

Summary
Lon, also known as protease La, is an ATP-dependent protease functioning to degrade many unstructured proteins. Currently, very little is known about the substrate determinants of Lon at the proteolytic site. Using synthetic peptides constituting different regions of the endogenous protein substrate λN, we demonstrated that the proteolytic site of Escherichia coli Lon exhibits a certain level of localized sequence specificity. Using an alanine positional scanning approach, we discovered a set of discontinuous substrate determinants surrounding the scissile Lon cleavage site in a model peptide substrate, which function to influence the kcat of the peptidase activity of Lon. We further investigated the mode of peptide interaction with the proteolytically inactive Lon mutant S679A in the absence and presence of ADP or AMPPNP by 2-dimensional nuclear magnetic resonance spectroscopy, and discovered that the binding interaction between protein and peptide varies with the nucleotide bound to the enzyme. This observation is suggestive of a substrate translocation step, which likely limits the turnover of the proteolytic reaction. The contribution of the identified substrate determinants towards the kinetics of ATP-dependent degradation of λN and truncated λN mutants by Lon was also examined. Our results indicated that Lon likely recognizes numerous discontinuous substrate determinants throughout λN such to achieve substrate promiscuity.
Keywords: ATP-dependent protease, fluorogenic peptidase assay, mechanistic probes
Introduction
Lon, also known as protease La, is an ATP-dependent serine protease that is localized in the mitochondria of eukaryotes and the cytosol of prokaryotes (1-7). It is a member of the AAA+ (ATPases associated with various cellular activities) family of proteases and is responsible for degrading regulatory and damaged or misfolded proteins in the cell into short peptides from 5-15 amino acids in length (3, 4, 6, 8-10). Lon is a homo-oligomer, comprised of six identical subunits, each containing an N-terminal domain which has, in some instances, been implicated in substrate binding, an ATPase domain, a proposed substrate sensor and discriminatory (SSD) domain, and a protease domain (11, 12). These subunits oligomerize into a barrel shape with the ATPase domain and protease domains at opposite ends. Upon hydrolysis of ATP, the protein is speculated to enter through the axial pore and be translocated to the protease domain where it is cleaved. Though optimal peptide bond cleavage occurs with hydrolysis of ATP, the ATPase and protease functions are not stoichiometrically linked, as degradation of certain unstructured protein and peptide substrates can occur in the presence of non-hydrolyzable ATP analogs such as AMPPNP (10, 13, 14).
Many in vivo substrates of bacterial Lon have been identified (SulA, RcsA, SoxS, λN) by genetic knock out studies (8, 15-17), but the mechanism by which the protease recognizes and initiates substrate degradation is not well understood. Currently, the accepted paradigm is that loosely folded or unstructured regions in a protein interact or bind to the proposed SSD domain of Lon followed by unfolding and translocation into the proteolytic chamber where the protein substrate is degraded (14, 18-23). Furthermore, synergistic interactions between Lon and different regions along an unfolded protein substrate lead to enhanced affinity of the enzyme substrate complex, thereby facilitating protein degradation (24). While these studies collectively present a global view of how Lon processes protein substrates, they do not address how the proteolytic site of Lon confers promiscuity in substrate cleavage specificity, nor do they reveal the extent to which the efficiency of peptide bond cleavage and ATP hydrolysis are affected by variation in the sequence of peptide substrate surrounding the scissile site.
Analysis of Lon cleavage profiles of protein substrates suggests that this protease prefers to cleave peptide bonds preceded by hydrophobic or aliphatic amino acids, but the presence of such residues alone does not warrant peptide bond cleavage (10, 25). As exemplified by the degradation of the intrinsically unstructured endogenous protein substrate, the λN protein (Fig 1), no consensus sequence could be discerned from the cleavage sites, suggesting that E. coli Lon displays promiscuity in the specificity of peptide bond cleavage. Another parameter that may dictate specificity of bond cleavage is which amino acid residues are located distal from the scissile site. An example from the λN protein substrate that supports this, is that only the peptide bond flanked by residues Ala16 – Gln17 and not Ala3 – Gln4 was cleaved (Fig 1) (10). Therefore, the substrate specificity at the proteolytic site of E. coli Lon likely constitutes the combined effects of enzyme interacting with substrate residues at and distal from the scissile sites. Previously, we developed a proteolytic site-director inhibitor of bacterial Lon by replacing the carboxyl terminal of a hydrolyzed peptide product with a boronic acid moiety. The resulting inhibitor exhibits a submicromolar inhibition constant against bacterial Lon (26). Therefore, an understanding of the mechanism of how Lon confers substrate specificity at the proteolytic site will likely benefit the next generation of specific active site-directed inhibitors, which could be further exploited as chemical agents to selectively inhibit bacterial Lon or as chemical genetic tools to identify the physiological functions of Lon in cells.
Figure 1. λN protein sequence and cleavage profile by E. coli Lon (10).
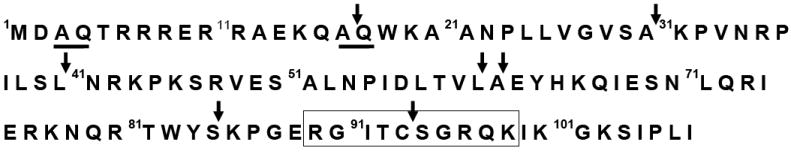
The λN protein is an intrinsically unstructured endogenous protein substrate of E. coli Lon. It has seven major Lon cleavage sites as indicated by the arrows. The amino acids that make up the FRETN89-98 peptide are highlighted in the rectangle. The underlined A-Q pairs highlight the lack of specificity of Lon, in that the amino acid residues flanking the scissile site alone do not warrant peptide bond cleavage, as 16A-17Q but not 3A-4Q was selectively cleaved by E.coli Lon.
In this study, we utilized the degradation profile of the endogenous E. coli Lon substrate, λN protein, to generate a panel of peptides to probe the substrate specificity of E. coli Lon (14). From the full length λN protein, we developed short peptides, eleven amino acids in length, that contain a single cleavage site and a fluorescent donor-quencher pair positioned on opposite termini, which allows us to monitor an increase in fluorescence upon cleavage and separation of the donor and quencher. An alanine scan of the model peptide FRETN 89-98 further identified the substrate determinants necessary for E. coli Lon cleavage. The substrate recognition residues are located at the cleavage site (P1) and 2 additional positions distal to the cleavage site (P3 and P3’ sites). After identifying these important residues, we synthesized analogs of FRETN 89-98 that were 13C and 15N labeled at the P1, P3 and/or P3’ positions and employed NMR spectroscopy to monitor the interaction of E. coli Lon and the peptide in the absence and presence of various nucleotides. The contribution of the peptide sequence constituting residues 89-98 of λN in directing protein degradation was also investigated. Collectively, our results implicate the existence of discontinuous substrate determinants located at the vicinity of the scissile sites of substrates. Interactions of these substrate determinants with E. coli Lon affect the kcat of peptide bond cleavage, which may be attributed to the ATP-dependent substrate translocation step in Lon.
Materials and Methods
Materials
ATP, ADP, AMPPNP, dansyl chloride, TPCK-treated Trypsin, HEPES, and PEI-cellulose TLC plates were purchased from Sigma and Fisher. All Fmoc-protected amino acids, Fmoc-protected Lys Wang resin, Boc-anthranilamide (Abz), and HBTU were purchased from Advanced ChemTech and EMD Biosciences. [α-P32]ATP was purchased from Perkin-Elmer. Ligate-IT Rapid Ligation Kit was purchased from USB. Primers were purchased from Integrated DNA Technologies. All enzyme concentrations are reported as E. coli Lon monomer concentrations, as determined by Bradford assay using BSA as a standard (27). All experiments were performed at 37° C. All reagents are reported as final concentrations. All peptides were synthesized using standard Fmoc solid-phase synthesis protocols as described previously (28-30). Fluorescent peptides corresponding to various regions of the λN protein are shown in Figure 2. All peptides contain the Y(NO2) - Abz internal fluorescence quenching pair used in the FRETN 89-98 peptide previously designed for the kinetic characterization of E. coli Lon (31). Nonfluorescent analogs, in which a tyrosine residue was used in place of the Y(NO2) and Bz was used in place of the Abz, were also synthesized (similar to the FRETN 89-98 peptide, (13)). FRETN 79-89 contains the Y(NO2) at the C-terminal and the Abz on the N-terminal due to the additional possibility of monitoring the cleavage using the W82 and Y(NO2) FRET pair. This puts both fluorescence donors on the N-terminal side of the cleavage site. Peptidase activity was detected by monitoring an increase in fluorescence upon excitation at 320 nm and emission at 420 nm.
Figure 2. Amino acid sequences of the fluorescent FRETN peptide substrates designed based upon the degradation profile of λN by E. coli Lon.

Peptides were synthesized corresponding to various regions of the λN protein cleaved by Lon. The Lon cleavage sites are underlined. With the exception of FRETN 79-89, all peptides contain the Y(NO2) on the N-terminal and the Abz on the C-terminal. The kinetic parameters for the cleavage of L60-A61 and A61-E63 in the peptide substrate (see Figure 1) could not be determined because the concentrations of substrate needed to obtain a velocity saturation plots exceeds the solubility limit of this peptide in the reaction buffer. Each peptide contains the same length and the same internal fluorescence quenching pair: Y(NO2) which quenches the fluorescence of Abz in the intact substrate. Though not native to the full length protein, in the peptides the Y(NO2) is assigned a number in the peptide sequence. Upon peptide bond cleavage, separation of Y(NO2) from Abz allows the detection of fluorescence emission generated by the latter at excitation 320 nm and emission 420 nm.
ATP-dependent peptidase assay
Peptidase activity was measured using a Fluoromax 3 spectrofluorimeter (Horiba Group) with a circulating water bath to maintain reactions at 37 °C and 3 mm path length microcuvettes from Hellma. Reactions contained 50 mM HEPES pH 8.0, 75 mM KOAc, 5 mM Mg(OAc)2, 5 mM DTT, 125 nM to 1 μM wild-type E. coli Lon and varying concentrations of peptide substrate. In all kinetic assays, a mixed substrate was utilized (10% fluorescent and 90% nonfluorescent or 1% fluorescent and 99% nonfluorescent when ≥ 10 mM substrate was used) to avoid problems from the inner filter effect where intermolecular quenching rather than intramolecular quenching occurs at high concentrations of Y-NO2 (13, 32). Previously, we demonstrated that the fluorescent and nonfluorescent analogs of FRETN 89-98 were degraded by E. coli Lon identically (13). Therefore, this study assumes that both analogs of the other λN peptides were processed by E. coli Lon identically. A 1% fluorescent and 99% nonfluorescent peptide mixture was used for the λN25-35 peptide due to the very high Km value and the need to employ very high peptide concentrations (25 μM – 2 mM) to determine kcat. For all other peptides a 10% fluorescent substrate mixture was used (25 μM – 1.5 mM) to determine the kinetic parameters. After equilibrating at 37°C for 1 minute, the reaction was initiated with 500 μM ATP. The amount of hydrolyzed peptide was measured by determining the maximum fluorescence generated per micromolar peptide after complete digestion by trypsin, chymotrypsin or elastase. The steady-state rate of the reaction was determined from the tangent of the linear portion of the time course. This rate was converted to an observed rate constant (kobs) by dividing the rate by the enzyme concentration used in the assay. Using KaleidaGraph (Synergy Software), all of the rate constants were plotted as a function of peptide concentration and fit with eq. 1
| (1) |
where kobs is the observed rate constant,kmax is the maximum rate constant, S is the concentration of peptide, n is the Hill coefficient. Km, the concentration of peptide required to reach 50% kmax, is calculated from the relationship log Km’ = n log Km. All kinetic data shown is a result of averaging at least 3 separate experiments.
ATPase Activity of E. coli Lon
Steady-state ATP hydrolysis activity was measured using the [α-32P]ATP as described previously (13). Briefly, each reaction contained 50 mM HEPES pH 8.0, 75 mM KOAc, 5 mM Mg(OAc)2, 5 mM DTT, 150 nM E. coli Lon, and the peptide stimulated reactions contained 1 mM peptide. The reactions were initiated with ATP and 5 μL aliquots were quenched at various time points with 10μL 0.5 N formic acid. The reaction was spotted onto a PEI-cellulose TLC plate and developed in 0.3 M potassium phosphate buffer (pH 3.4). The radiolabeled ADP was quantified using the Packard Cyclone storage phosphor screen Phosphor imager from Perkin-Elmer Life Science. The concentration of ADP at each time point was determined using eq. 2
| (2) |
where ADPdlu is the amount of ADP quantified, ATPdlu is the amount of ATP quantified, and [ATP] is the initial amount of ATP. The initial rates of ATP hydrolysis were defined by the [ADP] produced over time, when < 10% of ATP was consumed in the reactions. Dividing the initial rates of ATP hydrolysis by enzyme concentration yielded the kobs values.
λN purification and generation of λN deletion mutants
PCR was used to amplify the His tag λN insert from λN DNA using oHF014 (5’-CGCGGATCCAATGGATGCACAA-3’) as the forward primer and oHF013 (5’-AATTAAAGCTTCTAGATAAGAGGAATCGA-3’) as the reverse primer. The pCOLA-Duet vector and the new insert were digested with BamHI and HindIII restriction enzymes, purified, ligated using the Ligate-IT Rapid Ligation Kit (USB) and transformed into DH5α competent cells (Invitrogen). The plasmid pHF012 containing an N-terminal 6x His tag was transformed into the BL21 (DE3) E. coli Lon cell strain and the cells grown to OD600 = 0.6 in 30 μg/mL kanamycin in SB (superbroth) and induced for 2 hours with 1 mM IPTG. The cells were pelleted by centrifugation and lysed by adding 5 mL of lysis buffer per gram of cells (lysis buffer: 100 mM sodium phosphate pH 8.0, 10 mM Tris, 1 mM beta-mercaptoethanol, 8 M urea) and stirring for 45 minutes. All cell debris was pelleted by centrifugation. The cleared lysate was loaded onto a Ni-NTA agarose column (Qiagen) equilibrated with lysis buffer. The column was then washed with lysis buffer to remove all excess proteins. The λN protein was eluted from the column with elution buffer (elution buffer: 100 mM sodium phosphate pH 4.5, 10 mM Tris, 1 mM beta-mercaptoethanol, 8 M urea). The eluted protein was extensively dialyzed into λN storage buffer to remove urea (λN storage buffer: 20 mM Tris pH 7.6, 50 mM sodium chloride, 1 mM beta-mercaptoethanol, 20% glycerol). A white precipitate formed during dialysis and was removed by centrifugation. The λN protein in λN storage buffer was analyzed by SDS-PAGE on a 12.5% acrylamide gel and stained with Coomassie Brilliant Blue to assess purity. The protein was quantitated using the extinction coefficient at A280 (ε = 14,060 M-1 cm-1).
λN deletion mutants (λNΔ89-98, λNΔ89-107 and λNΔ99-107) were generated using basic cloning techniques. The forward primer used for all the mutants was oHF014 (5’-CGCGGATCCAATGGATGCACAA-3’). The reverse primers used were oJW103 (5’-ATGCTATAAGCTTCTAGATAAGAGGAATCGATTTTCCCTTAATTTCGCCAGGCTT-3’) for λNΔ89-98, oJW102 (5’-ATGCTATAAGCTTCTATTCGCCAGGCTT-3’) for λNΔ89-107 and λNΔ99-107rev (5’-ATGCTATAAGCTTCTATTTCTGGCGTCCACT-3’) for λNΔ99-107. PCR was used to amplify the λN gene with the desired deletions from plasmid pHF012. The PCR products were gel purified using a 0.8% agarose gel. The pCOLA-Duet vector (Novagen) and the purified PCR products (insert) were digested with Bam HI and Hind III restriction enzymes. The digested vector was gel purified and the digested inserts were purified by phenol chloroform extraction. The vector and insert were ligated using the Ligate-IT Rapid Ligation kit (USB) and transformed into XL1 Blue competent cells (Stratagene). The λN protein deletion mutant plasmids were verified by DNA sequencing analysis and transformed into the BL21 (DE3) E. coli cell strain.
λN degradation assay
λN degradation assays contained 50 mM HEPES pH 8.0, 15 mM magnesium acetate, 5 mM DTT, 10 μM wild-type or mutant λN protein, 1 μM E. coli Lon and the reaction was initiated with 5 mM ATP. At various time points, reaction aliquots were quenched with 5x SDS-PAGE loading dye and incubated at 90°C for 1 minute. The aliquots were loaded and run on a 12.5% SDS-PAGE gel and stained with Coomassie Brilliant Blue to detect the protein.
Results
Examining E. coli Lon substrate specificity using λN peptides
To examine the substrate specificity of E. coli Lon, we generated a panel of fluorescent λN peptides based on the cleavage profile of λN by E. coli Lon (Fig 1). The peptide sequences are shown in Figure 2. Despite the addition of the internal fluorescence quenching groups Y(NO2) and K(Abz), all the peptides have the same length but different amino acid sequences. Therefore comparing the kinetic parameters of ATP-dependent peptide cleavage and the extent of ATPase stimulation at saturating peptide concentration allows the evaluation of the contribution of substrate sequences towards the peptidase as well as the ATPase activities of Lon.
With the exception of FRETN 55-65, complete steady-state kinetic analyses of E. coli Lon degrading the FRETN 1-21, FRETN 35-45, FRETN 79-89 and FRETN 89-98 peptides were performed using the continuous fluorescence based assay. The steady-state rate (kobs) of peptide hydrolysis was determined under conditions of saturating ATP and varying concentrations of peptide. A complete kinetic study of the FRETN 55-65 peptide could not be completed due to peptide solubility issues. Nevertheless, ATP-dependent peptide cleavage occurred up to 25 μM of FRETN 55-65 (the solubility limit of this peptide), but the rate of the reaction was significantly slower compared to the one observed for FRETN 89-98 under identical substrate concentration (data not shown). Therefore FRETN 55-65 is likely a poorer substrate than the FRETN 89-98 substrate (data not shown).
As shown in Fig 3, sigmoidal plots are observed when the kobs (observed rate/[Lon]) values of ATP-dependent peptidase reactions are plotted as a function of peptide concentration. The data are best fit with eq. 1 and the results are summarized in Table 1. Comparing the kcat and Km values for the various peptides (Fig 2) in Table 1, reveals that FRETN 89-98 has the highest kcat/Km value, suggesting that it is a more “optimized” substrate compared to the others shown in Fig 2. There is also a difference in the Km and kcat values for peptide cleavage, but not in the extent of ATPase stimulation (Table 2) among the substrates examined. The observed rate constants for ATPase activity with the various FRETN peptides (with the exception of FRETN 55-65 due to problems with solubility) as well as the enhancement over the intrinsic ATPase activity without peptide are reported in Table 2. All the peptides stimulate the ATPase activity of E. coli Lon to approximately the same extent as FRETN 89-98. The relative sensitivity of the peptidase, but not ATPase activity towards variations in peptide substrate sequence, implicates the existence of “localized” substrate determinants for the proteolytic site of E. coli Lon within the ~ 9 amino acid substrates.
Figure 3. Steady-state kinetics of ATP-dependent hydrolysis of FRETN peptide substrates by E. coli Lon.
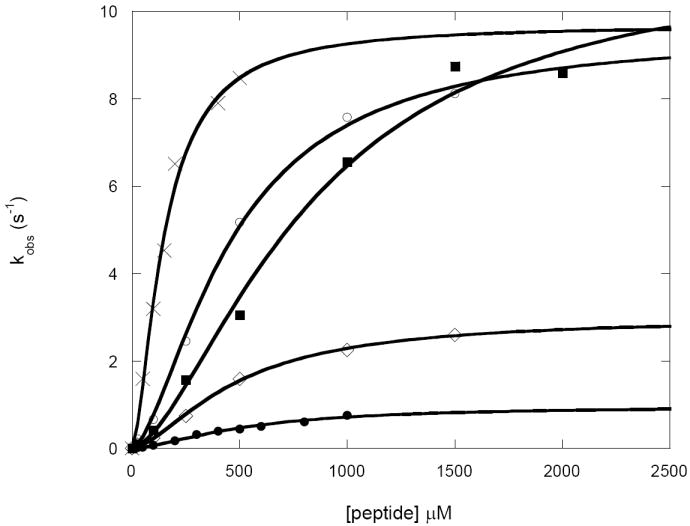
The steady-state rate constants (kobs) of peptide hydrolysis with varying concentrations of FRETN11 21 (●), FRETN25-35 (■), FRETN35-45 (○), FRETN79-89 (◊),and FRETN89-98 (X) were determined using the continuous fluorescence based peptidase assay as described in materials and methods (13). The kobs values, determined by dividing reaction rates with enzyme monomer concentration, were plotted as a function of peptide concentration. The data were best fit with eq 1 (materials and methods) to yield the kinetic parameters summarized in Table 3. The fit of the data is illustrated by solid lines.
Table 1. Steady-state kinetic parameters for peptidase activity with λN peptides.
The steady-state rates of peptide cleavage of the following peptides were monitored by fluorescence spectroscopy as described in the peptidase section of materials and methods to yield the velocity saturation plots of the corresponding substrates. The velocity data were best fit with eq 1 to provide a Hill coefficient (n = 1.6) and the kcat and Km values as summarized below.
| kcat (s-1) | Km (μM) | kcat/Km (× 104 M-1s-1) | |
|---|---|---|---|
| FRETN 11-21 | 0.97 ± 0.06 | 524 ± 149 | 0.18 |
| FRETN 25-35 | 11.3 ± 0.7 | 836 ± 290 | 1.3 |
| FRETN 35-45 | 9.5 ± 0.2 | 461 ± 92 | 2.0 |
| FRETN 55-65 | ND | ND | ND |
| FRETN 79-89 | 3.0 ± 0.1 | 479 ± 87 | 0.6 |
| FRETN 89-98 | 9.0 ± 0.5 | 102 ± 30 | 8.8 |
ND: not determined due to solubility problem
Table 2. Steady-state kinetic parameters for ATPase activity with λN peptides.
The reported kobs values for the ATPase activity of Lon in the absence and presence of various peptides were obtained by dividing the rate of ATP hydrolysis determined using the radiolabeled ATPase assay described in materials and methods by the concentration of enzyme monomer used. Stimulation values show the degree to which the rate of ATP hydrolysis was increased in the presence of peptide by dividing the kobs in the presence of peptide by the kobs in the absence of peptide. All peptides with the exception of FRETN 55-65 stimulated the ATPase activity of Lon to approximately the same extent.
| kobs (s-1) | Stimulation | |
|---|---|---|
| intrinsic | 0.34 ± 0.02 | - |
| FRETN 11-21 | 1.00 ± 0.25 | 2.9 |
| FRETN 25-35 | 1.35 ± 0.29 | 4.0 |
| FRETN 35-45 | 1.47 ± 0.39 | 4.3 |
| FRETN 55-65 | ND | - |
| FRETN 79-89 | 1.08 ± 0.02 | 3.2 |
| FRETN 89-98 | 1.21 ± 0.06 | 3.6 |
ND: not determined due to solubility problem
Alanine scan of FRETN 89-98 peptide
We performed an alanine scan of FRETN 89-98 to examine which residues within the substrate are important for recognition by the enzyme. Each amino acid in FRETN 89-98 was systematically substituted by an alanine residue to yield the Class I peptides (Table 3). The ATPase activity of E. coli Lon in the presence of the alanine scan peptides was measured using the radiolabeled ATPase assay previously described (13). All the peptides stimulated the ATPase activity of E. coli Lon to approximately the same extent as the FRETN 89-98 peptide (data not shown).
Table 3. Steady-state kinetic parameters for peptidase activity with λN 89-98 peptide variants.
From the FRETN 89-98 model substrate, additional peptides were synthesized to identify the residues important for activity. Steady-state kinetic parameters for peptidase activity were determined using the fluorogenic peptidase assay as described in materials and methods. Class I peptides represent the alanine scan, in which each residue, in turn, was replaced by an alanine while other residues remained the same. Of the Class I peptides, alanine substitutions at I91 (P3), C93 (P1), and R96 (P3’) showed a large decrease in the kcat/Km values, identifying these residues as important for maximal peptidase activity. Class II peptides contain substitutions of the three residues that were identified as integral to peptidase activity. Maximum activity occurred in C2-005, where the only non-alanine residues were at I91, C93 and R96. In the peptide C2-003, with alanine substitution at all three residues, peptidase activity was practically abolished. Class III peptides contained substituted at the cleavage site, indicating the importance of the C93 (P1).
| Class I peptides | kcat (s-1) | Km (μM) | kcat/Km (×104 M-1s-1) |
|---|---|---|---|
| FRETN 89-98 R89A | 8.5 ±0.4 | 165 ± 38 | 5.1 |
| FRETN 89-98 G90A | 7.2 ± 0.6 | 104±36 | 6.9 |
| FRETN 89-98 I91A | 2.1 ±0.3 | 168±69 | 1.2 |
| FRETN 89-98 T92A | 6.2 ±0.5 | 121±41 | 5.1 |
| FRETN 89-98 C93A | 3.3±0.3 | 146±47 | 2.2 |
| FRETN 89-98 S94A | 6.7±0.8 | 80±40 | 8.4 |
| FRETN 89-98 G95A | 8.7±0.6 | 123±38 | 7.0 |
| FRETN 89-98 R96A | 2.5±0.2 | 117±39 | 2.1 |
| FRETN 89-98 Q97A | 5.6±0.8 | 99±51 | 5.6 |
| Class II peptides | |||
| FRETN 89-98 I91R, R96I (C2-001) | 0.33±0.03 | 156±58 | 0.21 |
| FRETN 89-98 I91A, R96A (C2-002) | 0.24±0.03 | 199±83 | 0.12 |
| FRETN 89-98 I91A, C93A, R96A (C2-003) | 0.16±0.05 | 325±200 | 0.04 |
| FRETN 89-98 All Ala except I91, R96 (C2-004) | 6.7±0.1 | 133±75 | 5.0 |
| FRETN 89-98 All Ala except I91, C93, R96 (C2-005) | 4.7±0.2 | 56±15 | 8.4 |
| Class III peptides | |||
| FRETN 89-98 C93G | 0.031±0.01 | 206±98 | 0.01 |
| FRETN 89-98 C93S | 0.051±0.01 | 208±92 | 0.02 |
| FRETN 89-98 S94G | 3.67±0.57 | 196±92 | 1.9 |
The steady-state rates of peptide hydrolysis for each peptide with saturating ATP and varying concentrations of peptide (25 – 200 μM) were measured using the continuous fluorescence based assay used previously for FRETN 89-98 (14). Calibration curves for each peptide were created by completely digesting known amounts of peptide with trypsin and measuring the maximum amount of fluorescence generated. At peptide concentrations greater than ~ 150 μM the linear change in fluorescence was decreased due to the inner filter effect (32). The inner filter effects were corrected at peptide concentrations greater than 150 μM by determining the percentage of fluorescence that is reduced relative to the linear calibration curve. The rates of peptide hydrolysis were corrected by dividing the rate at a specific peptide concentration by the percentage of fluorescence reduction at the same peptide concentration (14). All of the rates were converted to observed rate constants (kobs) by dividing by the enzyme concentration. The observed rate constants were plotted as a function of peptide concentration and the data were fit best with eq. 1 to yield the kinetic parameters summarized in Table 3. Comparing the kcat and Km values among the Class I peptides reveal that the latter values are relatively insensitive to positional alanine substitution. The kcat values of the FRETN 89-98 I91A, FRETN 89-98 C93A, and FRETN 89-98 R96A peptides are ~ 3-to 4-fold reduced compared to the rest of the Class I and FRETN 89-98 peptides (Fig 4 and Table 1), suggesting that these positions, which are I91 (P3), C93 (P1), and R96 (P3’) contribute to the peptidase specificity.
Figure 4. Graphical comparison of the kcat values of FRETN 89-98 alanine-substituted peptides degradation by E. coli Lon.
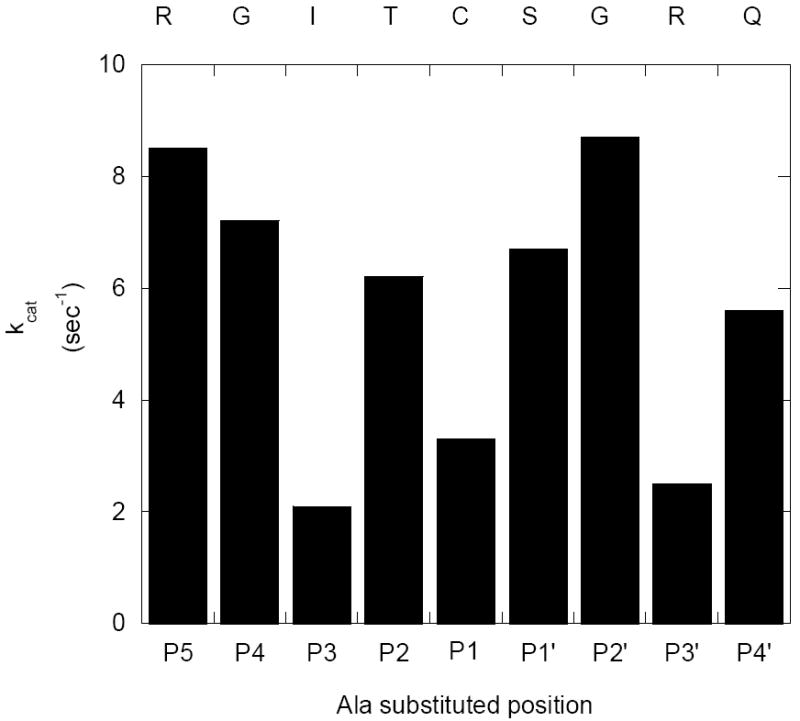
The kcat values for the ATP-dependent cleavage of the Class I peptides by E. coli Lon were determined using the fluorogenic peptidase assay described in methods and materials, in the presence of 150 nM Lon monomer and various concentrations of the indicated peptide at saturating ATP (500 μM). The velocity data were best fit with eq 1 to yield a Hill coefficient of 1.6 and the corresponding kcat and Km values. The kcat values of the respective peptides are compared. The kcat values for the peptides containing Ala substitution at the P3, P1 and P3’ positions are relatively lower than the others. Additional kinetic parameters are summarized in Table 3, under Class I peptides.
To further evaluate the involvement of I91, C93 and R96 in FRETN 89-98 (Fig 2), the steady-state kinetic parameters of peptide cleavage in the Class II substrates were determined (Table 3). Within the errors of analyses, the Km values of the Class II substrates were comparable. However, switching the positions of I91 and R96 reduces the kcat of peptide cleavage by almost 30-fold (C2-001 in Table 3). In addition, the kcat values of the peptides containing alanine replacement at > 1 of the aforementioned recognition sites (C2-002 to C2-003 in Table 3) differ from FRETN 89-98 by 30-to 50-fold. By contrast, the kcat values of peptides retaining 2 or more of the recognition sites (C2-004 and C2-005) are comparable to those determined for FRETN 89-98 or the Class I substrates. Taken together, these results indicate that the P3, P1 and P3’ positions are crucial for determining the substrate cleavage efficiency as manifested in the kcat of E. coli Lon protease, and a set of localized but discontinuous substrate determinants exist in the proteolytic site of E. coli Lon.
Importance of the C-S cleavage site in the FRETN 89-98 peptide
The FRETN 89-98 peptide is cleaved by E. coli Lon at a single site between the Cys93 and Ser94 residues (13). Results from the alanine scan experiment (Table 3, Class I peptides) indicate that replacing Cys93 with Ala reduces the kcat of peptide cleavage by approximately 3 fold, but replacing Ser94 with Ala imposes little effect. To further explore the importance of the P1 and P1’ positions in conferring peptide cleavage efficiency in the proteolytic site of E. coli Lon, we synthesized and determined the steady-state kinetic parameters for ATP-dependent cleavage of the Class III peptides (Table 3). According to Table 3, the Km values of the FRETN 89-98 C93G and FRETN 89-98 C93S peptides are similar to the Km of FRETN 89-98 (Table 1); but the kcat values of peptide cleavage are reduced by 180-to 300-fold. On the contrary, replacing the Ser94 (at the P1’ position) with a Gly residue (FRETN 89-98 S94G) results in very little effect on Km, and a 3-fold reduction in kcat.
Taken together, our results suggest that P1 has a lower tolerance than P1’ in accommodating substrate diversity. The significant reduction of the kcat in the Gly-and Ser-but not Ala-substituted C93 in FRETN 89-98 could be attributed the hydrophobicity of the substituted amino acid. Black and Mould calculated the logarithm of the partition coefficient (log P) for transfer from a polar to nonpolar phase (a measure of hydrophobicity) (33). Using the log P values they assigned a value from 0 (most polar) to 1.0 (most hydrophobic) to all 20 amino acids. The peptides with the most hydrophobic amino acid at the P1 position are the best substrates for Lon: Cys (0.680) > Ala (0.616) > Gly (0.501) > Ser (0.359). In addition, Gly contains a hydrogen atom as side chain, and is thus structurally less constrained than the other amino acids. As such, it is plausible the scissile bond in the FRETN 89-98 C93G peptide could not be oriented productively in the proteolytic site of E. coli Lon, which leads to the observed low kcat value.
Examine peptide binding to E. coli Lon by NMR spectroscopy
Previously, we demonstrated that the proteolytically inactive E. coli Lon mutant S679A displays wild type-like ATPase activity (34). The kinetic mechanism of peptide interaction with S679A has been determined. Upon defining the importance of the P3, P1 and P3’ sites in this study, we examined the extent to which nucleotide binding to E. coli Lon affects enzyme interaction with these three sites. To accomplish this, we utilized HSQC NMR spectroscopy to probe changes in the interaction between S679A Lon with the respective substrate recognition sites within the peptide containing residues 89-98 of λN. Three peptides, which contained uniformly labeled 13C (at the α-carbon and side chains), and 15N at the amide nitrogen at the P3 Ile and P3’ Arg, 13C and 15N labels at the P1 Leu, and 13C and 15N labels at the P3 Ile, were synthesized and used as probes to monitor peptide interaction with Lon upon binding to the protease inhibitor ADP and activator AMPPNP.
The 1H-15N HSQC spectra for the Ile/Arg and Leu isotopically labeled peptides are shown in Fig 6. The spectra of the Ile labeled peptide (not shown) were used as references to assign the Ile and Arg signals in the Ile/Arg peptide spectra shown in Fig 6A to E. The 1H-15N HSQC spectra, which detects 1H atoms directly attached to 15N atoms, is a standard NMR experiment that provides structural information about the backbone of peptides and proteins. As can be seen in Fig 6, significant signal broadening is observed in the peaks of both peptides in the presence of nucleotide (ADP or AMPPNP) and E. coli Lon. Interestingly, in the presence of E. coli Lon alone, the Ile/Arg exhibit a slight chemical shift change but little signal broadening (compare Figs 6A, 6C and 6E), whereas the Leu has no significant change in chemical shift, but signal broadening is observed (compare Figs 6F, 6H and 6J). The signal broadening phenomenon reflects that the peptide/enzyme interaction is occurring relatively fast on the NMR timescale, and is correlated with an increase in binding affinity. On the other hand, changes in the chemical shifts of the labeled peptides reflect a relatively rapid binding equilibrium on the NMR data acquisition timescale, and a weaker ligand binding scenario compared to the signal broadening situation. Analysis of the 1H-13C HSQC spectra showed similar broadening in the α-carbon and no peak movement in the side chains (data not shown). Differences in the spectral signals are further detected in the peptide samples containing E. coli Lon and ADP or AMPPNP (compare Figs 6B, 6D and 6E; Figs 6G, 6I and 6J), suggesting that the extent of peptide interaction with E. coli Lon is dictated by the specific nucleotide present. The signal broadening observed at the Leu residue in the presence of AMPPNP (Fig 6G) is more pronounced than that of the spectra corresponding to the presence of ADP (Fig 6I). Furthermore, the relative weakening of signal detected in Fig 6I compared to Fig 6G suggests that the association of peptide to E. coli Lon is probably stronger in the presence of AMPPNP.
Figure 6. Detect binding interaction between peptide substrate interacting with the proteolytically inactive Lon mutant S679A in the absence and presence of ADP or AMPPNP.
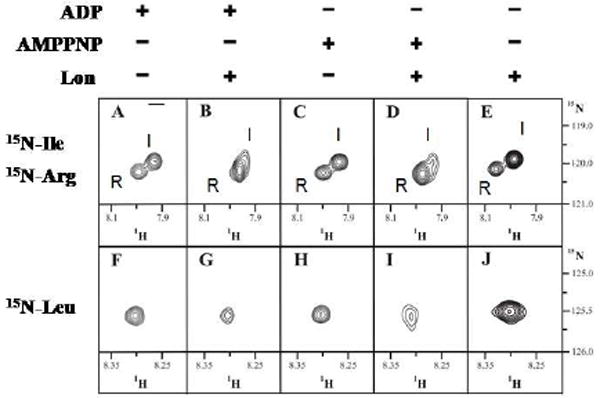
1H-15N HSQC spectra of synthetic peptide with 15N-labels on the backbone nitrogens of Ile and Arg (A-E) or Leu (F-J) in the presence of 0.1 mM ADP without (A, F) and with (B, G) Lon, 0.1 mM AMPPNP without (C, H) and with (D, I) Lon and Lon alone (E, J). The data suggests that the peptide-enzyme association is stronger in the presence of AMPPNP than ADP and Lon appears to interacts more with P1-Leu than P3-Ile and P3’-Arg.
It is also interesting to note that in the absence of nucleotide, the different regions of the peptide behave differently with the enzyme alone, as can be seen in Figs 6E and 6J. The change in chemical shift seen in 6E suggests that the Ile and Arg residues are in a different chemical environment, but may not be interacting as strongly with the enzyme as the Leu (Fig 6J) residue, where the detection of signal broadening is consistent with the binding equilibrium strongly shifted to the S679A:peptide complex.
Wild type λN and λN deletion mutant protein purification and degradation
E. coli Lon cleaves the λN protein at seven major cleavage sites (10). We synthesized and tested model peptides corresponding to these cleavage sites and found that the region spanning residues 89-98 of the λN protein is kinetically favored compared to the others (Fig 2 and Table 1). As some substrates contain a “recognition tag sequence”, which functions to aid the binding of substrates to Lon, we question whether residues 89-98 of λN confer a similar function. To evaluate this possibility, three λN mutants (λNΔ89-98, λNΔ89-107, and λNΔ99-107) were generated and purified using the same procedure as for the wild-type enzyme. The protein NΔ89-98 lacks residues 89-98, λNΔ89-107 lacks residues 89-107 and λNΔ99-107 lacks residues 99-107 (Fig. 7). The ATP-dependent degradation of mutant λN by E. coli Lon was monitored like wild type λN using a discontinuous protease assay, where each time point was quenched by gel loading dye containing 0.1% SDS that denatured the enzyme. The time courses of protein degradation are shown in Fig 7. It is discerned from Fig 7 that λNΔ89-98 is degraded with comparable efficiency as the wild type protein, but λNΔ89-107 and λNΔ99-107, which both lack the carboxyl terminus containing residues 99-107, are degraded at a slower rate. These results indicate that residues 99-107 rather than 89-98 are required for optimal λN protein degradation by E. coli Lon. The λN mutant study suggests that the carboxyl terminus of the λN protein contains some sort of Lon recognition sequence that functions to promote substrate degradation. However, additional signals located beyond residues 99-107 in λN likely contribute to substrate recognition, as the λNΔ89-107 and λNΔ99-107 mutants are still degraded by E. coli Lon, albeit with reduced efficiency.
Figure 7. Compare the time course of λN deletion mutants degradation by E. coli Lon.
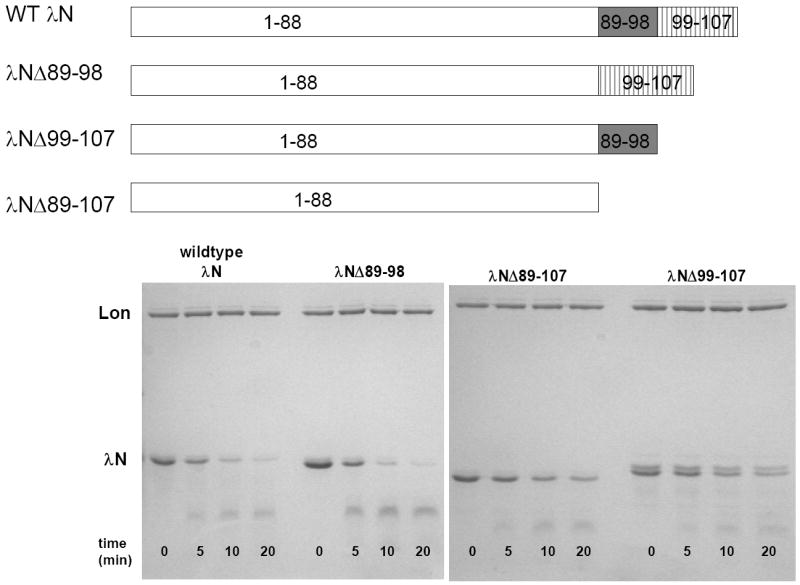
Purified wild-type and truncated λN lacking residues 89-98 (λNΔ89-98), 99-107 (λNΔ99-107) and 89-107 (λNΔ89-107) were digested with E. coli Lon in the presence of 1 mM ATP at the indicated times. The progress of the degradation reactions were monitored by 12.5% SDS-PAGE as described in materials and methods. All three mutants are degraded by Lon, but the wild type and λNΔ89-98 proteins are degraded faster than the λNΔ89-107 and λNΔ99-107 proteins, indicating that the presence of residues 99-107 promotes protein degradation by Lon.
Discussion
Lon catalyzes the hydrolysis of proteins into small peptides ranging from 5 – 15 amino acids in length (3). No consensus sequence has been determined which allows the prediction of cleavage sites a priori, however, some preference has been seen for cleavage after hydrophobic and non-polar residues (10, 35-37). Examining the degradation profile of the λN protein by the E. coli enzyme implicates the involvement of residues distal to the scissile bond in cleavage site selection, as sites with identical amino acids sequences are not cleaved (Fig 1) (10). On the basis that the kcat and Km values in the peptidase activity of E. coli Lon vary with peptide sequences (Table 1), we conclude that the cleavage specificity at the proteolytic site of E. coli Lon extends beyond the scissile site (P1-P1’). Through a positional alanine scan of FRETN 89-98 as substrates of E. coli Lon (Table 2, Classes I and II peptides), we further demonstrated the existence of multiple discontinuous substrate determinants located distal from the scissile site. In the case of FRETN 89-98, the substrate determinants are located at P3, P1 and P3’. As evident from the kcat values in the Class II peptide (Table 2), removal of side chain interaction by Ala at any one of these three positions modestly affects the cleavage of the respective substrate, elimination of multiple interactions between peptide with E. coli Lon significantly hampers substrate efficacy. Previously, we employed transient kinetic techniques to demonstrate that an ATP-dependent substrate delivery step constitutes the rate-limiting step in the first peptidase turnover in E. coli Lon (38). Therefore, the interaction between E. coli Lon and the three substrate determinants may be attributed to reduction in the substrate delivery step, which is manifested in the reduction in kcat of the peptidase activity of E. coli Lon.
The interactions between E. coli Lon and the substrate determinants in FRETN 89-98 were further investigated by monitoring the HSQC signals between peptides isotopically labeled at the P3, P1 and P3’ sites. In the case of the P1 site-labeled peptide, the Cys residue (Fig 2) was replaced by 13C and 15N labeled Leu. This replacement did not affect the kinetics of peptide cleavage (data not shown) and greatly simplified the peptide synthesis procedure. As suggested by the data presented in Fig 6, the Lon, Lon:ADP and Lon:AMPPNP complexes exhibit differences in their mode of interaction with the labeled peptides; and Lon:AMPPNP, which represent the ATP bound enzyme form, appears to bind peptide at higher affinity than Lon:ADP. The detection of variation in substrate affinity associated with different nucleotide binding to Lon is reminiscent of the “alternating affinity” mechanism proposed to account for the processive protein degradation activity of the heterosubunit ATP-dependent protease ClpAP (39). In the alternating affinity mechanism, the ATP and ADP bound enzyme forms exhibit different affinity for the polypeptide substrate such that repeated cycles of ATPase leads to processive translocation of the substrate to the proteolytic subunit. As Lon is also a processive protease, it is conceivable a similar alternating mechanism exists. Therefore, it is plausible that the P3, P1 and P3’ sites in the FRETN 89-98 substrate are important for substrate translocation, the rate-limiting step of the peptidase reaction catalyzed by E. coli Lon. Removal of these interactions, such as by alanine scan, will thus likely reduce the kcat of peptide cleavage, as has been observed in the Class I and II peptides (Table 3).
Comparing the HSQC data in Fig 6E and 6J also reveals that E. coli Lon interacts more with Leu at P1 than with Ile at P3 and Arg at P3’. This result explains why the C93G and C93S replacements in the Class III peptides (Table 3) almost abolish substrate efficacy, which results in significantly low kcat values. Possibly, the lack of productive side chain interaction at the P1 position hinders proper positioning of the scissile bond for attack by the proteolytic site S679 in Lon. Additional experiments concerning the investigation of uniformly-labeled FRETN 89-98 interacting with E. coli Lon by NMR spectroscopy are currently underway to further elucidate the mechanism of peptide interaction with Lon.
In some Lon substrates, certain regions within the substrate interact more strongly with Lon to increase the affinity of substrate binding, thereby facilitating subsequent degradation. In light of the relatively higher kcat and lower Km of FRETN 89-98 (Table 1), we question whether residues 89-98 in λN bear such a function. To this end, we compared the time courses of λN and truncated λN mutants lacking residues 89-98 and/or 99-107 degradation by E. coli Lon (Fig 7), and discovered that residues 99-107 rather than 89-98 of λN promote substrate degradation. Therefore, our results indicate that favorable kinetic parameters within a peptide substrate do not necessary constitute an “affinity tag” in a substrate. Interestingly, residues 99-107 in λN, which was previously shown by Maurizi to be relatively resistant to cleavage by E. coli Lon (10), facilitate λN degradation. Conceivably, this stretch of peptide may act as an affinity tag to promote substrate binding to Lon. As the deletion of residues 89-98 alone does not affect the efficiency of λN degradation (Fig 7), E. coli Lon likely binds to different regions of λN during protein degradation such that eliminating the interaction between residues 89-98 and the enzyme does not hinder substrate binding and degradation efficiency. Additional experiments will be needed to further elucidate the function of residues 99-107 of λN.
In conclusion, this work demonstrates that the proteolytic site of E. coli Lon exhibits sequence specificity by interacting with multiple substrate residues in a discontinuous manner. Previously, it has been shown that E. coli Lon interacts synergistically with a protein substrate at multiple sites (10, 25). It appears that the strategy is also employed in a more localized area to confer cleavage efficiency and/or specificity at the proteolytic site. As Lon is a stress-induced protease that needs to process damaged proteins of diverse sequences, the aforementioned substrate sampling strategy justifies the promiscuity of the peptide sequences that are processed by Lon. We propose that the use of synthetic peptides as mechanistic probes will be beneficial strategy for the development of proteolytic site-directed inhibitors for Lon. Recently, studies have indicated that Lon protease is required for systemic infection of the bacterial pathogen Salmonella enterica Typhimurium causing gastroenteritis in humans (40, 41). Therefore the production of specific inhibitors against bacterial Lon could potentially benefit antibiotic development. The work presented here provides a starting point for such an endeavor.
Figure 5. The P3, P1 and P3’ positions are crucial for determining substrate specificity.
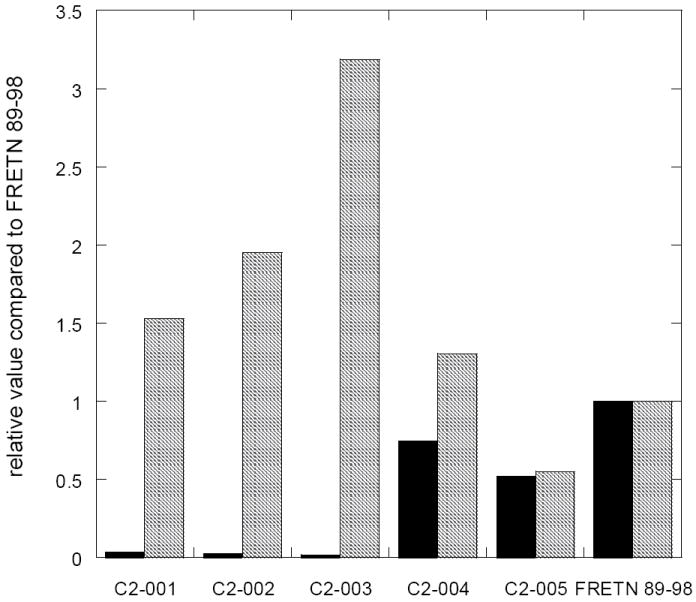
The kcat (■) and Km ( ) values for ATP-dependent cleavage of the Class II peptides (Table 3) by E. coli Lon are compared. The rates of peptide cleavage were determined using the fluorogenic peptidase assay described in materials and methods in the presence of 150 nM Lon monomer, various concentrations of the indicated peptides and 500 μM ATP. The Km values vary slightly among the peptide substrates but the kcat values exhibit significant reduction when > 1 Ala substitution are introduced into the peptide substrate.
) values for ATP-dependent cleavage of the Class II peptides (Table 3) by E. coli Lon are compared. The rates of peptide cleavage were determined using the fluorogenic peptidase assay described in materials and methods in the presence of 150 nM Lon monomer, various concentrations of the indicated peptides and 500 μM ATP. The Km values vary slightly among the peptide substrates but the kcat values exhibit significant reduction when > 1 Ala substitution are introduced into the peptide substrate.
Acknowledgments
Jason Hudak and Kirsten McNamara were recipients of the summer program in undergraduate research (SPUR) fellowship, a Howard Hughes Medical Institute sponsored program. This work was supported in part by the NIH grant GM067172.
Abbreviations
- Abz
anthranilamide
- ATP
adenosine tri-phosphate
- ADP
adenosine di-phosphate
- AMPPNP
adenylyl 5-imidodiphosphate
- Boc
tert-butoxycarbonyl
- BSA
bovine serum albumin
- Bz
benzoic acid amide
- Dansyl
5-(dimethylamino)naphthalene-1-sulfonyl
- Dlu
density light units
- DTT
dithiothreitol
- Fmoc
9H-fluoren-9-ylmethoxycarbonyl
- FRET
fluorescence resonance energy transfer
- HBTU
O-benzotriazole-N,N,N’,N’-tetramethyluroniumhexafluorophosphate
- HEPES
N-2-hydroxyethylpiperzaine-N’-ethanesulphonic acid
- IPTG
isopropyl-beta-D-thiogalactopyranoside
- KOAc
potassium acetate
- λN
also known as the lambda N protein, a lambda phage protein that allows E. coli RNA polymerase to transcribe through termination signals in the early operons of the phage
- Mg(OAc)2
magnesium acetate
- NO2
nitro
- PEI-cellulose
polyethyleneimine-cellulose
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TLC
thin layer chromatography
- TPCK
tosyl phenylalanyl chloromethyl ketone
- Tris
2-amino-2-hydroxymethyl-1,2-propanediol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Charette MF, Henderson GW, Markovitz A. ATP hydrolysis-dependent protease activity of the lon (capR) protein of Escherichia coli K-12. Proc Natl Acad Sci U S A. 1981;78:4728–32. doi: 10.1073/pnas.78.8.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung CH, Goldberg AL. The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proc Natl Acad Sci U S A. 1981;78:4931–5. doi: 10.1073/pnas.78.8.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg AL, Moerschell RP, Chung CH, Maurizi MR. ATP-dependent protease La (lon) from Escherichia coli. Methods Enzymol. 1994;244:350–75. doi: 10.1016/0076-6879(94)44027-1. [DOI] [PubMed] [Google Scholar]
- 4.Maurizi MR. Proteases and protein degradation in Escherichia coli. Experientia. 1992;48:178–201. doi: 10.1007/BF01923511. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg AL, Waxman L. The role of ATP hydrolysis in the breakdown of proteins and peptides by protease La from Escherichia coli. J Biol Chem. 1985;260:12029–34. [PubMed] [Google Scholar]
- 6.Gottesman S, Gottesman M, Shaw JE, Pearson ML. Protein degradation in E. coli: the Ion mutation and bacteriophage lambda N and cll protein stability. Cell. 1981;24:225–33. doi: 10.1016/0092-8674(81)90518-3. [DOI] [PubMed] [Google Scholar]
- 7.Goff SA, Goldberg AL. Production of abnormal proteins in E. coli stimulates transcription of lon and other heat shock genes. Cell. 1985;41:587–95. doi: 10.1016/s0092-8674(85)80031-3. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- 9.Goldberg AL. The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur J Biochem. 1992;203:9–23. doi: 10.1111/j.1432-1033.1992.tb19822.x. [DOI] [PubMed] [Google Scholar]
- 10.Maurizi MR. Degradation in vitro of bacteriophage lambda N protein by Lon protease from Escherichia coli. J Biol Chem. 1987;262:2696–703. [PubMed] [Google Scholar]
- 11.Amerik A, Chistiakov LG, Ostroumova NI, Gurevich AI, Antonov VK. Cloning, expression and structure of the functionally active shortened lon gene in Escherichia coli. Bioorg Khim. 1988;14:408–11. [PubMed] [Google Scholar]
- 12.Chin DT, Goff SA, Webster T, Smith T, Goldberg AL. Sequence of the lon gene in Escherichia coli. A heat-shock gene which encodes the ATP-dependent protease La. J Biol Chem. 1988;263:11718–28. [PubMed] [Google Scholar]
- 13.Thomas-Wohlever J, Lee I. Kinetic characterization of the peptidase activity of Escherichia coli Lon reveals the mechanistic similarities in ATP-dependent hydrolysis of peptide and protein substrates. Biochemistry. 2002;41:9418–25. doi: 10.1021/bi0255470. [DOI] [PubMed] [Google Scholar]
- 14.Lee I, Berdis AJ. Adenosine triphosphate-dependent degradation of a fluorescent lambda N substrate mimic by Lon protease. Anal Biochem. 2001;291:74–83. doi: 10.1006/abio.2001.4988. [DOI] [PubMed] [Google Scholar]
- 15.Gottesman S, Maurizi MR. Regulation by proteolysis: energy-dependent proteases and their targets. Microbiol Rev. 1992;56:592–621. doi: 10.1128/mr.56.4.592-621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottesman S, Zipser D. Deg phenotype of Escherichia coli lon mutants. J Bacteriol. 1978;133:844–51. doi: 10.1128/jb.133.2.844-851.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci U S A. 1983;80:358–62. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their susbtrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–37. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 19.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CK, Baker TA, Sauer RT. Lon and Clp family proteases and chaperones share homologous substrate-recognition domains. Proc Natl Acad Sci U S A. 1999;96:6678–82. doi: 10.1073/pnas.96.12.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–20. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 22.Reid BG, Fenton WA, Horwich AL, Weber-Ban EU. ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci U S A. 2001;98:3768–72. doi: 10.1073/pnas.071043698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navon A, Goldberg AL. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell. 2001;8:1339–49. doi: 10.1016/s1097-2765(01)00407-5. [DOI] [PubMed] [Google Scholar]
- 24.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22:2267–77. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishii W, Maruyama T, Matsuoka R, Muramatsu T, Takahashi K. The unique sites in SulA protein preferentially cleaved by ATP-dependent Lon protease from Escherichia coli. Eur J Biochem. 2002;269:451–7. doi: 10.1046/j.0014-2956.2001.02668.x. [DOI] [PubMed] [Google Scholar]
- 26.Frase H, Lee I. Peptidyl boronates inhibit Salmonella enterica serovar Typhimurium Lon protease by a competitive ATP-dependent mechanism. Biochemistry. 2007;46:6647–57. doi: 10.1021/bi7002789. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and senstive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Bodanszky M. Peptide Chemistry: A Practical Textbook 2nd Edition. 2. Springer-Verlag; New York: 1993. [Google Scholar]
- 29.Wellings DA, Atherton E. Standard Fmoc protocols. Methods Enzymol. 1997;289:44–67. doi: 10.1016/s0076-6879(97)89043-x. [DOI] [PubMed] [Google Scholar]
- 30.Chan WC, W PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press; New York: 2003. [Google Scholar]
- 31.Lee I, Berdis AJ. Adenosine triphosphate-dependent degradation of a fluorescent lambda N substrate mimic by Lon protease. Anal Biochem. 2001;291:74–83. doi: 10.1006/abio.2001.4988. [DOI] [PubMed] [Google Scholar]
- 32.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Plenum; New York: 1999. [Google Scholar]
- 33.Black SD, Mould DR. Development of hydrophobicity parameters to analyze proteins which bear post-or cotranslational modifications. Anal Biochem. 1991;193:72–82. doi: 10.1016/0003-2697(91)90045-u. [DOI] [PubMed] [Google Scholar]
- 34.Patterson-Ward J, Huang J, Lee I. Detection and characterization of two ATP-dependent conformational changes in proteolytically inactive Escherichia coli lon mutants by stopped flow kinetic techniques. Biochemistry. 2007;46:13593–605. doi: 10.1021/bi701649b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishii W, Suzuki T, Nakada M, Kim YT, Muramatsu T, Takahashi K. Cleavage mechanism of ATP-dependent Lon protease toward ribosomal S2 protein. FEBS Lett. 2005;579:6846–50. doi: 10.1016/j.febslet.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 36.Ondrovicova G, Liu T, Singh K, Tian B, Li H, Gakh O, Perecko D, Janata J, Granot Z, Orly J, Kutejova E, Suzuki CK. Cleavage site selection within a folded substrate by the ATP-dependent lon protease. J Biol Chem. 2005;280:25103–10. doi: 10.1074/jbc.M502796200. [DOI] [PubMed] [Google Scholar]
- 37.Van Melderen L, Thi MH, Lecchi P, Gottesman S, Couturier M, Maurizi MR. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J Biol Chem. 1996;271:27730–8. doi: 10.1074/jbc.271.44.27730. [DOI] [PubMed] [Google Scholar]
- 38.Vineyard D, Patterson-Ward J, Berdis AJ, Lee I. Monitoring the Timing of ATP Hydrolysis with Activation of Peptide Cleavage in Escherichia coli Lon by Transient Kinetics. Biochemistry. 2005;44:1671–82. doi: 10.1021/bi048618z. [DOI] [PubMed] [Google Scholar]
- 39.Farbman ME, Gershenson A, Licht S. Single-molecule analysis of nucleotide-dependent substrate binding by the protein unfoldase ClpA. J Am Chem Soc. 2007;129:12378–9. doi: 10.1021/ja074168x. [DOI] [PubMed] [Google Scholar]
- 40.Takaya A, Suzuki M, Matsui H, Tomoyasu T, Sashinami H, Nakane A, Yamamoto T. Lon, a stress-induced ATP-dependent protease, is critically important for systemic Salmonella enterica serovar typhimurium infection of mice. Infect Immun. 2003;71:690–6. doi: 10.1128/IAI.71.2.690-696.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takaya A, Tomoyasu T, Tokumitsu A, Morioka M, Yamamoto T. The ATP-dependent lon protease of Salmonella enterica serovar Typhimurium regulates invasion and expression of genes carried on Salmonella pathogenicity island 1. J Bacteriol. 2002;184:224–32. doi: 10.1128/JB.184.1.224-232.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]