1581. doi: 10.1084/jem.194.11.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baum J, Ward RH, Conway DJ. Natural selection on the erythrocyte surface. Mol Biol Evol. 2002;19:223–229. doi: 10.1093/oxfordjournals.molbev.a004075. [DOI] [PubMed] [Google Scholar]
- 38.Morisita K. Taiwan ni okeru mararia no yobou no kenkyun [Malaria Epidemiology in Taiwan]: Mararia no ekigaku to yobou [Epidemiology and Prevention of Malaria] Tokyo, Japan: Kikuoku Shobou; 1976. p. 51. [Google Scholar]
- 39.Day NP, Phu NH, Mai NT, et al. The pathophysiologic and prognostic significance of acidosis in severe adult malaria. Crit Care Med. 2000;28:1833–1840. doi: 10.1097/00003246-200006000-00025. [DOI] [PubMed] [Google Scholar]
- 40.Marsh K, Forster D, Waruiru C, et al. Indicators of life-threatening malaria in African children. N Engl J Med. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}