

Abstract
Chronic myelogenous leukemia (CML) is driven by Bcr-Abl, a constitutively active protein-tyrosine kinase that stimulates proliferation and survival of myeloid progenitors. Global inhibition of myeloid Src family kinase (SFK) activity with the broad-spectrum pyrrolo-pyrimidine inhibitor, A-419259, blocks proliferation and induces apoptosis in CML cell lines, suggesting that transformation by Bcr-Abl requires SFK activity. However, the contribution of Hck and other individual SFKs to Bcr-Abl signaling is less clear. Here, we developed an A-419259-resistant mutant of Hck by replacing the gatekeeper residue (Thr-338; c-Src numbering) in the inhibitor-binding site with a bulkier methionine residue (Hck-T338M). This substitution reduced Hck sensitivity to A-419259 by more than 30-fold without significantly affecting kinase activity in vitro. Expression of Hck-T338M protected K-562 CML cells and Bcr-Abl-transformed TF-1 myeloid cells from the apoptotic and antiproliferative effects of A-419259. These effects correlated with persistence of Hck-T338M kinase activity in the presence of the compound, and were accompanied by sustained Erk and Stat5 activation. In contrast, control cells expressing equivalent levels of wild-type Hck retained sensitivity to the inhibitor. We also show for the first time that A-419259 induces cell-cycle arrest and apoptosis in primary CD34+ CML cells with equal potency to imatinib. These data suggest that Hck has a nonredundant function as a key downstream signaling partner for Bcr-Abl and may represent a potential drug target in CML.
Keywords: Bcr-Abl, CML, Hck, Stat5, pyrrolo-pyrimidine, Src-family kinase inhibitor
Introduction
Chronic myelogenous leukemia (CML) is a genetic hematological malignancy that affects 1 in 100 000 people each year and is characterized by clonal expansion of transformed multipotent hematopoietic stem cells (Sawyers, 1999). The hallmark genetic anomaly of CML is the Philadelphia chromosome (Ph+) that results from a reciprocal translocation between the c-abl locus on chromosome 9 and the bcr locus on chromosome 22 (Rowley, 1973; Ben-Neriah et al., 1986). Bcr-Abl, the protein product of this translocation, is a 210 kDa chimeric tyrosine kinase that transforms fibroblasts, growth factor-dependent hematopoietic cell lines and primary bone marrow cells in culture (McLaughlin et al., 1987; Lugo and Witte, 1989; Ren, 2002), and induces a myeloproliferative disorder that closely resembles CML in mice (Daley et al., 1990; Kelliher et al., 1990).
Constitutive tyrosine kinase activity and cytoplasmic relocalization allow Bcr-Abl to activate numerous signal transduction pathways that promote cell proliferation and survival (Sattler et al., 1996). For example, Bcr-Abl activates the Stat5 transcription factor, inducing its nuclear translocation and transcription of cell growth and survival genes, such as Cyclin-D1 and Bcl-XL (Carlesso et al., 1996; Shuai et al., 1996; de Groot et al., 2000; Gesbert and Griffin, 2000). Bcr-Abl also activates the PI3K/Akt pathway upon phosphorylation of Tyr-177 in the Bcr region and recruitment of Grb2/Gab2 adapter proteins (Sattler et al., 1996). PI3K is also activated by Bcr-Abl through other adapter proteins, such as Shc (Harrison-Findik et al., 1995; Raffel et al., 1996), Crkl and c-Cbl (Sattler et al., 1996) or by direct binding of the p85 subunit of PI3K to Bcr-Abl (Skorski et al., 1997; Neshat et al., 2000). Activation of PI3K leads to the suppression of programmed cell death through Akt and other pathways. Similarly, the Ras/Erk pathway is activated by Bcr-Abl either upon phosphorylation of Tyr-177 and direct Grb2/Sos recruitment or through the Shc adaptor protein (Goga et al., 1995; Sattler et al., 2002). By stimulating the Ras/Erk pathway, Bcr-Abl increases growth factor-independent cell proliferation.
Members of the Src kinase family have been strongly linked to Bcr-Abl signaling and leukemogenesis (Lionberger et al., 2000; Klejman et al., 2002; Hu et al., 2004). Bcr-Abl binds to multiple Src family members including Hck, Lyn and Fyn, leading to their activation (Danhauser-Riedl et al., 1996; Warmuth et al., 1997; Lionberger et al., 2000; Meyn et al., 2006). Activation of SFKs may have a positive feedback effect on Bcr-Abl signaling, as SFKs directly phosphorylate Bcr-Abl on tyrosine residues critical for regulation (Pendergast et al., 1993; Warmuth et al., 1997; Million and Van Etten, 2000; Zhang et al., 2001; Smith et al., 2003; Meyn et al., 2006). SFKs may also serve as key intermediates linking Bcr-Abl with downstream effectors. Hck has been shown to couple Bcr-Abl to Stat5 activation in myeloid leukemia cells, which may contribute to survival (Klejman et al., 2002). Furthermore, global inhibition of SFK activity with the ATP-competitive pyrrolo-pyrimidine A-419259 blocks Stat5 and Erk signaling, leading to growth arrest and apoptosis in CML cell lines (Wilson et al., 2002). Here, we show for the first time that A-419259 also blocks proliferation and induces apoptosis in primary CD34+ CML cells as well. Other studies have shown that the SFKs Hck and Lyn are overexpressed and activated in CML blast crisis patients, and upregulation correlates with disease progression and drug resistance (Dai et al., 2004; Donato et al., 2004; Wu et al., 2008).
Consistent with the critical function of Bcr-Abl in CML, the Abl kinase inhibitor imatinib produces dramatic remission in most chronic phase CML cases (Sawyers, 1999; Kantarjian et al., 2002). However, patients with advanced disease often acquire drug resistance and continue to progress despite imatinib therapy. In addition, residual Bcr-Abl+ primitive progenitor cells can persist in patients achieving complete cytogenetic remission (Bhatia et al., 2003), highlighting the need for additional therapeutic targets. Given their important function in Bcr-Abl signaling and in imatinib resistance, SFKs have recently emerged as novel targets for CML treatment. However, the relative contribution of individual Src family members to Bcr-Abl signaling is not fully understood.
In this study, we investigated the contribution of Hck to Bcr-Abl signaling using a mutant (Hck-T338M) with engineered resistance to the broad-spectrum SFK inhibitor, A-419259. Expression of this mutant in the CML cell line, K562 and in TF-1 myeloid cells acutely transformed with Bcr-Abl allowed persistence of Hck kinase activity in the presence of A-419259 at concentrations that inhibited all endogenous Src family kinase activity. Remarkably, Hck-T338M rescued both K562 and TF-1/Bcr-Abl cells from the apoptotic effects of A-419259 treatment. This result correlated with sustained activation of Stat5 in the presence of the inhibitor and provides direct evidence that Hck alone is able to transmit antiapoptotic signals from Bcr-Abl. In addition, our data demonstrate the utility of engineered inhibitor-resistant mutants to dissect the roles of individual members of a closely related family of protein kinases in oncogenic signaling, and point to Hck as a clinically relevant target for CML therapy.
Results
Design of an inhibitor-resistant mutant of Hck
Many ATP-competitive kinase inhibitors access a hydrophobic pocket adjacent to the ATP-binding site in the kinase domain. Accessibility of inhibitors to this pocket is controlled by a nonconserved amino acid often referred to as the ‘gatekeeper’ residue (Liu et al., 1998), and natural variation in the gatekeeper residue is one of the main structural determinants of inhibitor sensitivity (Eyers et al., 1998; Bishop et al., 2000; Gorre et al., 2001; Blencke et al., 2003, 2004). Tyrosine kinases that possess a threonine at this position are sensitive to various classes of inhibitors that access the hydrophobic pocket (Table 1). However, replacement of the gatekeeper residue with more bulky amino acids, such as methionine or isoleucine often induces resistance to these inhibitors.
Table 1.
Peptide sequences surrounding the gatekeeper threonine at the ATP-binding site of several tyrosine kinases
| Kinase | Gatekeeper residue | Gatekeeper mutation | Inhibitor | Class | References |
|---|---|---|---|---|---|
| c-Abl | FYIIT315EFMTYGN | T→I | Imatinib | 2-phenylaminopyrimidine | Gorre et al. (2001) |
| Kit | TLVIT670EYCCYGD | T→I | Imatinib | 2-phenylaminopyrimidine | Tamborini et al. (2004) |
| PDGFR-α | IYIIT674EYCFYGD | T→I | Imatinib | 2-phenylaminopyrimidine | Cools et al. (2003) |
| EGFR | VQLIT766QMPFGD | T→M | Gefitinib | anilinoquinazoline | Kobayashi et al. (2005) |
| Src | IYIVT338EYMSKGS | T→I | PP58/PP1 | pyrido/pyrazolopyrimidine | Liu et al. (1999); Blencke et al. (2004) |
| Hck | IYIIT338EFMAKGS | T→M | A-419259 | pyrrolopyrimidine | This study |
Mutations of this residue to methionine or isoleucine account for a common mechanism of resistance to inhibitors of various classes as described in the text.
To determine whether we could artificially engineer resistance to A-419259 in Hck by increasing the size of the gatekeeper residue, we examined the crystal structure of Hck in complex with a related pyrazolopyrimi-dine inhibitor, A-420983 (Figure 1). This structure revealed close contact between the gatekeeper residue (Thr-338; human c-Src crystal structure numbering (Xu et al., 1999)) and the inhibitor, suggesting that the replacement of Thr-338 with a bulkier methionine residue would result in A-419259 resistance due to steric clash. To test this idea, wild-type Hck and Hck-T338M were expressed in Sf9 insect cells and purified to homogeneity in their downregulated conformations. Normally, Hck as well as other SFKs adopt an inactive, downregulated conformation in vivo due to the phosphorylation of a conserved tyrosine residue in the C-terminal tail (Tyr-527) by the regulatory kinases Csk and Chk (Boggon and Eck, 2004). To attain this conformation in the recombinant kinases, while avoiding coexpression of Csk, both Hck sequences were altered at their C-terminal tails from Tyr-527-Gln-Gln-Gln-Pro to Tyr-527-Glu-Glu-Ile-Pro (referred to hereafter as Hck-YEEI). This modification promotes autophosphorylation of the tail independently of Csk and enhances SH2-tail interaction, stabilizing the down-regulated conformation (Songyang et al., 1993; Schindler et al., 1999; Boggon and Eck, 2004). Importantly, Hck-YEEI undergoes autophosphorylation and exhibits substrate phosphorylation kinetics similar to wild-type Hck (Porter et al., 2000; Boggon and Eck, 2004). The sensitivity of recombinant Hck-YEEI and Hck-T338M-YEEI to A-419259 were compared in an in vitro kinase assay using a peptide substrate. As shown in Figure 2, the T338M mutation induced dramatic resistance to A-419259, increasing the IC50 value by almost 30-fold from 11.26±1.23 nM for wild-type Hck to 315.6±80.3 nM for the T338M mutant. Kinetic analysis showed a modest difference in the Km for ATP in the T338M mutant compared to wild-type Hck (10.7±2.5 μM for wild-type vs 3.9±0.8 μM for T338M).
Figure 1.

(a) Orientation of A-420983, an analog of A-419259, in the Hck nucleotide-binding pocket. The overall structure of Hck is shown on the left, with the SH3 domain in red, the SH2 domain in blue, and the kinase domain in gray. The side chain of the gatekeeper residue (T338; c-Src numbering) is highlighted in magenta. The relationship of the gatekeeper residue to the pyrazolopyrimidine moiety of A-420983 is enlarged on the right. This model was produced using PyMol and the Protein Data Bank (PDB) file 2C01. (b) Chemical structures of A-419259 and A-420983.
Figure 2.
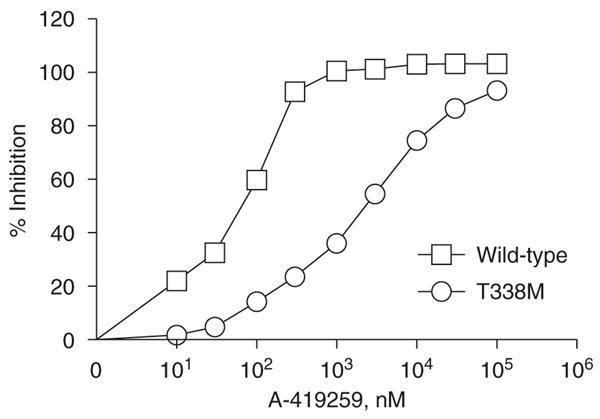
In vitro kinase assay of recombinant wild type and T338M forms of Hck. Recombinant wild-type Hck and Hck-T338M were purified from Sf9 insect cells in their downregulated conformations and assayed for kinase activity with a peptide substrate in vitro in the presence or absence of the indicated concentrations of A-419259. A representative experiment is shown and the extent of inhibition is expressed as mean±s.d. of four assays. The entire experiment was repeated twice with comparable results. Data from two independent experiments were best fit by nonlinear regression analysis and yielded IC50 values of 11.26±1.23 nM for wild-type Hck and 315.6±80.3 nM for Hck-T338M.
Hck-T338M retains its activity and A-419259-resistance in fibroblasts
We next investigated whether the T338M mutation influenced Hck biological activity and whether resistance to A-419259 was maintained in intact cells. To address these questions, both wild-type Hck and Hck-T338M were activated by replacing the regulatory C-terminal tail tyrosine (Tyr-527) with phenylalanine (YF mutation). This mutation upregulates Hck kinase activity and induces oncogenic transformation of fibroblasts (Briggs et al., 1997; Briggs and Smithgall, 1999; Lerner and Smithgall, 2002; Lerner et al., 2005). Rat-2 fibroblasts expressing Hck-YF, Hck-T338M-YF, as well as wild-type Hck or the neo-resistance marker as negative controls were plated in soft agar in the presence or absence of increasing concentrations of A-419259. As shown in Figure 3a, the T338M mutation did not impair biological activity, as cells expressing Hck-YF or Hck-T338M-YF produced similar numbers of transformed colonies in the absence of the inhibitor. To determine whether transformation correlated with constitutive activation of the kinase, cell lysates were tested for reactivity with the phosphospecific antibody pY418, which recognizes the phosphotyrosine residue in the Hck activation loop (Lerner and Smithgall, 2002; Schreiner et al., 2002; Wilson et al., 2002). Consistent with the transformation results, both Hck-YF and Hck-T338M-YF reacted strongly with this phosphospecific antibody (Figure 3b). Furthermore, activation loop phosphorylation correlated with phosphorylation of the endogenous Hck substrate protein, pp40 (Briggs et al., 1997; Lerner and Smithgall, 2002; Figure 3b). In contrast, wild-type Hck did not exhibit kinase or transforming activity. Taken together, these results show that the T338M mutation is functionally silent and does not influence Hck biological activity in cells.
Figure 3.

Hck-T338M maintains its biological activity and A-419259 resistance in fibroblasts. Rat-2 fibroblasts were infected with recombinant Hck-YF and Hck-T338M-YF retroviruses. Cells infected with a virus carrying only the selection marker (neo) or wild-type Hck served as negative controls. Upon G418 selection, cells were plated in soft agar in the presence or absence of the indicated concentrations of A-419259 for 10–14 days. Transformed colonies were visualized using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) staining. (a) Colony numbers for each cell line were determined using scanned images of the plates and BioRad QuantityOne colony-counting software. Results from a representative experiment are shown as the mean number of colonies±s.d. The entire experiment was performed twice and yielded comparable results. (b) Lysates from each of the cell lines shown in panel a were probed with phosphospecific antibodies against the Hck activation loop phosphotyrosine residue (pHck). Replicate membranes were probed with a general antiphosphotyrosine antibody to determine phosphorylation of the endogenous Hck substrate pp40, with an anti-Hck antibody to determine Hck expression levels, and with anti-actin as a loading control.
Next, we investigated the sensitivity of Rat-2 cells transformed by each form of Hck to A-419259. As shown in Figure 3a, 0.3 μM A-419259 almost completely blocked colony formation by cells expressing the Hck-YF mutant. This was accompanied by complete inhibition of Hck-YF autophosphorylation and pp40 substrate phosphorylation (Figure 3b). In contrast, the same concentration of A-419259 induced only a 25% decrease in colony formation in cells expressing Hck-T338M-YF mutant with no detectable change in kinase activity. Interestingly, the activity of Hck-T338M-YF was also unaffected by 1 μM A-419259, despite a further decrease in colony-forming activity. This finding suggests that endogenous A-419259-sensitive SFKs may cooperate with Hck to induce the transformed phenotype.
Hck-T338M expression in K562 cells confers resistance to A-419259-induced growth arrest and apoptosis
The in vitro and cell-based assays described above show that the T338M mutation induces Hck resistance to A-419259 without affecting kinase activity, providing a unique probe to test the contribution of Hck to Bcr-Abl signaling in CML cells. For these studies, we first used K562 cells, a Ph+ human CML cell line in which endogenous Hck expression is not detectable by immunoblot (Figure 4). K562 cells were infected with recombinant retroviruses carrying wild-type Hck, the Hck-T338M mutant or a control virus carrying only the G418 selection marker. These three cell populations (K562-neo, K562-Hck and K562-Hck-T338M) were treated with A-419259, and the effect of inhibitor treatment on Hck activity was assessed with the phosphospecific antibody, pY418. As shown in Figure 4, A-419259 caused partial inhibition of wild-type Hck at 0.1 μM and complete inhibition at 0.3 and 1.0 μM in K562-Hck cells. This observation is consistent with our previous findings that A-419259 inhibits overall SFK activity in K562 and other CML cell lines with an IC50 value of 0.1–0.3 μM (Wilson et al., 2002). In contrast, little change in Hck-T338M pY418 phosphorylation was observed at these A-419259 concentrations, although partial inhibition was observed at 1 μM.
Figure 4.

Hck-T338M is resistant to inhibition by A-419259 in K562 cells. (a) Wild-type and Hck-T338M proteins were expressed in K562 cells using recombinant retroviruses. K562-Hck wild-type and K562-Hck-T338M cells were treated with A-419259 at the indicated concentrations for 5 h. Hck activity was assessed by immunoprecipitation of Hck from clarified cell lysates and immunoblotting with phosphospecific antibodies against the Hck activation loop phosphotyrosine residue (pHck). Duplicate blots of the immunoprecipitates were blotted with the anti-Hck antibody to insure equal loading. Representative blots are shown at the top. Phosphotyrosine signal intensities from the blots of two independent experiments were normalized to the levels of Hck. Results are presented as percent of control levels±s.d. in the bar chart. (b) A-419259 does not affect Bcr-Abl activation loop autophosphorylation. Lysates from A-419259-treated cells in part A were immunoblotted with a phosphospecific antibody to the Abl activation loop phosphotyrosine residue (pY412; upper panels). Control blots were performed with an antibody to the Abl protein (Bcr-Abl; lower panels). (c) Inhibition of Bcr-Abl activation loop autophosphorylation by imatinib. K-562 cell populations from part A were treated with imatinib at the concentrations shown. Cell lysates were then immunoblotted with a phosphospecific antibody to the Abl activation loop phosphotyrosine residue (pY412; upper panels) as well as an antibody to the Abl protein (Bcr-Abl; lower panels).
Our previous work has shown that A-419259-dependent inhibition of SFK activity in K562 cells induces growth arrest and apoptosis (Wilson et al., 2002). In addition, we found that A-419259 is 300–1000 times more potent against SFKs compared with c-Abl in vitro. To determine whether A-419259 has a direct inhibitory effect on Bcr-Abl in our experimental system, we probed the lysates from the inhibitor-treated K562 cell populations with a phosphospecific antibody against the phosphotyrosine residue in the activation loop of Bcr-Abl (pY412). As shown in Figure 4b, A-419259 did not inhibit Bcr-Abl phosphorylation at the autoactivation site. In contrast, the Abl-selective inhibitor imatinib caused a dose-dependent decrease of the pY412 signal (Figure 4c). These data support the selectivity of A-419259 for SFKs vs Bcr-Abl in K562 cells, in agreement with our previous data in this cell line (Wilson et al., 2002).
To investigate the contribution of Hck to Bcr-Abl-induced cell growth, proliferation of K562-neo, K562-Hck, and K562-Hck-T338M cells was assessed in the presence of A-419259. Figure 5 shows that 0.1 μM A-419259 induced 75–80% growth arrest of K562-Hck and K562-neo cells. Conversely, K562-Hck-T338M cells exhibited only a 50% reduction in cell proliferation at this inhibitor concentration. This result correlates with sustained Hck-T338 kinase activity compared to wild-type Hck in the presence of the inhibitor (Figure 4). Given that 0.1–0.3 μM A-419259 completely inhibits endogenous Src family kinase activity in CML cells (Wilson et al., 2002), this result suggests that Hck alone is able to partially sustain Bcr-Abl-induced cell proliferation (see Discussion).
Figure 5.

Hck-T338M expression in K562 cells confers resistance to A-419259-induced growth arrest. K562 cells expressing wild-type Hck, the T338M mutant, as well as the vector control cells (neo) were treated with A-419259 at the indicated concentrations. Cell proliferation was monitored 24, 48 and 72 h later using the CellTiter-Blue cell viability assay as described under ‘Materials and methods’. Three replicate wells were monitored for each dose in three independent experiments and gave comparable results; a representative example is shown.
We next investigated whether the expression of Hck-T338M also confers resistance to A-419259-induced apoptosis in CML cells. K562-neo, K562-Hck and K562-Hck-T338M cells were treated with A-419259 for 72 h and apoptosis was measured by staining the cells with a fluorescent antiphosphatidylserine antibody and flow cytometry. As shown in Figure 6, A-419259 induced apoptosis in K562-neo and K562-Hck cells at 0.3 and 1.0 μM. In contrast, Hck-T338M expression completely rescued K562 cells from apoptosis induced by 0.3 μM A-419259. Furthermore, K562-Hck-T338M cells also displayed significant resistance to apoptosis induced by 1 μM A-419259. These data correlate with Hck-T338M kinase activity, which is not affected by 0.3 μM and only partially inhibited by 1 μM A-419259 (Figure 4).
Figure 6.

Hck-T338M protects K562 cells against the apoptotic effect of the Src family kinase (SFK) inhibitor A-419259. K562 cells expressing wild-type Hck, the T338M mutant, as well as the vector control (neo) cells were plated in the absence or presence of the indicated concentrations of A-419259 for 72 h. Apoptotic cells were detected by anti-phosphatidylserine-Alexa Fluor 488-conjugated antibody staining and flow cytometry. (a) Representative experiment with the percentage of apoptotic cells in each population shown above the bar. (b) Bar graph showing the average of three independent experiments and plotted±s.d.
Expression of Hck-T338M in K562 cells rescues the inhibitory effects of A-419259 on Stat5 and Erk activation
Previous reports suggest that Hck may couple Bcr-Abl to Stat5 and Erk activation in myeloid leukemia cells (Warmuth et al., 1997; Klejman et al., 2002). In addition, work in our laboratory has shown that the inhibition of SFK activity in K562 cells using A-419259 induces apoptosis and growth arrest that correlates with decreased Ras/Erk and Stat5 activation (Wilson et al., 2002). To assess the contribution of Hck to these growth and survival pathways downstream of Bcr-Abl, we determined whether Hck-T338M could rescue Ras/Erk and Stat5 activation from the inhibitory effects of A-419259. K562-neo, K562-Hck, and K562-Hck-T338M cells were treated with A-419259 and cell lysates were probed with phosphospecific antibodies for activated Erk by immunoblotting. As shown in Figure 7, A-419259 induced a dose-dependent inhibition of Erk phosphorylation in K562-neo and K562-Hck cells. In both cases, the phospho-Erk signal was partially reduced at 0.3 μM and completely absent at 1.0 μM. However, expression of Hck-T338M completely rescued Erk activation at 0.3 μM A-419259, and showed a partial effect at 1.0 μM. Although these data are consistent with the partial reversal of A-419259 growth arrest observed in K562-Hck-T338 cells (Figure 4), they suggest that Hck- and Erk-independent pathways also contribute to proliferation of K562 cells.
Figure 7.

Expression of Hck-T338M in K562 cells reverses the inhibitory effects of A-419259 on Erk activation. K562 cells expressing wild-type Hck, the T338M mutant, as well as the vector control cells (neo) were treated with the indicated concentrations of A-419259 for 5 h. (a) Cell lysates were prepared and analysed for the presence of active Erk by immunoblotting with phosphospecific antibodies (pErk). Duplicate blots were probed with antibodies to Erk2 as a loading control. Representative blots are shown. (b) Phospho-Erk signal intensities from the blots of two independent experiments were normalized to the levels of Erk, and are presented as percent of control levels±s.d.
To examine the effect of Hck-T338M on Stat5 activation, Stat5 was immunoprecipitated from lysates of A-419259-treated cells and probed with antiphosphotyrosine antibodies. As shown in Figure 8, A-419259 induced a dose-dependent inhibition of Stat5 activation in K562-neo and K562-Hck cells. In contrast, only a modest change in Stat5 phosphorylation was observed in K562-Hck-T338M cells, indicating that Hck has a major function in coupling Bcr-Abl to Stat5 activation. This result correlates with the levels of Hck-T338M activity observed in the presence of A-419259 (Figure 4a) and with the ability of Hck-T338M to rescue K562 cells from A-419259-induced apoptosis (Figure 6).
Figure 8.

Expression of Hck-T338M in K562 cells opposes the inhibitory effects of A-419259 on Stat5 activation. K562 cells expressing wild-type Hck, the T338M mutant, as well as the vector control cells (neo) were treated with the indicated concentrations of A-419259 for 5 h. (a) Stat5 tyrosine phosphorylation was assessed by immunoprecipitation of Stat5 from clarified cell lysates and immunoblotting with antiphosphotyrosine antibodies (pStat5). Duplicate membranes were blotted with an anti-Stat5 antibody to insure equal loading (Stat5). Representative blots are shown. (b) Stat5 phosphotyrosine signal intensities from the blots of two independent experiments were normalized to the levels of Stat5 protein, and the results are presented as percent of control levels±s.d.
Expression of Hck-T338M in Bcr-Abl-transformed TF-1 cells confers resistance to the apoptotic effects of A-419259 and correlates with sustained Stat5 activity
All of the data presented thus far are derived from the K562 CML cell line. To rule out the possibility that our observations are unique to this particular system, we turned to the human myeloid cell line, TF-1 (Kitamura et al., 1989; Nakajima et al., 2001), which requires granulocyte macrophage colony-stimulating factor (GM-CSF) for growth and survival. Expression of Bcr-Abl in TF-1 cells results in cytokine-independent survival and proliferation (Nakajima et al., 2001), providing a model system that lacks the secondary genetic aberrations present in K562 cells. TF-1 cells were first infected with a Bcr-Abl retrovirus, which resulted in transformation to a cytokine-independent phenotype as expected (Figure 9a). These TF-1/Bcr-Abl cells were then infected with the wild-type and T338M-Hck retroviruses as well as the control virus carrying only the drug selection marker. Expression of these Hck proteins did not significantly affect the cytokine-independent growth of these cells (Figure 9a). We also expressed the Hck proteins alone in TF-1 cells, and observed that they were unable to stimulate GM-CSF-independent proliferation on their own (Figure 9a). All of the cell lines grew in the presence of GM-CSF (data not shown).
Figure 9.

Expression of Hck-T338M protects TF-1/Bcr-Abl cells against the apoptotic effects of A-419259. (a) Transformation of TF-1 cells to cytokine independence with Bcr-Abl. TF-1 cells were first transduced with Bcr-Abl retroviruses or the corresponding vector control and selected with G-418. The resulting populations were then transduced with wild-type Hck, Hck-T338M or vector control retroviruses and selected with puromycin. The six resulting cell populations were then tested for growth in the absence of GM-CSF using the CellTiter Blue assay (see Materials and methods). The average fold increase in the relative number of cells from three replicate assays is shown±s.d. Two separate experiments from independently derived cell populations gave comparable results; a representative example is shown. (b) TF-1/Bcr-Abl cells expressing the wild-type and T338M Hck proteins along with vector control cells were plated in the absence or presence of the indicated concentrations of A-419259 for 72 h. Apoptotic cells were detected by antiphosphatidylserine-Alexa Fluor 488-conjugated antibody staining and flow cytometry as described in ‘Materials and methods’. The bargraph shows the average of two independent experiments plotted±s.d. (c) TF-1/Bcr-Abl cells expressing wild-type and T338M-Hck along with the vector control were treated with the indicated concentrations of A-419259 for 5 h. Hck and Stat5 were immunoprecipitated from clarified cell lysates and immunoblotted with an anti-Hck phosphospecific pY418 antibody (pHck) or an antiphosphotyrosine antibody (pStat5), respectively. Duplicate membranes were blotted with an anti-Hck or anti-Stat5 antibody to insure equal loading. Representative blots are shown.
Next, we examined the effect of A-419259 treatment on survival of TF-1/Bcr-Abl cells. As shown in Figure 9b, incubation of the TF-1/Bcr-Abl vector control cells with A-419259-induced apoptosis in a dose-dependent manner. Expression of wild-type Hck partially reversed apoptosis in response to 0.3 and 1 μM A-419259 in the TF-1/Bcr-Abl population. However, TF-1/Bcr-Abl/Hck-T338M cells were much more resistant to A-419259-induced apoptosis, with almost complete protection at 0.3 μM. This effect closely parallels that observed in K-562 cells expressing Hck-T338M (Figure 6). Note that parental TF-1 cells are completely unresponsive to A-419259 in this assay (Wilson et al., 2002).
We next assessed Hck and Stat5 activity in each TF-1/Bcr-Abl cell population following A-419259 treatment. As shown in Figure 9c, wild-type Hck activity was partially inhibited with 0.1 and 0.3 μM A-419259 and completely blocked at 1 μM. Conversely, Hck-T338M showed significant resistance to A-419259, with partial inhibition observed only at 1 μM A-419259. Expression of wild-type Hck partially rescued Stat5 activation at 0.1 μM A-419259 compared with control TF-1/Bcr-Abl cells, whereas Hck-T338M rescued Stat5 activation at 0.3 and 1 μM. Taken together, these data are consistent with the partial reversal of A-419259-induced apoptosis by wild-type Hck and the more pronounced effect observed with Hck-T338M (Figure 9b).
A-419259 inhibits proliferation and induces apoptosis in CML CD34+ progenitor cells
In a final series of experiments, we determined whether selective inhibition of SFK activity affects the growth and survival of primary CML cells. Purified CD34+ progenitors from three chronic phase CML patients were incubated with A-419259 in the presence of cytokines (see Supplementary materials and methods for details). A-419259 treatment induced a dose-dependent inhibition of cell proliferation in all three patient samples, with caspase activation evident as early as 16 h (Figure 10). The extent of growth inhibition and caspase activation is comparable to that observed with 1 μM imatinib treatment. These results provide the first evidence that inhibition of SFK activity is sufficient to induce cell-cycle arrest and apoptosis in primary CD34+ CML cells.
Figure 10.

A-419259 induces growth arrest and apoptosis in CD34+ progenitor cells from chronic myelogenous leukemia (CML) patients. Purified CD34+ cells from three chronic phase CML patients were plated in triplicate in 96-well plates at 105 cells/ml and incubated for 16 or 48 h in the presence of A-419259 or imatinib at the concentrations indicated. Cell proliferation was monitored using the CellTiter-Blue viability assay as described in ‘Materials and methods’. The average fold increase in the relative number of cells from three replicate assays is shown±s.d. (left panels). Apoptosis was measured in each well using the Apo-One Caspase-3/-7 assay as described in ‘Materials and methods’. The average fold increase in caspase-3/-7 signal intensity from three replicate wells is shown±s.d. (right panels).
Discussion
Src family kinases have been implicated as important therapeutic targets for the treatment of CML (Kantarjian et al., 2007) with the success of dual Abl/Src inhibitors, such as dasatinib (Martinelli et al., 2005; Olivieri and Manzione, 2007). However, dasatinib has a broad range of molecular targets that include not only Bcr-Abl and the SFKs Fgr, Fyn, Hck, Lck, Lyn and Yes (Das et al., 2006), but also c-Kit, the PDGF receptor and the Ephrin receptor tyrosine kinase (Melnick et al., 2006). Although target promiscuity may be critical to the efficacy of many approved drugs, this property has traditionally been regarded as undesirable due to possible adverse effects. This concept points to a need for drugs that have ‘controlled promiscuity’—ones that specifically inhibit only disease-relevant targets. In the case of CML, one key to effective progress in designing such drugs is a more in-depth understanding of the relative contribution of individual members of the Src kinase family to Bcr-Abl signaling. Work presented here provides a novel approach to dissect the individual roles of myeloid Src family kinases to Bcr-Abl signaling and provides new evidence supporting a unique role for Hck in Bcr-Abl-induced proliferation and survival.
In this study, we exploited engineered inhibitor resistance to elucidate the role of Hck in Bcr-Abl signaling. Inhibition of protein kinases by ATP-competitive compounds often depends on the presence of a relatively small threonine residue at the gatekeeper position adjacent to the hydrophobic pocket in the catalytic site. Substitution of the gatekeeper residue with amino acids bearing bulkier side chains can dramatically reduce inhibitor potency (Table 1). Some of the first drugs developed against protein-tyrosine kinases include imatinib, which inhibits Abl, c-Kit and the PDGFR, as well as and gefitinib and erlotinib, which target the EGF receptor. Each of these compounds targets the hydrophobic pocket, and clinical resistance to these inhibitors can arise from mutations that replace the gatekeeper residue with bulkier amino acids (Blencke et al., 2004). On the basis of these observations, we engineered an A-419259-resistant Hck allele by replacing its gatekeeper residue, Thr-338, with methionine. This mutant allowed us to test the hypothesis that the expression of inhibitor-resistant Hck alone is sufficient to rescue CML cells from the antiproliferative and apoptotic effects of this broad-spectrum SFK inhibitor (Wilson et al., 2002).
Using both in vitro and cellular model systems, we first provide proof-of-principle evidence that the T338M mutation renders Hck resistant to A-419259. Thus, Hck-T338M was 30-fold less sensitive to A-419259 than wild-type Hck in an in vitro kinase assay (Figure 2). In addition, the expression of active forms of wild-type Hck or the Hck-T338M mutant in Rat-2 fibroblasts induced similar levels of transformation, suggesting that T338M mutation does not significantly affect the biological function of Hck in a cell-based system (Figure 3a). The Hck-T338M-YF mutant was at least 10-fold less sensitive to A-419259 than Hck-YF in the fibroblast transformation assay (Figure 3). More importantly, we show that Hck-T338M also maintains resistance to A-419259 upon expression in K562 cells and Bcr-Abl-transformed TF-1 cells, allowing us to test the individual contribution of Hck to Bcr-Abl transformation (Figures 4 and 9). The Hck-T338M mutation did induce a subtle decrease in the Km for ATP relative to the wild-type kinase in vitro. Although this difference did not appear to impact the biological functions of Hck examined here, this observation represents a possible caveat of the engineered inhibitor-resistant mutant approach.
Previous experiments with selective inhibitors as well as an Hck dominant-negative mutant suggest that SFKs contribute to Bcr-Abl-induced cell proliferation (Warmuth et al., 1997; Abram and Courtneidge, 2000; Lionberger et al., 2000; Wilson et al., 2002). A-419259 treatment also led to the suppression of Erk activity, a critical component of proliferative signaling downstream of Bcr-Abl (Hoover et al., 2001; Wilson et al., 2002). In this study, we show that Hck-T338M expression has a moderate protective effect on the A-419259-induced inhibition of cell proliferation (Figure 5), and this effect correlated with a rescue of Erk activity (Figure 7). The observation that Hck-T338M does not fully reverse A-419259-induced growth arrest and Erk inhibition suggests that other pathways contribute to Bcr-Abl-mediated cell proliferation.
Previous work in our laboratory has shown that collective SFK inhibition in K562 cells using A-419259 induces apoptosis that correlates with decreased Stat5 activation (Wilson et al., 2002). Here, we show that Hck-T338M, but not wild-type Hck, fully protects K562 cells against the apoptotic effects of 0.3 μM A-419259 (Figure 6). This new finding supports a nonredundant role for Hck in Bcr-Abl survival signaling in CML cells. In addition, Hck-T338M expression rescued Stat5 activation, which is completely blocked by A-419259 in control cells as well as cells expressing wild-type Hck (Figure 8). These results identify Hck as an important target for the development of apoptosis-inducing drugs for the treatment of CML. However, our data do not exclude the possibility that additional Src family members may participate in Bcr-Abl antiapoptotic signaling in other experimental systems. For example, siRNA against Lyn induces apoptosis of CML blast crisis cells, especially of lymphoid origin (Ptasznik et al., 2004). Furthermore, Lyn overexpression has been identified as an alternative mechanism of imatinib resistance in the absence of Bcr-Abl mutations (Dai et al., 2004; Wu et al., 2008). Taken together, these findings suggest that differences may exist in the roles of various SFKs within different hematopoietic lineages transformed by Bcr-Abl.
There is increasing evidence that patients achieving complete cytogenetic remission on imatinib have persistent residual CML stem cells (nonproliferating CD34+ cells), which might be responsible for relapse upon imatinib cessation (Cortes et al., 2004; Mauro et al., 2004; Holtz et al., 2005). These quiescent CD34+ cells represent less than 1% of total CD34+ cells (Copland et al., 2006). Here, we show for the first time that selective inhibition of SFK activity by A-419259 blocks proliferation and induces apoptosis in primary CML CD34+ progenitor cell populations as effectively as imatinib. This observation suggests that SFKs have a major function in Bcr-Abl signaling in primary CML progenitors. Future work will address the effect of selective SFK inhibition on the nonproliferating CD34+ cell population.
By pairing an inhibitor-resistant SFK mutant with a broad-spectrum SFK inhibitor, we have established new pharmacological evidence for Hck in Bcr-Abl survival signaling. This work validates Hck as a specific target for anti-CML drugs. More generally, our studies demonstrate the utility of mutants with engineered inhibitor resistance to address the contributions of individual members of a highly homologous kinase family to specific signaling pathways.
Materials and methods
Cell culture
K562, TF-1 and Rat-2 cells were obtained from the American Type Culture Collection and cultured as described elsewhere (Lerner and Smithgall, 2002; Wilson et al., 2002). Primary CD34+ CML cells were isolated using an immunomagnetic column separation method (Miltenyi Biotech, Auburn, CA, USA) following the manufacturer’s instructions.
Hck protein purification and kinase assay
Human Hck-YEEI and Hck-T338M-YEEI were expressed in Sf9 insect cells and purified as described by Schindler et al. (1999) and Trible et al. (2006). Kinase assays were performed using the Z′-Lyte method according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA) and our published protocols (Trible et al., 2006).
Rat-2 fibroblast transformation assay
Active forms of Hck and Hck-T338M were expressed in Rat-2 fibroblasts and transformation was assessed as colony formation in soft agar (Briggs and Smithgall, 1999; Schreiner et al., 2002). Hck expression and autophosphorylation were assessed by immuoblotting of clarified cell protein extracts.
Retroviral transduction of leukemia cell lines
Wild type and T338M mutant forms of Hck were expressed in K562 CML cells using the retroviral expression vector pMSCV-IRES-neo (Clontech, Mountain View, CA, USA). Transformation of TF-1 cells with a Bcr-Abl retrovirus carrying a G418 resistance marker is described elsewhere (Meyn et al., 2006). These cells were then infected with retroviruses carrying the wild type and T338M forms of Hck and a second antibiotic selection marker (puromycin resistance). Hck was immunoprecipitated from transduced cell lysates and immunoblotted for Hck protein and activation loop auotphosphorylation. Activation of the Stat5 and Erk pathways was evaluated with phosphospecific antibodies as described by Wilson et al. (2002).
Proliferation and apoptosis assays
Proliferation was assessed using the CellTiter-Blue Cell viability assay (Promega, Madison, WI, USA) and the manufacturer’s protocol. Apoptosis was measured using an antiphosphatidylserine antibody conjugated to Alexa Fluor 488 (Millipore, Billerica, MA, USA), and flow cytometry. Caspase activation was measured using the Apo-One Caspase-3/-7 assay (Promega) and the manufacturer’s instructions (Supplementary information).
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA101828 and GM077629 to TES). We thank Dr David Calderwood of the Abbott Bioresearch Center for the generous gift of the Src family kinase inhibitor, A-419259 and Dr Elisabeth Buchdunger of Novartis, Inc. for providing imatinib mesylate.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Abram CL, Courtneidge SA. Src family tyrosine kinases and growth factor signaling. Exp Cell Res. 2000;254:1–13. doi: 10.1006/excr.1999.4732. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific p210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701–4707. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Blencke S, Ullrich A, Daub H. Mutation of threonine 766 in the epidermal growth factor receptor reveals a hotspot for resistance formation against selective tyrosine kinase inhibitors. J Biol Chem. 2003;278:15435–15440. doi: 10.1074/jbc.M211158200. [DOI] [PubMed] [Google Scholar]
- Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- Briggs SD, Sharkey M, Stevenson M, Smithgall TE. SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J Biol Chem. 1997;272:17899–17902. doi: 10.1074/jbc.272.29.17899. [DOI] [PubMed] [Google Scholar]
- Briggs SD, Smithgall TE. SH2-kinase linker mutations release Hck tyrosine kinase and transforming activities in rat-2 fibroblasts. J Biol Chem. 1999;274:26579–26583. doi: 10.1074/jbc.274.37.26579. [DOI] [PubMed] [Google Scholar]
- Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- Cortes J, O’Brien S, Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004;104:2204–2205. doi: 10.1182/blood-2004-04-1335. [DOI] [PubMed] [Google Scholar]
- Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J Biol Chem. 2004;279:34227–34239. doi: 10.1074/jbc.M402290200. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the p210bcr-abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56:3589–3596. [PubMed] [Google Scholar]
- Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl] amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49:6819–6832. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- de Groot RP, Raaijmakers JA, Lammers JW, Koenderman L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. 2000;3:299–305. doi: 10.1006/mcbr.2000.0231. [DOI] [PubMed] [Google Scholar]
- Donato NJ, Wu JY, Stapley J, Lin H, Arlinghaus R, Aggarwal BB, et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res. 2004;64:672–677. doi: 10.1158/0008-5472.can-03-1484. [DOI] [PubMed] [Google Scholar]
- Eyers PA, Craxton M, Morrice N, Cohen P, Goedert M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single aminoacid substitution. Chem Biol. 1998;5:321–328. doi: 10.1016/s1074-5521(98)90170-3. [DOI] [PubMed] [Google Scholar]
- Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96:2269–2276. [PubMed] [Google Scholar]
- Goga A, McLaughlin J, Afar DEH, Saffran DC, Witte ON. Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell. 1995;82:981–988. doi: 10.1016/0092-8674(95)90277-5. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik D, Susa M, Varticovski L. Association of phosphatidylinositol 3-kinase with SHC in chronic myelogeneous leukemia cells. Oncogene. 1995;10:1385–1391. [PubMed] [Google Scholar]
- Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. 2005;19:1034–1041. doi: 10.1038/sj.leu.2403724. [DOI] [PubMed] [Google Scholar]
- Hoover RR, Gerlach MJ, Koh EY, Daley GQ. Cooperative and redundant effects of STAT5 and Ras signaling in BCR/ABL transformed hematopoietic cells. Oncogene. 2001;20:5826–5835. doi: 10.1038/sj.onc.1204549. [DOI] [PubMed] [Google Scholar]
- Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, Giles F, Quintas-Cardama A, Cortes J. Important therapeutic targets in chronic myelogenous leukemia. Clin Cancer Res. 2007;13:1089–1097. doi: 10.1158/1078-0432.CCR-06-2147. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci USA. 1990;87:6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, et al. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3 or erythropoietin. J Cell Physiol. 1989;140:323–334. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson MB, Smithgall TE, et al. The Src family kinase Hck couples Bcr-Abl to Stat5 activation in myeloid cells. EMBO J. 2002;21:5766–5774. doi: 10.1093/emboj/cdf562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Lerner EC, Smithgall TE. SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat Struct Biol. 2002;9:365–369. doi: 10.1038/nsb782. [DOI] [PubMed] [Google Scholar]
- Lerner EC, Trible RP, Schiavone AP, Hochrein JM, Engen JR, Smithgall TE. Activation of the Src family kinase Hck without SH3-linker release. J Biol Chem. 2005;280:40832–40837. doi: 10.1074/jbc.M508782200. [DOI] [PubMed] [Google Scholar]
- Lionberger JM, Wilson MB, Smithgall TE. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is suppressed by kinase-defective Hck. J Biol Chem. 2000;275:18581–18585. doi: 10.1074/jbc.C000126200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, et al. Structural basis for selective inhibition of Src family kinases by PP1. Chem Biol. 1999;6:671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shah K, Yang F, Witucki L, Shokat KM. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem. 1998;6:1219–1226. doi: 10.1016/s0968-0896(98)00099-6. [DOI] [PubMed] [Google Scholar]
- Lugo TG, Witte ON. The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol. 1989;9:1263–1270. doi: 10.1128/mcb.9.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli G, Soverini S, Rosti G, Baccarani M. Dual tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2005;19:1872–1879. doi: 10.1038/sj.leu.2403950. [DOI] [PubMed] [Google Scholar]
- Mauro MJ, Druker BJ, Maziarz RT. Divergent clinical outcome in two CML patients who discontinued imatinib therapy after achieving a molecular remission. Leuk Res. 2004;28(Suppl 1):S71–S73. doi: 10.1016/j.leukres.2003.10.017. [DOI] [PubMed] [Google Scholar]
- McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci USA. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci USA. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyn MA, III, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, et al. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem. 2006;281:30907–30916. doi: 10.1074/jbc.M605902200. [DOI] [PubMed] [Google Scholar]
- Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96:664–670. [PubMed] [Google Scholar]
- Nakajima A, Tauchi T, Ohyashiki K. ABL-specific tyrosine kinase inhibitor, STI571 in vitro, affects Ph-positive acute lymphoblastic leukemia and chronic myelogenous leukemia in blastic crisis. Leukemia. 2001;15:989–990. doi: 10.1038/sj.leu.2402137. [DOI] [PubMed] [Google Scholar]
- Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20:1179–1186. doi: 10.1128/mcb.20.4.1179-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri A, Manzione L. Dasatinib: a new step in molecular target therapy. Ann Oncol. 2007;18(Suppl 6):vi42–vi46. doi: 10.1093/annonc/mdm223. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- Porter M, Schindler T, Kuriyan J, Miller WT. Reciprocal regulation of Hck activity by phosphorylation of Tyr(527) and Tyr(416). Effect of introducing a high affinity intramolecular SH2 ligand. J Biol Chem. 2000;275:2721–2726. doi: 10.1074/jbc.275.4.2721. [DOI] [PubMed] [Google Scholar]
- Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat Med. 2004;10:1187–1189. doi: 10.1038/nm1127. [DOI] [PubMed] [Google Scholar]
- Raffel GD, Parmar K, Rosenberg N. In vivo association of v-Abl with Shc mediated by a non-phosphotyrosine-dependent SH2 interaction. J Biol Chem. 1996;271:4640–4645. doi: 10.1074/jbc.271.9.4640. [DOI] [PubMed] [Google Scholar]
- Ren R. The molecular mechanism of chronic myelogenous leukemia and its therapeutic implications: studies in a murine model. Oncogene. 2002;21:8629–8642. doi: 10.1038/sj.onc.1206090. [DOI] [PubMed] [Google Scholar]
- Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and G Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1:479–492. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, et al. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene. 1996;12:839–846. [PubMed] [Google Scholar]
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330–1340. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- Schreiner SJ, Schiavone AP, Smithgall TE. Activation of Stat3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem. 2002;277:45680–45687. doi: 10.1074/jbc.M204255200. [DOI] [PubMed] [Google Scholar]
- Shuai K, Halpern J, ten Hoeve J, Rao XP, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska MN, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3K/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12:27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–299. doi: 10.1053/j.gastro.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Trible RP, Emert-Sedlak L, Smithgall TE. HIV-1 Nef selectively activates SRC family kinases HCK, LYN, and c-SRC through direct SH3 domain interaction. J Biol Chem. 2006;281:27029–27038. doi: 10.1074/jbc.M601128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmuth M, Bergmann M, Priess A, Hauslmann K, Emmerich B, Hallek M. The Src family kinase Hck interacts with Bcr-Abl by a kinase- independent mechanism and phosphorylates the Grb2-binding site of Bcr. J Biol Chem. 1997;272:33260–33270. doi: 10.1074/jbc.272.52.33260. [DOI] [PubMed] [Google Scholar]
- Wilson MB, Schreiner SJ, Choi H-J, Kamens JS, Smithgall TE. Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene. 2002;21:8075–8088. doi: 10.1038/sj.onc.1206008. [DOI] [PubMed] [Google Scholar]
- Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, et al. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. 2008;100:926–939. doi: 10.1093/jnci/djn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol. 2001;21:840–853. doi: 10.1128/MCB.21.3.840-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]