

Abstract
The evaluation of the solvation entropies is a major conceptual and practical challenge. On the one hand, it is interesting to quantify the factors that are responsible for the solvation entropies in solutions, while on the other, it is essential to be able to assess the contributions of the solvation entropies to the binding free energies and related properties. In fact, the solvation entropies are neglected in almost all the studies of the binding entropies. The main problem is that widely used approaches, such as the quasiharmonic (QH) approximation do not provide reliable results particularly, in cases of shallow potential and multidimensional surfaces while brute force evaluations of the entropic effects by simulating temperature dependence of the free energy converges very slowly. This paper addresses the above issue by starting with an analysis of the factors that are responsible for the negative solvation entropy of ions, showing that it is not due to the change in the solvent vibration modes or to the solvent force constant but to the changes in the solvent configurational space upon change in the solute charges. We begin by clarifying that when one deals with aqueous solutions, it is easy to evaluate the corresponding entropic effect by the Langevin dipole(LD) treatment. However, in this work we are interested in developing a general microscopic tool that can be used to study similar effects in the proteins. To this end, we explore the ability of our restraint release (RR) approach to evaluate the solvation entropy. We start this analysis by reviewing the foundation of this approach and in particular, the requirements of minimizing the enthalpy contribution to the RR free energy. We then establish that our approach is not a specialized harmonic treatment but a rather powerful approach. Moving to the main topic of this work, we demonstrate that the RR approach provides quantitative results for the solvation entropies of monovalent and divalent ions and effectively captures the physics of these entropic effects. The success of the current approach indicates that it should be applicable to the studies of the solvation entropies in the proteins and also, in examining hydrophobic effects. Thus, we believe that the RR approach provides a powerful tool for evaluating the corresponding contributions to the binding entropies and eventually, to the binding free energies. This holds promise for extending the information theory modeling to proteins and protein-ligand complexes in aqueous solutions and consequently, facilitating computer-aided drug design.
Keywords: Langevin dipole, Restraint release, Molecular dynamics, Quasiharmonic approximation, Drug design
I. Introduction
Entropic effects play a crucial role in biological and chemical processes contributing to folding,1–6 binding processes 3,7–11 and sometimes, modulating catalytic effects.12–15 Therefore, it is important to be able to assess the magnitude of the entropic contributions in a quantitative way. Unfortunately, this task is extremely challenging and popular approaches such as the quasiharmonic (QH) and related treatments are quite inaccurate.16,17 Several approaches have been proposed in the recent years for more accurate calculation of configurational entropies1,6,12,13,17–27 and of these, some have been validated in the actual studies of biological systems.12,13,17 Furthermore, it has been shown that the entropic effects can be evaluated by brute force approaches but these require an extremely large computer time.15,17,28 Interestingly however, most of the attempts to study the entropic contributions to binding and catalysis conveniently ignored the effect of the solvation entropies.10,17,29–32 In some case s, it was found that the solvation entropy for the given process in water can be determined quite accurately by the phenomenological approach of Florian and Warshel33 who calibrated the polarization of their Langevin dipole(LD) model to reproduce the solvation entropies (see below). While the LD approach is extremely effective in solution studies, it is not clear how to implement this type of approach in the studies of the solvation entropies in the proteins. In such a case, one needs a fully microscopic approach and the development of a proper strategy which is the subject of the present work. This paper begins by considering the physical basis of the solvation entropies. It is shown that the main contributions are not associated with the solvent vibrations but with the solvent configurations. We also explore the ability of the restraint release (RR) approach to evaluate the solvation entropies. It is found that the RR approach provides an effective tool for the quantitative evaluation of the solvation energies of the charged molecules.
II. Physical Considerations
In studying the solvation entropies, it is important to have a working hypothesis regarding the origin of these effects. The simplest idea is that somehow the vibrational frequencies of the first solvation shell are higher near uncharged solute than near the solute in its charged form. One way to explore this idea is to look at the solvent vibrations that are associated with the solute-solvent interactions whilst the solute is charged and uncharged. This type of analysis can be done conveniently by the dispersed polaron (Spin Boson) method34 which provides the projection of the solvent modes along the solvation coordinates. The results of such an analysis for sodium and calcium ion in water gave the results summarized in Fig 1. Evidently, the vibrational frequencies are similar and thus, unlikely to account for the solvation entropy.
Fig 1.

The Fourier transform of the time dependence of the energy gap (ΔVab) between the charged (state a) and uncharged (state b) states of (a) Na+ ion (b) Ca2+ ions. The dimensionless coordinate displacement (Δ) values are obtained from a simple Fourier transform on the time dependent energy gap obtained from classical trajectories on the potential surface of Va.
Attempts to use the calculated spectrum in the evaluation of the vibrational entropy S(v), ΔSvib =∫S (v)dv (see ref 35 for a related treatment of free energy), gave basically the same results for the charged and the uncharged systems. These results were obtained regardless of the length of the simulation or resolution of the low frequencies. Now, this finding may appear counterintuitive considering the expectations of the larger solvent force constant for both the charged and the uncharged states. Therefore, we estimated the solvent force constants by evaluating the microscopic Marcus parabolas using our well tested and widely used formulation. 36 It was found that the curvature of the charged and the uncharged parabolas are very similar (also see ref 36 for a related study). This indicated that the solvent vibrations cannot account for the solvation entropy.
The above finding tells us to look in the same direction as explored in the LD treatment. That is, it is more likely that the solvation entropies reflect the tendency of the dipoles of the first solvation shell to be oriented toward the charged solute and to assume random conformation when the solute is uncharged. This has been in fact the idea beyond our LD treatment33 which will be discussed below.
According to the LD model, the hydration entropy for charged solutes is dominated by the solvent immobilization in the presence of the solute electrostatic field, ξ. This entropy contribution can be estimated using the concept of accessible configurational volumes introduced by Frank and Evans37,38 and expressed as
| (1) |
where kB is the Boltzmann constant while Vpl and Vsl are the accessible configurational volumes of the solvent dipoles in the pure (bulk) liquid and in the presence of the solute, respectively. The accessible configurational volumes can be expressed as the products of the volumes corresponding to the translational, rotational, and vibrational degrees of freedom of the solvent molecules. The use of the LD solvation model, in which the positions of the solvent dipoles are fixed, corresponds to the assumption that the translational contributions to the hydration entropy are negligible. Although this contribution is not negligible, it is certainly small compared to the changes in rotational degrees of freedom of the solvent molecules. Similarly, the vibrational entropy of water is supposed to be unaffected by the presence of the dissolved ions. Using the above assumptions and the fact that the direction of a solvent dipole in large electrostatic field is bound to lie between 0 and 2α (Fig. 2) one obtains the relationship
Fig 2.
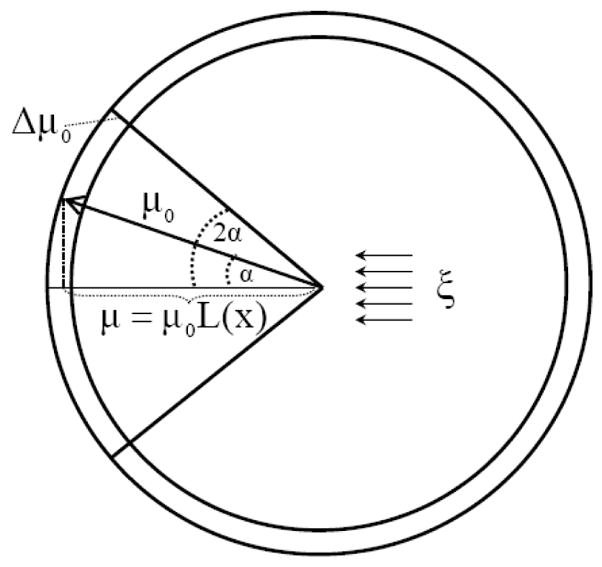
Schematic representation of the restricted configurational volume of a grid-centered solvent dipole in the presence of an external electrostatic field, ξ. In the pure liquid, the rotation of the solvent dipole is assumed to be free and the accessible volume corresponds to the outer shell of a sphere of a radius μ0. In the presence of the external field, the end point of the solvent dipole becomes confined, on the average, to the outer shell determined by the solid angle between the direction of the field and the maximum deviation of the solvent dipole from this direction.
| (2) |
The angle α corresponds to the average angle between the direction of the solvent dip ole and the external field. In the LD solvation model, it is assumed that the average solvent polarization is determined even in solution by the Langevin function L(x)39:
| (3) |
| (4) |
In Eq 4, μ0, ξ, kB, C and T denote, respectively, the magnitude of the solvent dipole, the total electrostatic field at the position of this dipole, the Boltzmann constant, a scaling constant and the thermodynamic temperature. The assumption that the LD model is valid in solution40 has been shown to be physically valid 41 and thus can be used in entropy studies. At any rate, using Eqs 1–3, the immobilization entropy of the jth Langevin dipole becomes
| (5) |
Eq 5 does not include explicitly the dipole-dipole interaction term. Thus, this equation loses its validity for solvent dipoles positioned in the regions where the magnitude of the solute electrostatic field is comparable to the field from other solvent dipoles. Consequently, we introduced an empirical correction function
| (6) |
that decreases the magnitude of ΔSimmob for small fields. For +1 or −1 charged solutes, this correction affects the solvent dipoles beyond the first hydration shell, whereas for +2 and −2 charged solutes this term affects dipoles outside the first two hydration shells. Finally, a multiplicative constant, Ci = 0.878, that incorporates entropy contributions that cannot be described by the dipolar solvent model, was introduced and adjusted by comparing the calculated and experimental hydration entropies of atomic ions. The resulting empirically corrected immobilization entropy is expressed as
| (7) |
The behavior of the uncorrected (Eq 5) and the corrected (Eq 7) immobilization entropy as a function of x is presented in Fig. 2 of ref33. Finally, the total hydration entropies are calculated as the sum of the hydrophobic and immobilization entropies
| (8) |
This expression can be applied to both neutral and ionic solutes. Note that it has been suggested that the hydration entropy of large positive ions may be significantly different from that of large negative ions of the same size.42,43 In our model, this entropy does not depend on the sign of the solute charge since we believe that the magnitude of this dependence is still an open question.
It should be mentioned here that ΔSphob could have also been expressed by considering the decrease in the number of configurations available for the solvent dipoles near the nonpolar solute surface relative to the configurations available in the bulk solvent.44 A rigorous implementation of this model would require a complicated treatment of the explicit interactions between each surface molecule and its environment, which involves both the neighboring solvent molecules and the solute surface.
In order to explore the above idea, we evaluated the polarization of the water molecules around Na+, K+ and Ca 2+ions and around the uncharged forms of these ions. The corresponding results are summarized in Figs 3–6.
Fig 3.

The time-dependent fluctuations of three water molecules at distances of 1.96Å (a), 4.90Å (b) and 6.21Å, respectively (c) from charged (left) and uncharged (right) Na+ ion during a 2ns molecular dynamics simulation.
Fig 6.

Histogram of the average orientation of three water molecules at distances of 1.96Å (a), 4.90Å (b) and 6.21Å, respectively (c) from charged (left) and uncharged (right) Ca2+ ion during a 2ns molecular dynamics simulation.
Figs 3 and 4 describe the fluctuations of the angle between the solvent field and the bisector of the two O-H vectors of the representative water molecules. Figs 5 and 6 provide histogram of the angle, θ. As expected, the figures indicate that the orientation of the molecules in the first solvation shell is more restricted in the charged solute than in the neutral solute. However, the trend in the case of molecules in the second and third solvation shell is less clear.
Fig 4.

The time-dependent fluctuations of three water molecules at distances of 1.96Å (a), 4.90Å (b) and 6.21Å, respectively (c) from charged (left) and uncharged (right) Ca2+ ion during a 2ns molecular dynamics simulation.
Fig 5.

Histogram of the average orientation of three water molecules at distances of 1.96Å (a), 4.90Å (b) and 6.21Å, respectively (c) from charged (left) and uncharged (right) Na+ ion during a 2ns molecular dynamics simulation.
While some qualitative aspects of the origin of the entropic effect are observed in Figs 3–6, we need a more quantitative way that can also be “imported” to similar studies in the proteins. Thus, we turn our attention to the RR approach which is described in the next section.
III. Using the RR Approach
The RR approach is based on the idea that with strong harmonic Cartesian restraints we have no entropy effects so, in some way, the free energy of releasing the restraints can tell us about the magnitude of the entropic effect. This general idea has been proposed by Herman and Wang45 for evaluating entropic effects in limited internal coordinates and is similar in some respects to that suggested in an early proposal.46 However, the problem is to formulate the RR idea correctly in a general set of coordinates and also, to take into account the fact recognized in ref 47, that the RR free energy can include a large enthalpic contribution. The resolution of this problem and the introduction of a practical general RR approach have been introduced in refs12,47 with a general cartesian treatment and an optimization approach that eliminates the enthalpic contribution from the RR free energy. The main points of this approach and the additional recent improvements will be discussed below.
III. 1. Formulation of the Restraint Release Approach
In order to obtain the solute entropic contribution to a transfer between two potential surfaces (UI→UII), one has to separate the corresponding partition functions to the solute and the solvent contributions (where in the present case, the ‘solute’ includes explicit solvent molecules and the ‘solvent’ is defined as the rest of the system) by writing
| (9) |
and
where UN is the potential surface for state N and δ (R − R̅) indicates that the corresponding function would be collected at ±ΔR/2. Here R and r are the solute and the solvent coordinate, respectively, qN (R̅) is the solute unnormalized probability distribution evaluated at the given R̅ averaged over all the solvent coordinates and WN (R̅) is the corresponding potential of mean force (PMF) which includes, of course, the solute and solvent contributions. Expanding this potential around its minimum gives
| (10) |
where we could use R but instead, took R̅ as our variable to indicate that this is our restraint coordinate. Here, Δgsol (R̅) is the solvation free energy at R̅, KW is the force constant of the quadratic term of our expansion and is the value of R̅ at the minimum of WN. The harmonic approximation of the PMF will be removed below.
For the sake of simplicity, we continue by examining a case where the PMF corresponds to a one-dimensional solute coordinate, although extension to many dimensions is straightforward. Our one-dimensional case is described by the thermodynamic cycle of Fig 7. In this cycle, we divide R to segments that can be defined by the delta function of Eq 9 or by introducing a strong quadratic constraint ((K′1/2)(R̅ − R̅′)2). In this case, we have
Fig 7.

A schematic illustration of the effect of R̅ and the enthalpic contribution to ΔG′. The figure considers one-dimensional potentials of mean force (W) for two systems (e.g. charged and uncharged solute) and the effect of confining the system to small segments of R by a strong constraint (K1). The figure also represents the effect of restraining the system in different R̅’s by the PMF of the restraint systems, (W′). The value of ΔG′ depends on R̅ and the ΔG′ obtained with the minimum of the corresponding W (i.e., R̅0) has the smallest absolute value. This ΔG′ is our −T ΔS′.
| (11) |
where the notation W′ designates the addition of the constraint.
Next, we use Eq 11 and our quadratic constraint and evaluate the relevant partition functions.
| (12) |
where we use Q′ or the partition function evaluated with W′N and where . Using the above equation we can write
| (13) |
where E̅ is the average energy and Ssol is the contribution from the surrounding of the specified system included in our explicit treatment. We also have
| (14) |
Now, we can write
| (15) |
| (16) |
Thus,
| (17) |
Finally, from Eq 13 and Eq 15 we have in the special case when and
| (18) |
The usage of this relationship however, requires knowing the values of and . This can be done, in principle, by minimizing the corresponding PMF (see section III. 4). Furthermore, using Eq 13 we can write
| (19) |
Thus, we estimate ΔG′(R0) by trying different R̅’s and taking the ΔG′(R̅) with the smallest absolute value. Now, we can use Eqs 17 and 18 to evaluate ΔS′I→II and . The R̅ that minimizes |ΔG′| is also used in evaluating the of Eq 17 and the total entropy ΔSI→II.
III. 2. Non-Harmonic Case
The validity of the inequality of Eq. 19 has been established above for harmonic systems but probably it is also valid for an harmonic potentials. Here, we only demonstrate the validity in some limiting cases. We first consider a square well potential (Fig. 8)
Fig 8.

Schematic representation of a square well potential.
In this case, we can define a unit length ΔR by
| (20) |
and then we have,
| (21) |
where n = L/ΔR and L is the width of the potential well. This leads to
| (22) |
Now, we also have
| (23) |
| (24) |
Using the above relationship for transition from I to II, with the same K1, we obtain
| (25) |
as long as we are in the region where R ≥ 0 and W = W0, and under the condition that the contributions for ΔGI from the region with are small. Once we move to the region where , where (e.g. at R < 0), we have
| (26) |
Hence,
thus, satisfying our condition of obtaining ΔSI→II from R′1 that minimizes ΔG′. The same treatment can be used for periodic systems such as the one in Fig. 9.
Fig 9.
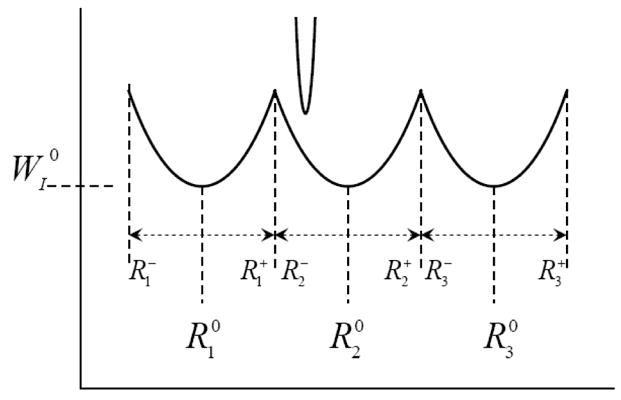
Schematic representation of a periodic system that may be utilized to obtain ΔSI→II from R′1 that minimizes ΔG′.
In this case
| (27) |
where θ(R)l = 0 when R is out of the lth range, and θ(R)l = 1 inside this range. This gives
| (28) |
where . Now, we can write
| (29) |
We also, obtain
| (30) |
where R′l is the center of the constraint potential. We also, have
| (31) |
Following the same consideration as in the previous case, and utilizing the fact that when , we will obtain the same inequality.
III. 3. Practical time saving by using the initial quasiharmonic treatment
The RR treatment is based on starting the simulations with a strong positional restraint where the entropic contributions are negligible. However, the free energy perturbation (FEP) release requires an enormous amount of computer time even where K1 is significantly large. Fortunately, we realized that the RR procedure is not really needed at the stage when K1 is large since the corresponding entropic contribution can be easily estimated by the QH approximations.48,49 In general, the QH tends to be valid when restraints are significant and only starts to become problematic when K1 is small (resulting in a range of very shallow and an harmonic potential energy surfaces).16,17,47 We exploit this fact by combining the QH and RR approximations by evaluating the entropic contribution at a moderate value of K1 by the QH approach followed by the RR procedure from this specific value. This treatment can be expressed by
| (32) |
where the −TΔS(K = K1)QH designates the entropy computed by the QH approximation, where K1 is the initial value of the restraint and ‘min’ indicates the minimum value of the indicated ΔGRR. This approach has been successfully exploited in the studies of ribosome13 and more recently, a series of substituted phosphate diesters.50
III. 4. Optimizing the Restraint Coordinates
Our next task is to optimize ΔGRR. This free energy term is obtained by a FEP/umbrella sampling approach using a mapping potential
| (33) |
where
| (34) |
where Usyst is the potential energy of the unconstraint system.
If we use the end point linear response approximation (LRA)instead of the full FEP treatment, we will have10
| (35) |
Now we have to find R0 that minimizes ΔGRR. This is done by
Now,
| (36) |
Thus,
| (37) |
We also know that the step from the current R to the optimal R is given by the Newton Rhapson relationship51
| (38) |
This gives
| (39) |
The LRA approximation of Eq 35 works the best when K(1) is small since this is when we get the largest contribution to ΔGRR. Since Eq 39 is not exact (due to the use of the LRA approach), we found it useful to scale it and to use
| (40) |
where α is a scaling factor.
III. 5 Actual Implementation
All calculations were performed using the MOLARIS software package, and the ENZYMIX force field.10,52
The constraint release free energy terms are evaluated by a free energy perturbation (FEP)53,54 (also see ref55). The FEP method evaluates the free energy associated with the change of the potential surface from U(1) to U(2) by gradually changing the potential surface using the relationship
where , U(2) = Usyst and λm is a mapping parameter that changes between (0 ≤ λm ≤ 1). The free energy increment, associated with the change of Um can be obtained by
where 〈 〉m indicates that the given average is evaluated by propagating trajectories over Um and β − (kBT)−1. The total free energy difference between the states A and B is obtained by changing λm in n equal increments and evaluating the sum of the corresponding δG.
The RR calculations require imposing strong positional constraints on the Cartesian coordinates of the solvent molecules in the presence of a solute (either in its charged or uncharged state) whose position is fixed by applying a moderate positional constraint K=10 kcal mol−1Å−2 and to subsequently evaluate the free energies associated with releasing these restraints using the following equation:
| (41) |
and denotes RR free energies of the systems with a fixed, charged and uncharged solute, respectively. All RR free energies contain a residual contribution from the enthalpy of the system. However, this contribution approaches zero for restraint coordinates that give the lowest RR energy.45,47
To calculate the optimized restraint coordinates (R̅) that would give the optimal ΔG for entropy calculations, the given system with the solute in its charged state, was subjected to MD simulations using a constraint force of 5.0 kcal mol−1Å−2 and a scaling factor (α) of 0.1 while averaging 〈R − R̅〉 over 1000 steps (see section III. 4). A total of 10 iterations were carried out in this procedure. Since Eq 39 is expressed in Cartesian coordinates and the application to water molecules resulted in some stretching of the bonding coordinates, we found it useful to relax the constraint coordinates obtained from Eq 40 in a 10 ps simulation run and generate nine different Rs(we report results for those with the lowest RR energy). Apparently, our exploratory studies indicated that using a small number of Rs may result in missing the one that minimizes the enthalpic contribution despite using Eq. 40. The best estimate of −TΔS is obtained by taking the corresponding values from the run with R that gives the smallest |ΔG|. This variational minimization reflects the fact that all the RR free energies contain enthalpic contributions and these contributions approach to zero for restraint coordinates that gives the lowest RR contribution.
An initial 30 ps molecular dynamics simulation was performed on the solvent molecules around the fixed charged and uncharged solute, with snapshots taken every 5 ps. MD simulations, 40 ps in length, were performed on each snapshot (with coordinates collected every 0.01 ps) to calculate the QH entropy for each snapshot, and this procedure was carried out for K= 3.0 kcalmol−1 in magnitude. This allows the calculation of the absolute entropy for this restraint. Subsequently, the free energy for the release of these restraints was evaluated using the RR approach employing Eq 32.
The RR-FEP calculations were performed with an 18Å simulation sphere of explicit water molecules subject to the surface constraint all-atom solvent (SCAAS) boundary conditions.56 The RR simulations, consisting of 42 windows each with a simulation time of 40ps, involved the release of the position restraints in 5 FEP stages, changing K1 from 3.0 to 1.0, from 1 to 0.3, from 0.3 to 0.03, from 0.03 to 0.003 and finally, from 0.003 to 0.0003, where all the values of K1 are given in kcal mol−1 Å−2. These simulations were done at 300K with 1fs time steps.
Simultaneously, the LD model33 implemented in the program ChemSol 2.057 was also used to evaluate the solvation entropy. This method evaluates the solvation entropy by estimating the restriction of the solvent motion due to the field from the solute.
IV. RESULTS
As stated above, we tried to explore in this work the ability of the RR approach to reproduce solvation entropies. The most obvious benchmark is provided by the solvation entropies of ions and this challenge has been addressed here by evaluating the solvation entropies of Na+, K+ and Ca2+ ions. The corresponding RR calculations were performed on the inner 29 water molecules that are closest to the ion, while the rest of the 18Å SCAAS system to move with a very small position constraint on the oxygen atoms in addition to the SCAAS polarization and distance restraints. In short, the entropic effect associated with the effect of the solute on the inner 29 water molecules was explored while representing rest of the system by the SCAAS model. In calculating the solvation entropy, one should also consider the hydrophobic energy of forming the cavity for the uncharged ion. This hydrophobic contribution is small for the ions considered here and were added to the final calculated results. This was done using the corresponding values obtained from ChemSol. In future, we hope to obtain such contributions from our RR calculations. The results of these calculations are summarized in Tables 1–3. Remarkably, we reproduce the observed trend in an almost quantitative way. The tables also give the quantitative LD results but as mentioned before, the LD treatment is empirical in nature and cannot be used within the protein active site. It should also be mentioned that the recent works of Aqvist and coworkers28 reproduced quantitative results for the same system studied here. However, this was done by evaluating the van’t Hoff approach that requires a major investment in terms of computer time.
Table 1.
Entropy calculation for a solvated Na+ ion, using the RR approacha
| Charged | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| RR3→0.0003 | −210.71 | −208.85 | −209.73 | −210.67 | −209.44 | −210.80 |
| RR1→0.0003 | −203.43 | −201.39 | −201.34 | −203.07 | −200.07 | −201.1 |
| RR0.3→0.0003 | −147.88 | −145.51 | −145.53 | −145.73 | −141.73 | −142.32 |
| RR0.03→0.0003 | −72.95 | −71.18 | −72.1 | −69.06 | −66.07 | −67.63 |
| QH3 | −144.46 | −146.35 | −147.43 | −145.71 | −146.11 | −147.28 |
| RR3→0.0003+QH3 | −355.17 | −355.2 | −357.16 | −356.38 | −355.55 | −358.08 |
|
Uncharged | ||||||
| RR3→0.0003 | −227.95 | −222.7 | −224.28 | −212.05 | −213.96 | −221.96 |
| RR1→0.0003 | −220.11 | −214.12 | −215.88 | −204.21 | −204.72 | −213.95 |
| RR0.3→0.0003 | −159.96 | −152.71 | −155.03 | −149.40 | −150.94 | −154.16 |
| RR0.03→0.0003 | −79.14 | −72.65 | −73.73 | −70.36 | −70.42 | −74.18 |
| QH3 | −148.97 | −153.06 | −155.15 | −145.98 | −146.88 | −147.24 |
| RR3→0.0003+QH3 | −376.92 | −375.76 | −379.43 | −358.03 | −360.84 | −369.2 |
| −T(ΔSRR) | 2.86 | |||||
| −T(ΔS)hyd | 2.90 | |||||
| −T(ΔS)calc | 5.76 (2.86 + 2.90) | |||||
| −TΔSexp | 5.838 | |||||
| 6.663 | ||||||
| 5.264 | ||||||
| −T(ΔSLD)calc | 5.8 | |||||
Energy values are given in kcal/mol. The table includes the results of six sets (1–6) of simulations with different restraint coordinates. −T(ΔS)hyd is the cavity formation contribution calculated using Chemsol. The value in bold signifies the smallest ΔG and thus, the value that satisfies Eq 32.
Table 3.
Entropy calculation for solvated Ca2+ ion, using RR approacha
| Charged | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| RR3→0.0003 | −215.2 | −211.94 | −215.52 | −211.63 | −216.17 | −215.83 |
| RR1→0.0003 | −205.45 | −204.44 | −208.5 | −204.62 | −204.11 | −207.71 |
| RR0.3→0.0003 | −147.87 | −147.65 | −149.06 | −142.91 | −143.53 | −148.69 |
| RR0.03→0.0003 | −70.43 | −67.13 | −70.69 | −70.74 | −70.73 | −71.85 |
| QHK=3 | −143.29 | −143.45 | −143.06 | −143.54 | −144.32 | −142.55 |
| RR3→0.0003+QH3 | −358.49 | −355.39 | −358.58 | −355.17 | −360.49 | −358.38 |
|
Uncharged | ||||||
| RR3→0.0003 | −219.61 | −233.24 | −232.47 | −227.5 | −220.93 | −232.61 |
| RR1→0.0003 | −211.93 | −223.56 | −223.4 | −218.28 | −211.94 | −223.93 |
| RR0.3→0.0003 | −150.63 | −158.36 | −161.32 | −156.25 | −150.72 | −153.06 |
| RR0.03→0.0003 | −72.25 | −73.39 | −76.51 | −74.11 | −71.25 | −72.44 |
| QH3 | −150.72 | −153.23 | −151.43 | −157.86 | −151.50 | −154.29 |
| RR3→0.0003+QH3 | −370.33 | −386.47 | −390.33 | −385.36 | −372.43 | −386.9 |
| −T(ΔSRR) | 15.16 | |||||
| −T(ΔS)hyd | 2.58 | |||||
| −T(ΔS)calc | 17.74 (15.16 + 2.58) | |||||
| −TΔSexp | 15.338 | |||||
| 16.663 | ||||||
| 15.164 | ||||||
| −T(ΔSLD)calc | 17.6 | |||||
Energy values are given in kcal/mol. The table includes the results of six sets (1–6) of simulations with different restraint coordinates. −T(ΔS)hyd is the cavity formation contribution calculated using Chemsol. The value in bold signifies the smallest ΔG and thus, the value that satisfies Eq 32.
The fact that the computational methodology employed in this work reproduces the observed solvation entropy indicates that the physics of this effect is captured by this approach and that the effect is associated with the change in the effective space available for the solvent molecules upon change in the solute charges.
The overall trend is clearly discussed in section II, but here we have a well-defined simulation approach that reproduces the entropic effect quantitatively. Furthermore, in order to clarify the nature of the challenge overcome by the RR calculations, we also report in Table 4 the QH results obtained with no restraint (K= 0 kcal mol−1 Å−2) for Na+ and Ca2+ ions. Although QH entropic estimation for Na+ ion seems fairly accurate, it provides completely erroneous results for Ca2+ ion. Thus, we may say that the QH is simply unable to capture the solvation entropies. This is probably one of the best illustrations of the fundamental problems with the widely used QH and related approaches.
Table 4.
QH calculations for solvation entropiesof the metal ions.a
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Na+ | ||||||
| Uncharged | −251.17 | −249.26 | −246.77 | −246.29 | −246.74 | −248.67 |
| Charged | −250.05 | −248.53 | −242.55 | −243.11 | −244.34 | −244.45 |
| −T(ΔSQH) | 2.64 | |||||
| −T(ΔS)hyd | 2.90 | |||||
| −T(ΔS)calc | 5.54 (2.64 + 2.90) | |||||
| Ca2+ | ||||||
| Uncharged | −251.12 | −249.03 | −250.03 | −246.38 | −246.15 | −247.77 |
| Charged | −245.81 | −242.79 | −239.91 | −241.62 | −241.79 | −243.0 |
| −T(ΔSQH)calc | 5.92 | |||||
| −T(ΔS)hyd | 2.58 | |||||
| −T(ΔS)calc | 8.5 (5.92 + 2.58) | |||||
Energy values are given in kcal/mol. The table includes the results of six sets (1–6) of simulations with K=0 kcal mol−1 Å −2 on the system. The final entropy value is obtained by taking an average of the difference between the charged and uncharged form of the six different conformations used.
V. DISCUSSION
The present work focuses on the calculations of the entropic effects, despite the fact that most questions about biophysics functions are related to the corresponding free energy surfaces.58,59 Additionally, we are well aware of the fact that free energy calculations converge much faster than the entropic calculations. However, in many cases complete understanding of the entropy-enthalpy compensation is useful for the deeper understanding of the underlying physics of the system and in particular, the origin of the temperature dependence of various processes. Moreover, the evaluation of the entropic effects is one of the utmost challenges of computational biophysics and in fact, the failure to reproduce entropic effects is frequently used by the experimental community in trying to diminish the important free energy calculations (also see discussion in ref60).
In any case, the accurate evaluation of the entropic contributions to the free energy changes in condensed phases is an extremely challenging task. Popular approaches such as the QH and related treatments have been widely used to extract the configurational entropy from molecular dynamics simulations.48,61,62 Unfortunately, although QH approximation functions quite well for simple systems with single energy wells, this approach provides a poor approximation of the configurational entropies of systems with multiple occupied energy wells, as it merges multiple narrow energy wells into one broad energy well.16,17 In fact, the validity of such approaches in systems with very shallow potentials is questionable. Therefore, the use of the QH approach in the studies of binding energies or related problems is problematic. Several approaches have been proposed for obtaining quantitative entropic contributions, but most of these approaches have not been able to provide quantitative results in general cases, except by brute force studies of the free energy change with temperature that requires enormous simulation time for convergence.28 In our view and experience, one of the most promising strategies is our RR approach that has been shown to give quantitative results for activation entropies.10,12,13,47,50 Nevertheless, this approach has not been validated in the studies of binding entropies.
One of the most serious problems in calculations of binding entropy is the evaluation of the solvation entropies in water and in the protein active site. This contribution has been generally neglected in key studies29–32 although it has been evaluated or included in calculations of activation free energies.13,15,47 The problem of activation entropies is also a problem of general interest both for studies of molecules in solutions and in proteins.
When one deals with molecules in solution, it is possible to use the LD models to obtain a relatively reliable estimate of the solvation entropies. Unfortunately, this approach has not been generalized to protein active sites where one must use a microscopic approach. The studies of ions in solution should provide the best benchmark for the development and validation of the corresponding approach since the corresponding values are known experimentally.
CONCLUSIONS AND FUTURE APPLICATIONS
The molecular simulations of the entropy contribution in the areas of protein-ligand binding and enzyme catalysis, is an extremely challenging task and the subject of intense research. The analyses of the role of solvent entropies in ligand binding and enzyme catalysis are still rare, although knowledge of their values would be of great utility considering that the binding processes can be relatively dominated by solvent entropy effects. Therefore, there is a strong motivation to obtain the solvation entropic contributions that can lead to a better understanding of the process under consideration. Although the solvent entropies can be extracted from the temperature dependence of the explicit solvent FEP simulations, these calculations are difficult to converge and their convergence tends to be considerably slower than that of the free energy itself. The present work focuses on the development of efficient microscopic model for the studies of the solvation entropies in aqueous solution. Our preliminary studies clarified that the solvation entropies are not associated with the solvent vibrations but rather with the availability of larger configurational space upon transition from the charged to the uncharged form of the solute. This effect is easily observed in the cases of divalent ions but in the case of monovalent ions it is restricted to the first solvation shell. With this in mind, we demonstrated that the solvent entropies can be successfully and accurately evaluated by our RR approach. This has been illustrated for the simple but relevant test case of monovalent and divalent metal ions. It is clear from the results that solvation entropies depend strongly on the magnitude of the charge of the solvated ion. This is obviously related to the corresponding restriction in the solvent fluctuations.
Although convergence problems may exist for larger and more complex simulation systems, a good agreement is obtained between the calculated and the experimental data for solvation entropies of monovalent and divalent metal ions. Furthermore, we also developed a systematic approach for the minimization of ΔGRR through the Newton-Rhapson minimization of the ΔGRR free energy, formulated within the LRA. Our previous studies have shown that the random search for the optimal R̅ may otherwise, require extensive simulations. Thus, this approach reduces computer cost and time while finding the optimal R̅ for our RR calculations. Overall, the use of the RR approach has been found to provide a powerful way of quantifying the solvation entropies. On the other hand, it has been demonstrated that the QH approach provides an entirely inadequate tool for this important purpose.
The success of the present approach in the evaluation of the electrostatic components of the solvation entropies indicates that the RR method will probably be useful in the studies of hydrophobic effects where the physics (of restricted orientation) is smaller. Efforts are underway in our laboratory to extend the utility of this approach in the studies of the contribution of the solvation entropies to the binding entropies, hoping to help advance the understanding of this important effect. The corresponding strategy will be similar to the one used in the present study and will require releasing the restraints on the protein residues and the water molecules within a given cut-off distance. However, this approach will require careful refinement since releasing the restraints on all of the protein residues may lead to computational instability while releasing the restraints on a few residues may not capture the full solvation entropy contribution. In future, it should be feasible to estimate the magnitude of the solvent entropic contributions involved in ligand binding to proteins. Generally, the binding affinity of a compound can be improved by generating a favorable binding enthalpy, favorable solvation entropy, or by minimizing the unfavorable conformational entropy. In some cases, it is much easier to optimize the entropy. For example, medicinal chemists have attempted to conformationally constrain the substrate thus, restricting the entropy loss upon binding. Further improvement may be obtained by trying to optimize the solvation entropies by choosing specialized functional groups. However, the main advancement in our approach is in obtaining more reliable predictions of the entropic contributions to ligand binding. Our RR approach should therefore, be viewed as a step towards the generation of algorithms capable of thermodynamic dissection particularly, those that can predict the entropic consequences of introducing different chemical functionalities in the molecular scaffold generated either de novo (during lead generation) or while being optimized (during lead optimization)for improving their binding affinity towards a target protein. Thus, this study may have implications towards our understanding of protein-ligand interactions, for theoretical modeling of molecular recognition, and for rational drug design.
Table 2.
Entropy calculation for a solvated K+ ion, using RR approacha
| Charged | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| RR3→0.0003 | −227.62 | −223.75 | −223.3 | −224.20 | −226.04 | −224.59 |
| RR1→0.0003 | −218.7 | −215.21 | −210.73 | −215.51 | −217.26 | −219.2 |
| RR0.3→0.0003 | −158.51 | −153.06 | −149.39 | −154.64 | −154.78 | −154.45 |
| RR0.03→0.0003 | −69.6 | −74.7 | −71.73 | −72.9 | −72.73 | −73.73 |
| QH3 | −136.98 | −135.56 | −136.6 | −135.55 | −135.86 | −135.25 |
| RR3→0.0003+QH3 | −364.6 | −359.31 | −359.90 | −359.75 | −361.9 | −359.84 |
|
Uncharged | ||||||
| RR3→0.0003 | −227.95 | −227.58 | −230.48 | −225.04 | −235.33 | −231.1 |
| RR1→0.0003 | −217.97 | −217.29 | −220.28 | −217.14 | −223.96 | −220.94 |
| RR0.3→0.0003 | −158.62 | −156.67 | −155.61 | −156.72 | −156.35 | −158.62 |
| RR0.03→0.0003 | −76.63 | −76.6 | −74.1 | −76.56 | −78.88 | −76.33 |
| QH3 | −137.37 | −136.40 | −138.32 | −135.11 | −135.17 | −138.1 |
| RR3→0.0003+QH3 | −365.32 | −363.98 | −368.8 | −360.15 | −370.5 | −369.2 |
| −T(ΔSRR) | 0.84 | |||||
| −T(ΔS)hyd | 3.87 | |||||
| −T(ΔS)calc | 4.71 (0.84 + 3.87) | |||||
| −TΔSexp | 3.338 | |||||
| 3.963 | ||||||
| 2.864 | ||||||
| −T(ΔSLD)calc | 4.67 | |||||
Energy values are given in kcal/mol. The table includes the results of six sets (1–6) of simulations with different restraint coordinates. −T(ΔS)hyd is the cavity formation contribution calculated using Chemsol. The value in bold signifies the smallest ΔG and thus, the value that satisfies Eq 32.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM40283. All computational work was supported by the University of Southern California High Performance Computing and Communication Center (HPCC). The authors are grateful to Drs. Z. T. Chu and Pankaz Sharmafor their technical assistance and insightful suggestions throughout this work.
References
- 1.Meirovitch H. Current Opinion in Structure Biology. 2007;17:181. doi: 10.1016/j.sbi.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 2.Amzel LM. Proteins: Structure, Function, and Genetics. 1997;28:144. [PubMed] [Google Scholar]
- 3.Brady GP, Sharp KA. Current Opinion in Structural Biology. 1997;7:215. doi: 10.1016/s0959-440x(97)80028-0. [DOI] [PubMed] [Google Scholar]
- 4.Privalov PL, Makhatadze GI. J Mol Biol. 1993;232:660. doi: 10.1006/jmbi.1993.1417. [DOI] [PubMed] [Google Scholar]
- 5.Creamer TP. Methods in Molecular Biology (Totowa, NJ, United States) 2001;168:117. doi: 10.1385/1-59259-193-0:117. [DOI] [PubMed] [Google Scholar]
- 6.Li DW, Khanlarzadeh M, Wang J, Huo S, Brüschweiler R. J Phys Chem B. 2007;111:13807. doi: 10.1021/jp075220e. [DOI] [PubMed] [Google Scholar]
- 7.Finkelstein A, Janin J. Protein Eng. 1989;3:1. doi: 10.1093/protein/3.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Murray C, Verdonk M. J Comput Aided Mol Des. 2002;16:741. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
- 9.Gilson MK, Given JA, Bush BL, McCammon JA. Biophys J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sham YY, Chu ZT, Tao H, Warshel A. Proteins: Struct Funct Genet. 2000;39:393. [PubMed] [Google Scholar]
- 11.Chang CE, Chen W, Gilson MK. Proc Natl Acad Sci U S A. 2007;104:1534. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villà J, Štrajbl M, Glennon TM, Sham YY, Chu ZT, Warshel A. Proc Natl Acad Sci USA. 2000;97:11899. doi: 10.1073/pnas.97.22.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma PK, Xiang Y, Kato M, Warshel A. Biochemistry. 2005;44:11307. doi: 10.1021/bi0509806. [DOI] [PubMed] [Google Scholar]
- 14.Warshel A. Annu Rev Biophys Biomol Struct. 2003;32:425. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- 15.Bjelic S, Brandsdal B, Aqvist J. Biochemistry. 2008;47:10049. doi: 10.1021/bi801177k. [DOI] [PubMed] [Google Scholar]
- 16.Chang CE, Chen W, Gilson MK. J Chem Theory Comput. 2005;1:1017. doi: 10.1021/ct0500904. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson J, Aqvist J. J Phys Chem B. 2005;109:6448. doi: 10.1021/jp046022f. [DOI] [PubMed] [Google Scholar]
- 18.Meirovitch H. Calculation of the free energy and entropy of macromolecular systems by computer simulation. Vol. 12 Wiley-VCH; 1998. [Google Scholar]
- 19.Hnizdo V, Fedorowicz A, Singh H, Demchuk E. J Comput Chem. 2003;24:1172. doi: 10.1002/jcc.10289. [DOI] [PubMed] [Google Scholar]
- 20.Cheluvaraja S, Meirovitch H. Proc Natl Acad Sci USA. 2004;101:9241. doi: 10.1073/pnas.0308201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darian E, Hnizdo V, Fedorowicz A, Singh H, Demchuk E. J Comput Chem. 2005;26:651. doi: 10.1002/jcc.20198. [DOI] [PubMed] [Google Scholar]
- 22.Cheluvaraja S, Meirovitch H. J Chem Phys. 2006;125:24905. doi: 10.1063/1.2208608. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Bruschweiler R. J Chem Theory Comput. 2006;2:18. doi: 10.1021/ct050118b. [DOI] [PubMed] [Google Scholar]
- 24.Hnizdo V, Darian E, Fedorowicz A, Demchuk E, Li S, Singh H. J Comput Chem. 2007;28:655. doi: 10.1002/jcc.20589. [DOI] [PubMed] [Google Scholar]
- 25.Hnizdo V, Tan J, Killian BJ, Gilson MK. J Comput Chem. 2008;29:1605. doi: 10.1002/jcc.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheluvaraja S, Mihailescu M, Meirovitch H. J Phys Chem B. 2008;112:9512. doi: 10.1021/jp801827f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy RM, Karplus M, Kushick J, Perahia D. Macromolecules. 1984;17:1370. [Google Scholar]
- 28.Carlsson J, Aqvist J. Phys Chem Chem Phys. 2006;8:5385. doi: 10.1039/b608486a. [DOI] [PubMed] [Google Scholar]
- 29.Lee MS, Olsson MA. Biophys J. 2006;90:864. doi: 10.1529/biophysj.105.071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen W, Chang CE, Gilson MK. Biophys J. 2004;87:3035. doi: 10.1529/biophysj.104.049494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruvinsky AM. Journal of Computer-Aided Molecular Design. 2007;21:361. doi: 10.1007/s10822-007-9116-0. [DOI] [PubMed] [Google Scholar]
- 32.Chang CE, Chen W, Gilson MK. Proc Natl Acad Sci U S A. 2007;104:1534. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Florián J, Warshel A. J Phys Chem B. 1999;103:10282. [Google Scholar]
- 34.Warshel A, Hwang JK. J Chem Phys. 1986;84:4938–4957. [Google Scholar]
- 35.Warshel A, Lifson S. Journal of Chemical Physics. 1970;53:582. [Google Scholar]
- 36.King G, Warshel J Chem Phys. 1990;93:8682. [Google Scholar]
- 37.Frank HS. J Chem Phys. 1945;13:478. [Google Scholar]
- 38.Frank HS, Evans MW. J Chem Phys. 1945;13:507. [Google Scholar]
- 39.Langevin P. Annales de Chimie Physique. 1905;8:70. [Google Scholar]
- 40.Warshel A, Levitt M. J Mol Biol. 1976;103:227. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 41.Papazyan A, Warshel A. J Phys Chem B. 1998;102:5248. [Google Scholar]
- 42.Rashin AA, Bukatin MA. Biophys Chem. 1994;51:167. doi: 10.1016/0301-4622(94)00060-3. [DOI] [PubMed] [Google Scholar]
- 43.Rashin AA, Bukatin MA. J Phys Chem. 1994;98:386. doi: 10.1016/0301-4622(94)00060-3. [DOI] [PubMed] [Google Scholar]
- 44.Luzhkov V, Warshel A. J Comp Chem. 1992;13:199. [Google Scholar]
- 45.Hermans J, Wang L. J Am Chem Soc. 1997;119:2707. [Google Scholar]
- 46.Warshel A. Office of Naval Research, Award Number N00014-91-J-1318. 1991. Computer simulation of chemical reactions in synthetic model compounds and genetically engineered active sites. [Google Scholar]
- 47.Štrajbl M, Sham YY, Villà J, Chu ZT, Warshel A. J Phys Chem B. 2000;104:4578. [Google Scholar]
- 48.Karplus M, Kushick JN. Macromolecules. 1981;14:325. [Google Scholar]
- 49.DiNola A, Berendsen H, Edholm O. Macromolecules. 1984;17:2044. [Google Scholar]
- 50.Rosta E, Kamerlin S, Warshel A. Biochemistry. 2008;47:3725. doi: 10.1021/bi702106m. [DOI] [PubMed] [Google Scholar]
- 51.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. John Wiley & Sons; New York: 1991. [Google Scholar]
- 52.Lee FS, Chu ZT, Warshel A. J Comp Chem. 1993;14:161. [Google Scholar]
- 53.Zwanzig RW. J Chem Phys. 1954;22:1420. [Google Scholar]
- 54.Valleau JP, Torrie GM. A guide to Monte Carlo for statistical mechanics. 2. Byways. In: Berne BJ, editor. Modern Theoretical Chemistry. Plenum Press; New York: 1977. pp. 5–169. [Google Scholar]
- 55.Warshel A. In Encyclopedia of Molecular Biology. John Wiley & Sons; 1999. [Google Scholar]
- 56.Warshel A, King G. Chem Phys Lett. 1985;121:124. [Google Scholar]
- 57.Florian J, Warshel A. ChemSol. 2.1. University of Southern California; Los Angeles: 1999. [Google Scholar]
- 58.Olsson MH, Parson WW, Warshel A. Chem Rev. 2006;106:1737. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 59.Warshel A, Parson WW. Quart Rev Biophys. 2001;34:563. doi: 10.1017/s0033583501003730. [DOI] [PubMed] [Google Scholar]
- 60.Olsson MH, Siegbahn PE, Warshel A. Journal of the American Chemical Society. 2004;126:2820. doi: 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- 61.Andricioaei I, Dinner AR, Karplus M. J Chem Phys. 2003;118:1074. [Google Scholar]
- 62.Schlitter J. Chem Phys Lett. 1993;215:617. [Google Scholar]
- 63.Marcus Y. Ion Properties. Marcel Dekker, Inc.; New York: 1997. [Google Scholar]
- 64.Noyes RM. J Am Chem Soc. 1962;84:513. [Google Scholar]