

Abstract
Symptomatic ischemia following aneurysmal subarachnoid hemorrhage is common but poorly understood and inadequately treated. Severe constriction of the major arteries at the base of the brain, termed vasospasm, traditionally has been thought to be a proximal event underlying these ischemias, although microvascular changes also have been described. The vast majority of studies aimed at understanding the pathogenesis of ischemic deficits and vasospasm have focused on the interaction of the “spasmogen” of the extravasated blood with the smooth muscle and endothelium of the arteries. This has led to a comparative neglect of the contribution of the central nervous system to the maintenance of cerebral perfusion. In the present study, we focused on the role of the rostral ventromedial medulla (RVM) in modulating cerebral perfusion at rest and following an experimental subarachnoid hemorrhage in the rat. Changes in cerebral blood flow (CBF) were measured using laser-Doppler flowmetry and three-dimensional optical microangiography. Focal application of a GABAA receptor agonist and antagonist were used to respectively inactivate and activate the RVM. We show here that the RVM modulates cerebral blood flow under resting conditions, and further, contributes to restoration of cerebral perfusion following a high-grade subarachnoid hemorrhage. Failure of this brainstem compensatory mechanism could be significant for acute perfusion deficits seen in patients following subarachnoid hemorrhage.
Keywords: cerebral blood flow, subarachnoid hemorrhage, brainstem, modulation
Symptomatic ischemic deficits remain poorly understood and inadequately treated sequelae of aneurysmal subarachnoid hemorrhage (SAH), and are seen in 25–30% of patients. Approximately half of those suffer permanent disability or death (Lombard and Borel, 2007). Vasospasm of the major arteries is thought to be a major contributor to the development of cerebral infarcts in these patients, although it is not necessary nor sufficient for all cases of ischemia (Macdonald et al., 2007).
The majority of studies on the sequelae of SAH have focused on arterial smooth muscle and endothelium. Abnormal contraction of smooth muscle, free radical release, endothelial factors, nitric oxide, endothelins, inflammatory mediators, neuronal cell death and structural changes in the arterial wall have all been implicated. However, the focus on the smooth muscle and endothelium has led to a comparative de-emphasis of a crucial element of large cerebral vessel walls, the perivascular nerves and of the associated mechanisms of the central nervous system (CNS). Extracranial arteries are innervated by sympathetic and parasympathetic fibers, and by sensory fibers, primarily from the trigeminal ganglion (Hamel, 2006), and there is evidence that this extrinsic innervation may contribute to ischemic deficits. A role for the sympathetic innervation is suggested by the finding that neuropeptide Y and noradrenaline are elevated in patients with SAH (Lambert et al., 2002). In addition, although early attempts at treating vasospasm with cervical sympathectomies failed (Wilkins, 1973), cervical ganglionic blockade and central α2-receptor agonists are reported to attenuate vasospasm after SAH (Treggiari et al., 2003). A role for sympathetic efferents should therefore not be dismissed. By contrast with sympathetic outflows, a possible role for parasympathetic control has received relatively little attention. However, stimulation of the sphenopalatine ganglion results in an increase in cerebral blood flow (CBF) under resting conditions and attenuates delayed vasospasm in experimental models of SAH (Seylaz et al., 1988; Goadsby, 1990; Toda et al., 2000; Yarnitsky et al., 2005; Henninger and Fisher, 2007). Finally, a possible role for sensory fibers is supported by the elevated levels of the sensory peptides calcitonin-gene related peptide (CGRP) and substance P in patients with vasospasm (Edvinsson et al., 1994; Juul et al., 1995) and by evidence for CGRP and trigeminal afferent involvement in animal models (Arienta et al., 1991; Edvinsson et al., 1994; Shiokawa and Svendgaard, 1994). Viewed collectively, these studies suggest that the perivascular innervation and associated CNS mechanisms contribute to ischemic deficits following SAH.
In this report, we test the hypothesis that the rostral ventromedial medulla (RVM) modulates the acute cerebrovascular response to SAH. Long known to modulate autonomic and sensorimotor circuits important in defense and homeostasis (Lovick, 1997), this region is well positioned to influence the extrinsic perivascular innervation through its interactions with both trigeminal sensory and autonomic pathways. We show for the first time that the RVM modulates cerebral blood flow under resting conditions, and plays an important role in restoring flow after experimental SAH. These findings raise the possibility that a failure of brainstem compensatory mechanisms including the RVM is significant for acute, and possibly delayed, ischemic deficits following SAH.
EXPERIMENTAL PROCEDURES
Animals and surgical preparation
All experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Oregon Health & Science University. Efforts were made throughout experiments to reduce the number of animals used, and to minimize animal discomfort. Experiments were performed on male Sprague-Dawley rats (Taconic, 250–350 g). Rats were initially anesthetized with pentobarbital (60mg/kg, i.p.), and a catheter was inserted into the external jugular vein for anesthetic administration. The right femoral artery was catheterized with PE-10 tubing, and the catheter was connected to a blood pressure monitor (Pressure Monitor BP-1, World Precision Instruments). Mean arterial pressure (MAP) was monitored throughout and the femoral catheter was flushed with approximately 0.2 ml of physiological saline at five minute intervals to clear the catheter of blood and prevent clotting. Rats were placed in a stereotactic apparatus and an area of the skull approximately 2 mm anterior to the coronal suture and 3 mm lateral to the midline was thinned with a drill until the superficial blood vessels became visible through the skull. A thin remnant of bone was left to ensure that the underlying dura and cortex were not injured. A craniotomy or craniotomies were drilled to allow placement of a needle for blood injection in the prechiasmatic cistern and/or a microinjection pipette in the RVM (see further description below). A circulating water pad was used to maintain body temperature at approximately 37 °C, and body temperature was monitored using a rectal temperature probe (Thermalert TH-5, Physitemp, Clifton, New Jersey).
As described previously (Barbaro et al., 1989), following the initial surgical preparation, the anesthetic level was allowed to lighten until a withdrawal reflex could be elicited by application of noxious heat using a feedback-controlled projector lamp focused on the ventral surface of the paw or by pinch. A continuous infusion of methohexital at a rate (15–30 mg/kg per h, i.v.) that allowed a stable withdrawal response latency and that prevented any signs of discomfort was then initiated. The rate was adjusted for each animal to allow a baseline withdrawal latency of 3–4 s. The experimental protocols (below) were begun after a stabilization period of at least 30 min, and infusion rate was not altered during the protocol.
Experimental model of subarachnoid hemorrhage (SAH)
We adapted a rodent model of SAH originally described by Prunell and colleagues (2002; 2003). In this model, autologous blood is injected into the prechiasmatic cistern. Unlike Prunell, we used a lightly anesthetized rat because this preparation has been extensively employed for functional analyses of brainstem circuitry in the context of autonomic control and pain modulation (Fields and Heinricher, 1985; Heinricher et al., 2009). A small craniotomy was drilled in the skull 7.5 mm anterior to bregma in the midline. Care was taken not to injure the underlying superior sagittal sinus. A blunted 27-gauge cannula was introduced stereotactically into the interhemispheric fissure at an angle of 30° posteriorly, and advanced just until the base of the brain was encountered, generally 9.5 mm below the surface. Fresh autologous blood (250 μl) was withdrawn immediately prior to use from the femoral catheter and infused slowly over 1 min through the cannula to minimize changes in intracranial pressure (Prunell et al., 2002; Prunell et al., 2003). We found that by adjusting the direction of the bevel of the cannula in the prechiasmatic cistern, we were able to create low- or high-“grade” hemorrhages analogous to the Fisher grading system (Fisher et al., 1980) which is based upon the amount of blood within the subarachnoid space versus the subdural space. Low-grade hemorrhages resulted if the bevel pointed up toward the base of the brain, whereas a high-grade hemorrhage was produced only if the bevel pointed directly lateral.
Following a 15-min baseline period, blood or saline was injected, and CBF and MAP recorded for an additional 60 min. Intracranial pressure was not monitored during the course of the experiment as intracranial pressure is known to be only transiently elevated following the blood injection and quickly returns to baseline (Prunell et al., 2002).
Blood gases were measured in four animals at baseline and at 5 and 60 min post-hemorrhage to ensure stability following the SAH. All parameters remained within the physiological range over the full 75 min protocol: pH, 7.39 ± 0.03; pCO2, 34.4 ± 4.1 mmHg; and pO2, 73.6 ± 7.0 mmHg (mean ± SD).
Microinjection and Drug Administration
An additional craniotomy followed by a small durotomy was made just posterior to lambda in the midline to allow the stereotactic placement of a microinjection pipette (usually glass, OD 75–100 μm, but in experiments utilizing optical microangiography, a 31g stainless-steel injector) into the RVM. To determine the effects of activating or blocking neuronal activity in RVM on CBF, the GABAA receptor antagonist bicuculline (50–200 pmol/200–400 nl) or the GABAA receptor agonist muscimol (8.8 pmol/200–400 nl) were respectively microinjected into the RVM. Extracellular single unit recordings have shown that at these doses, bicuculline produces a profound activation of all known RVM cell classes for a period of 45 min or more, with a peak 10–15 min after the injection (Heinricher and Tortorici, 1994). This method of activation is selective for cell bodies and avoids problems with electrical stimulation that recruit fibers of passage. Conversely, muscimol completely suppresses RVM cell activity for a similar period (Martenson et al., 2009). The micropipette was connected to a 1-μl Hamilton syringe via PE-50 tubing. Controls included an equivalent volume of saline vehicle and injections into surrounding brainstem areas. Early experiments used a 400 nl injection volume, but the injection volume was reduced to 200 nl to limit the spread of injectate to the RVM for most experiments.
To determine the effect of RVM manipulations on basal CBF, the drug or vehicle was microinjected into the RVM following a 15-min baseline period, and CBF and MAP then monitored for an additional 60 min. The effect of RVM muscimol (n = 5) and bicuculline (n = 7) on arterial blood gases were measured in additional animals at the times of peak effects on cerebral blood flow (for muscimol, 40–50 min post-injection; for bicuculline, 10–20 min post-injection). To determine the effects of RVM manipulations on the CBF response to the experimental hemorrhage, baseline CBF and MAP were determined (15 min) and drug or vehicle then microinjected into the RVM 10 min prior to experimental hemorrhage. CBF and MAP were then recorded for the subsequent 60 min.
Histology
The RVM was defined as the nucleus raphe magnus and adjacent reticular formation medial to the lateral edge of the pyramids at the level of the facial nucleus (Fields et al., 1983; Fields and Heinricher, 1985; Fields et al., 1991). It does not extend caudally beyond the facial nucleus (over the inferior olive), and is distinct from the more caudal region referred to by Strack and colleagues (1989) as the rostral ventral medulla. At the conclusion of each experiment, microinjection sites in the RVM were marked with an injection of pontamine sky blue dye. Rats were overdosed with methohexital, and perfused intracardially with physiological saline followed by 10% formalin. Distribution of blood on the surface of the brain was examined to assign a grade to the hemorrhage. Microinjection sites were histologically verified and plotted on standardized sections (Paxinos and Watson, 1997).
Measurement of CBF
In the majority of experiments, blood flow was measured using a laser Doppler probe placed directly onto the area of thinned skull, just anterior to the coronal suture and approximately 1 cm lateral to the sagittal sinus, and connected to a laser Doppler monitor (Laser Blood Flow MBF3D, Moor Instruments, Axminster, England).
A recently developed imaging modality, three-dimensional optical microangiography (OMAG, Wang, 2007; Wang et al., 2007) was used in four animals to image dynamic CBF in the right cerebral hemisphere before and after activation of the RVM. In brief, OMAG uses the laser Doppler effect to determine dynamic blood flow in both macro- and micro-circulatory beds by separating static light scattering elements, i.e. the microstructural features, from dynamic scattering elements, such as moving red blood cells. The signal strength of the blood flow in OMAG (luminance in the OMAG flow image) is proportional to the blood flow velocity, and it can thus be used to calculate absolute blood flow within the scanned volume. Here, total luminance was calculated from the dynamic scattering elements in the baseline OMAG scan and then compared to the post-injection OMAG images. Vessel lumen diameters were measured directly from the spatially resolved luminance from dynamic blood flow structures, which represent vessel walls. In these experiments, a larger area of the skull (roughly from the coronal to the lambdoidal suture and from the midline to 4 mm lateral) was thinned to expose the blood vessels. A metal cannula was inserted into the brain to a depth of 2 mm dorsal to the RVM and cemented to the skull. Baseline OMAG CBF images were obtained and a metal injector was inserted into the cannula, with the tip extending into the RVM. Bicuculline was then injected as in the experiments using laser Doppler flowmetry, and additional OMAG images were taken.
Analysis
After histological verification of drug injection sites, animals were grouped based on whether the injection was in or out of the RVM. For each animal, CBF and MAP measurements were normalized relative to baseline for that animal. The mean percent change at each post-injection time point was calculated for each group. Data are presented as mean ± SEM. Two-way repeated measures ANOVA was used for comparing the mean percent change between groups at each post-manipulation measurement point. Blood gas parameters before and after manipulation of the RVM were compared using t-tests for correlated means. P < 0.05 was considered significant.
RESULTS
Acute reduction in CBF is dependent on experimental grade of hemorrhage
Injection of 250 μl of fresh autologous blood into the pre-chiasmatic cistern resulted in an experimental SAH that could be defined as low or high “grade” as determined by the distribution of blood in the subarachnoid space. Examples of the different grades are shown in Fig. 1. Low-grade hemorrhages were defined as minimal or moderate blood, restricted to the basal cisterns. A high-grade hemorrhage was defined by blood that also tracked out of the basal cisterns into the subarachnoid space around the cerebral cortices.
Fig 1. Representative photographs illustrating different grades of experimental subarachnoid hemorrhage.
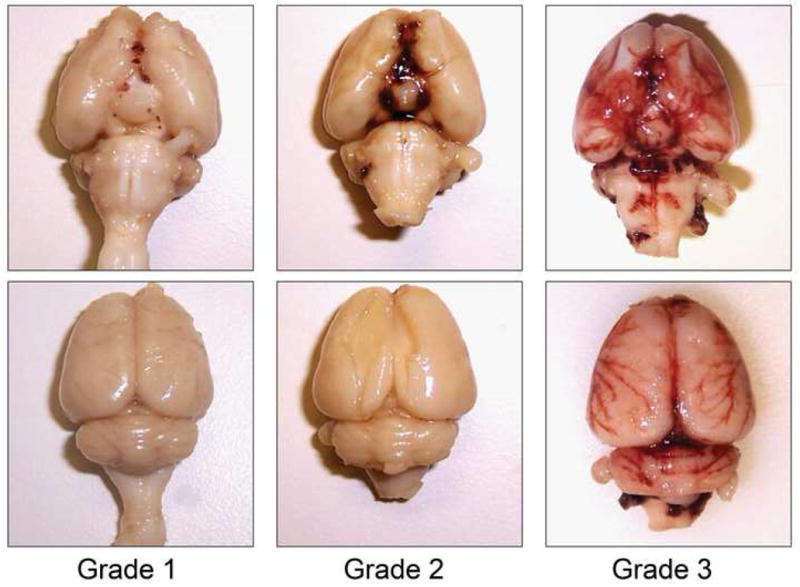
Blood is confined to the basal cisterns in low-grade hemorrhages. In hemorrhages assigned to the “high-grade” group, blood was found to extend beyond the basal cisterns around the lateral convexities.
We next investigated the effect of the experimental hemorrhage on CBF using Laser Doppler flowmetry. Consistent with a previous report of an acute global decrease in cerebral blood flow in this model (Prunell et al., 2004), high-grade hemorrhages in our animals resulted in a significant reduction in CBF that lasted 60–90 min. Early (1 min) and later (40 min) responses to intracisternal blood injection as a function of hemorrhage grade are shown in Fig. 2. A transient rapid fall in CBF that quickly rebounded within 1 to 2 min of starting the injection was seen with both low- and high-grade hemorrhages (Fig. 2A and B), and with injections of warmed saline as a volume control (not shown). This transient fall approximates the time course of changes in intracranial pressure previously described in this model (Prunell et al., 2002). With saline injection and with low-grade hemorrhages, this initial fall was followed in all cases by a small transient rebound hyperemia before returning to baseline (e.g. Fig. 2A). With high-grade hemorrhages, a brief rebound was seen in some cases (3 of 8 animals, e.g. Fig. 2B), but CBF then dropped to approximately 20% below baseline for at least an hour. Group data comparing the early transient and later sustained response to intracisternal blood injection as a function of hemorrhage grade are shown in Fig. 2C.
Fig 2. Changes in CBF with experimental SAH.
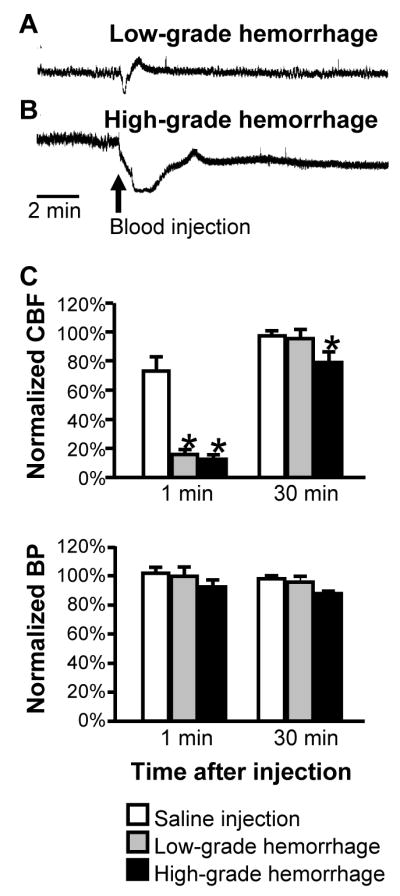
A and B: Initial response to hemorrhage illustrates transient drop in CBF in animals sustaining either low-grade (A) or high-grade (B) hemorrhage. Raw laser Doppler flowmeter traces show flow (arbitrary perfusion units) in initial 15 min period following blood injection. In both animals, there was a transient sharp drop in flow. In the animal sustaining the low-grade hemorrhage, flow immediately rebounded above baseline and then recovered to baseline levels. By contrast, recovery was slower and incomplete after the high-grade hemorrhage. Scale bar indicates 2 minutes.
C: Group data show average drop in laser Doppler perfusion measured at one and forty minutes after injection of saline or blood into the basal cisterns. Immediately following the injection, flow was depressed to a significantly greater degree in animals receiving blood compared to saline, irrespective of whether the resulting hemorrhage was considered low-grade or high-grade. Only with high-grade hemorrhage was flow still depressed at forty min after the injection. (*p < 0.05 ANOVA followed by Dunnett’s test for comparison to saline, n = 6–9/group).
Blood pressure in both hemorrhage groups declined gradually over the hour following blood injection (approximately 10% decrease by one hour post-injection in both groups), but this difference was not statistically significant compared to saline-injected animals (ANOVA, p > 0.05).
Role of the RVM in changes in cerebral blood flow (CBF) following experimental SAH
Focal application of the GABAA receptor agonist muscimol was then used to investigate the role of the RVM in the response to experimental hemorrhage. When the RVM was inactivated, the long-lasting drop in flow exhibited by animals sustaining a high-grade hemorrhage was significantly potentiated (Fig. 3). Importantly, there was no difference between the two groups in post-hemorrhage blood pressure (RVM intact: 88.2 ± 3.8% of baseline, RVM block: 81.5 ± 7.3, p = 0.39, t-test for independent groups).
Fig 3. Fall in CBF following high-grade hemorrhage is potentiated in animals in which the RVM has been inactivated using the GABAA receptor agonist muscimol.
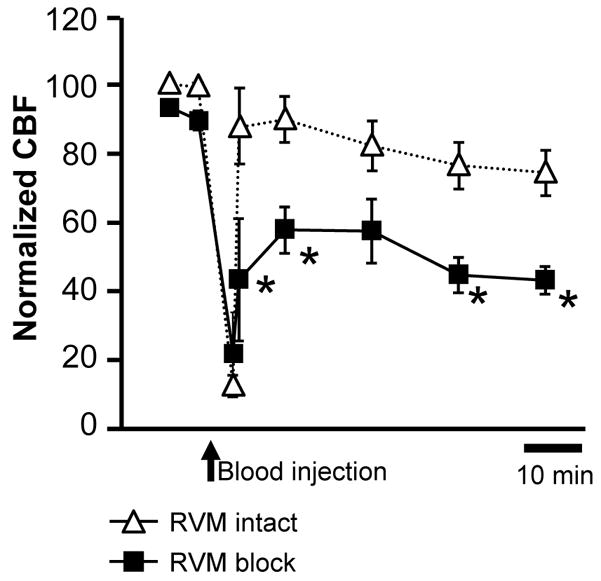
This persisted for the duration of the experiment. *p < 0.05 compared to SAH with RVM intact, two-way repeated measures ANOVA followed by Bonferroni post-hoc tests; hemorrhage alone, n = 5; RVM muscimol/hemorrhage, n = 8.
In animals sustaining a low-grade hemorrhage following RVM block, we observed a modest decline in CBF over the course of the experiment, to 90.3 ± 9.0% of baseline (p > 0.05 compared to animals sustaining a low-grade hemorrhage with the RVM intact). This decline should possibly be attributed to the overall effect of RVM inactivation on resting CBF (see next section).
Effects of RVM manipulation on resting CBF
We next determined whether the RVM contributes to CBF under baseline conditions. Muscimol or the GABAA receptor antagonist bicuculline were microinjected at sites shown in Fig. 4 to respectively inactivate and activate the RVM. Injections adjacent to the RVM (grey symbols) served as anatomical controls.
Fig 4. Plot of the muscimol and bicuculline injection sites in the set of experiments measuring resting CBF.

Grey filled circles are considered anatomical placement controls, and injection in these animals was associated with no change in resting CBF. Black circles indicate sites within the RVM, and were associated with altered resting CBF. Inset shows representative microinjection site dorsal to the pyramids at the level of the facial nucleus.
Fig. 5 summarizes the effects of pharmacological inactivation and stimulation of the RVM on CBF measured using laser-Doppler flowmetry (Fig. 5A, top). CBF was not altered by control injections of saline vehicle into the RVM (example in Fig. 5B, Saline). Blocking activity in the RVM using focal microinjection of the GABAA receptor agonist muscimol led to a modest but significant and prolonged decline in resting CBF, to approximately 80% of baseline (example in Fig. 5B, Muscimol). Activating RVM neurons using the GABAA receptor antagonist bicuculline resulted in a 20–30% increase in flow (example in Fig. 5B, Bicuculline). The timing of the CBF responses to muscimol and bicuculline parallel the known duration of RVM neuronal responses to these compounds determined using single-cell recording (Heinricher and Tortorici, 1994; Martenson et al., 2009). Injections in areas surrounding the RVM had no effect on CBF (data not shown), localizing the site of action to the RVM.
Fig 5. Inactivation or activation of the RVM lead to decreases and increases respectively in resting CBF.

A. Block of RVM activity using focal application of the GABAA receptor agonist muscimol (8.8 pmol/200 nl, n = 9) resulted in a reduction in resting CBF that was maintained over the 60 min post-injection time course, consistent with the known kinetics of microinjected muscimol in this region (Martenson et al., 2009). Disinhibition of the RVM using the GABAA receptor antagonist bicuculline (50–200 pmol/200–400 nl, n = 8) resulted in an increase in flow that peaked at 10–20 min, again consistent with the known time course of bicuculline effects in the RVM (Heinricher and Tortorici, 1994). Saline injections had no effect (n = 6). Middle and lower panels show blood pressure (BP, mean arterial pressure) and calculated cerebrovascular resistance (CVR) in the same animals. Blood pressure was significantly reduced following RVM activation with bicuculline, and unchanged following RVM inactivation using muscimol. The respective decrease and increase in cerebral perfusion in these animals was not therefore secondary to a change in perfusion pressure. CBF expressed as percent of baseline. For muscimol, values are mean of 40–50 min post-injection; for bicuculline and saline, mean of 10–20 min post-injection. *p < 0.05, ANOVA followed by Dunnett’s test for comparison to saline-treated animals.
B. Laser-Doppler traces illustrate typical time course of the CBF response to RVM microinjection of the GABAA receptor agonist muscimol, the GABAA receptor antagonist bicuculline, and saline vehicle in different animals. Drug was injected at the end of a 30 min baseline period, and CBF followed for the next 60 min.
C. Muscimol and bicuculline microinjection in the RVM did not alter blood gas parameters. pH, pCO2 and pO2 of arterial blood in baseline and following microinjection of muscimol (MUS) or bicuculline (BIC).
Changes in CBF cannot be explained by parallel changes in perfusion pressure, as arterial pressure was reduced following both RVM block and stimulation, although this achieved significance only in the case of RVM stimulation (Fig. 5A, middle). Calculated cerebrovascular resistance was increased following muscimol injection, and reduced following bicuculline injection, although the change was statistically significant only for bicuculline (Fig. 5A, bottom). In addition, activation and inactivation of the RVM failed to significantly alter blood gas parameters (pH, pCO2, pO2, p > 0.05, t-tests for correlated means, Fig. 5C).
The effects of RVM activation (bicuculline microinjection) on CBF obtained using laser Doppler flowmetry were confirmed in four animals using three-dimensional OMAG, which provided an absolute measure of flow over the entire distal territory of the middle cerebral artery (Wang, 2007; Wang et al., 2007). Because OMAG provides depth-resolved imaging, the blood flow within the meninges was separately evaluated from the blood flow within the cerebral cortex. Volumetric OMAG blood flow images demonstrated that both meningeal and cortical arteries dilated by approximately 25% at the time of peak effect and followed a time course similar to that observed using conventional laser Doppler flowmetry (Fig. 6). Absolute blood flow in a 2×2×2 mm block of cortex increased by 42%. This confirms that the increased CBF evoked by RVM stimulation is in fact the result of vasodilation.
Fig 6.

OMAG imaging of cerebral blood flow with RVM activation. (A) Volumetric rendering of OMAG blood flow image over the distal middle cerebral artery territory, in which the blood vessels situated in meninges (green arrow) and cortex (white arrow) can be separately identified. Size of the scanned volume = 2×2×2 mm3 (x-y-z). Shown also are the OMAG x-y projection image captured before the activation of the RVM (B), and at the time of peak effect (C) after the activation of the RVM. Scale bar 500μm.
Anatomical controls in which areas outside of the RVM were stimulated failed to elicit an effect (not shown). Video images showing changes in CBF and vessel diameter with RVM stimulation can be found in Supplementary Figure 1.
DISCUSSION
Over the past decade, there has been growing acceptance of the notion that the RVM integrates aspects of defense in response to a range of internal and external threats to homeostasis and bodily integrity (Lovick, 1997; Heinricher et al., 2009; Martenson et al., 2009). However, this region has not previously been demonstrated to play a role in CBF. We show here that the RVM is an important brainstem site for modulation of CBF under basal conditions: disinhibition of the RVM produced a significant increase in CBF, whereas inhibition of RVM neurons resulted in a decrease in CBF. Optical microangiography (OMAG), a novel optical imaging technique based on Fourier domain optical coherence tomography (Wang, 2007; Wang et al., 2007), confirmed that RVM disinhibition resulted in cerebral vasodilation.
We also studied the role of the RVM in the context of subarachnoid hemorrhage. Following experimental subarachnoid hemorrhage, we saw an acute reduction of CBF that was markedly potentiated by inactivation of the RVM. This finding indicates that the RVM contributes to the restoration of cerebral perfusion in the face of a high-grade experimental SAH, and suggests that a pronounced decrease in cerebral perfusion in patients following SAH could be in part be due to a failure of brainstem compensatory mechanisms.
Model of subarachnoid hemorrhage
In these studies, we adapted a rodent model of SAH in which blood is injected into the pre-chiasmatic cistern (Prunell et al., 2002; Prunell et al., 2003). Although animal models of SAH may not fully represent the disease course seen in humans (Titova et al., in press), the model employed here has been associated with a global decrease in cerebral perfusion, and with both transient (< 90 min duration) and late (48 h) hypoperfusion, measured using laser-Doppler flowmetry, PET or iodoantipyrine autoradiography (Yamamoto et al., 1998; Prunell et al., 2002, 2004). Changes in intracranial pressure are transient (< 5 min) and do not predict subsequent reductions in perfusion (Prunell et al., 2002; Prunell et al., 2003). Despite the fact that there is no direct vascular damage in this model, the distribution of blood is similar to that often seen after clinical SAH (Prunell et al., 2002), which most commonly involves the anterior circulation. Loss of forebrain cholinergic neurons and markers of inflammation and cell death are found in at least some animals up to a week after the experimental hemorrhage (Prunell et al., 2005a, b; Lohr et al., 2008). Further, a subset of animals show neurological deficits (Prunell et al., 2004). Although changes in vessels have also been demonstrated (Hansen-Schwartz et al., 2003a; Hansen-Schwartz et al., 2003b; Ansar et al., 2007; Ansar and Edvinsson, 2008), the global decrease in cerebral perfusion points to a neural or neurohumoral mechanism as playing at least some role (Yamamoto et al., 1998; Prunell et al., 2004).
These studies focused on the acute hemodynamic changes following the experimental SAH. However, delayed vasospasm and associated clinical ischemic deficits are by definition not seen until 3 to 14 days after the initial hemorrhage. Nevertheless, acute ischemic events in patients are not uncommon (Schmidt et al., 2007), and these are an important predictor of increased mortality and long-term morbidity (Schubert and Thome, 2008). Certainly, it is reasonable to think that delayed ischemia has its foundation in earlier pathophysiological processes (Sehba and Bederson, 2006; Cahill and Zhang, 2008). There is, in addition, increasing evidence for acute reversible vasoconstriction as a clinical problem in its own right (Ducros et al., 2007). The acute animal model therefore has significant translational relevance.
The RVM and modulation of CBF
To our knowledge these experiments provide the first demonstration that manipulation of the RVM gives rise to alterations in cerebral blood flow. This region has long been known to modulate sensorimotor processing important for nocifensor behaviors (Heinricher and Ingram, 2008), and has also been implicated in regulation of some sympathetic outflows, based on both anatomical and functional studies (Ter Horst et al., 1996; Blessing, 1997; Horiuchi et al., 2004; Nakamura et al., 2004; Kerman, 2008; Morrison et al., 2008; Salo et al., 2009). Positive and negative modulation of nociception and various aspects of autonomic regulation appear to be mediated by distinct RVM cell populations, but are likely recruited in a coordinated manner to orchestrate behavioral and physiological responses to internal and external threats (Lovick, 1997; Heinricher et al., 2009; Martenson et al., 2009). The present findings extend this idea to CBF, and indicate that the RVM contributes to maintenance of normal CBF under basal conditions and following SAH. It would therefore be of significant interest to go beyond the effects of non-specific stimulation and inactivation of this region as a whole (as was done here) to ask what specific RVM neural populations and output pathways are involved in the effects on cerebral blood flow.
CBF was not altered by infusions of muscimol or bicuculline into areas dorsal, ventral, or caudal to the RVM. This lack of effect is consistent with previous work using this same microinjection technique in which we demonstrated that placements as little as 500 μm away from the RVM had no effect on nocifensor reflexes (Heinricher and Kaplan, 1991; Heinricher et al., 1994). These anatomical controls thus allow us to localize drug action to the RVM (Yaksh and Rudy, 1978; Wise and Hoffman, 1992), and rule out diffusion to brainstem sympathetic and parasympathetic premotor areas previously implicated in regulation of cerebral blood flow, such as the superior salivatory nuclei or rostral ventrolateral medulla (Underwood et al., 1992; Golanov and Reis, 1994; Talman et al., 2007; Boysen et al., 2009).
Potential mechanisms through which the RVM could modulate cerebral blood flow
These studies demonstrate that the RVM contributes to both resting CBF and recovery of cerebral perfusion after a high-grade SAH, raising the question of how neuronal activity in the RVM is transduced into cerebrovascular changes. The RVM is well positioned to modulate circuits important for the extrinsic perivascular innervation, including sympathetic, parasympathetic, and sensory fibers, which is increasingly recognized as important in times of stress. The RVM includes sympathetic premotor neurons (Strack et al., 1989; Kerman et al., 2003; Kerman et al., 2006), has connections with the parasympathetic outflow to the pterygopalatine ganglion (Spencer et al., 1990), and projects directly to the trigeminal sensory nucleus (Lovick and Robinson, 1983), providing potential anatomical substrates for interactions of this region with the known perivascular innervation. Alternatively, the RVM could interact not with the extrinsic perivascular innervation, but with intrinsic systems. Ascending projections have been documented from the RVM, but these projections are not serotonergic, and there is no evidence at present that they influence intrinsic vessels (Bowker, 1986; Hermann et al., 1996).
The RVM is also interconnected with other brainstem regions previously implicated in regulation of cerebral blood flow, including the rostral ventrolateral medulla (caudal and lateral to the rostral ventromedial medulla which is our focus in the present set of experiments, Spencer et al., 1990; Zagon, 1993; Babic et al., 2008). Stimulation of the rostral ventrolateral medulla and the associated medullary cerebrovasodilator area increases resting cerebral blood flow without altering cerebral metabolism (Underwood et al., 1992; Golanov and Reis, 1996; Golanov et al., 2000).
Another possibility is that altered CBF was due to changes in ventilation, since the RVM contains chemosensitive neurons and participates in parallel with other medullary sites to modulate respiratory drive (Nattie and Li, 2001; Mulkey et al., 2007; Guyenet et al., 2008). However, the fact that RVM manipulations did not alter arterial blood gases argues against this possibility.
Changes in arterial blood pressure also cannot account for the changes in CBF evoked by RVM manipulation. Increased CBF produced by activation of RVM neurons was associated with a significant decrease in arterial blood pressure, and a substantial decrease in calculated cerebrovascular resistance, consistent with the vasodilation revealed using optical microangiography. The reduction in CBF following RVM injection of muscimol was also associated with decrease in blood pressure, but an increase in cerebrovascular resistance, albeit smaller and not statistically significant. The observation that arterial blood pressure was not significantly lowered following experimental SAH, whether the RVM was intact or blocked, argues that the observed reduction in CBF following SAH was similarly not secondary to reduced perfusion pressure.
Inactivation of the RVM has been reported to increase cutaneous blood flow (tail and pinna), and to reduce sympathetic cutaneous vasoconstrictor activity, whereas stimulation in this region increases sympathetic activity in the tail, but not renal, outflow (Blessing et al., 1999; Rathner and McAllen, 1999; Korsak and Gilbey, 2004; Rathner et al., 2008). The skin blood flow response to RVM manipulation is thus the reverse of what we observed here with cerebral blood flow, and indicates that the RVM has the potential to exert opposing influences on cerebral and cutaneous blood flows. This may be related to the coordinated cutaneous vasoconstriction and diversion of blood to the brain triggered by stimuli that alert the organism or threaten cerebral homeostasis (Reis et al., 1997; Blessing and Nalivaiko, 2000; Ootsuka and Blessing, 2005).
Implications for ischemic events following subarachnoid hemorrhage
An important output center for the brainstem autonomic core, the RVM has long been implicated in coordinating physiological and behavioral aspects of defense in response to both internal and external challenges to homeostasis (Lovick, 1997). This has been studied most intensively in the context of pain modulation, where the RVM has been shown to exert both inhibitory and facilitatory influences mediated by distinct cell populations. In the period immediately following a noxious insult or during acute inflammation, the facilitating outflow from the RVM enhances vigilance and recuperative mechanisms as part of a positive feedback loop. This is generally considered beneficial, and the inhibitory output from the RVM is suppressed under these conditions (Porreca et al., 2002; Fields, 2004; Heinricher et al., 2009). However, chronic activation of this system is now known to contribute to pathological pain states, and alterations of neuronal excitability and phenotype have been demonstrated under chronic conditions (Miki et al., 2002; Porreca et al., 2002; Ren and Dubner, 2002; Ossipov et al., 2004; Carlson et al., 2007; Heinricher et al., 2009)
The pathophysiology of aneurysmal subarachnoid hemorrhage remains poorly understood. This is due in part to its biphasic course, but also to the large number of changes that occur in multiple body and brain systems. Despite the general correlation between clinical grade and the amount of blood within the subarachnoid space, some patients die immediately from modest hemorrhages while others are coherent and even conversant. Indeed, a better predictor of long-term outcome may be the initial neurological condition at presentation (Macdonald et al., 2007; Losiniecki and Zuccarello, 2008). Clinical reports of patients presenting with acute ischemic deficits after SAH demonstrate that such patients are much more likely to have either lost consciousness at the time of aneurysm rupture, or to be in a coma at presentation (Schmidt et al., 2007), findings consistent with a failure of brainstem mechanisms. Taken together with the present observation that inactivation of the RVM contributes to acute restoration of cerebral perfusion following a high-grade experimental hemorrhage, these observations raise the possibility that deficits in intrinsic brainstem homeostatic mechanisms contribute to the acute reduction in CBF and neurological function seen in some patients with high grade hemorrhages.
Supplementary Material
Supplementary Figure 1. Dynamic OMAG blood flow imaging in vivo after microinjection of bicuculline into the RVM with a temporal time resolution of 1 minute. The changes in flow can readily be seen over the course of 30 minutes. (See video). (B) Measured changes of the diameter of the vessel indicated in (A) over 30 minutes after activation of the RVM. Scale bar = 500μm.
{kind=link}
Acknowledgments
This work was supported by grants from the Neurosurgery Research and Education Foundation from the American Association of Neurological Surgeons to JSC, from NINDS (NS052364 to MMH; NS044313 to NJA); and NIHLB (HL093140 to R-K.W). We thank Shirley McCartney, Ph.D. for manuscript processing.
LIST OF ABBREVIATIONS
- BP
blood pressure
- CBF
cerebral blood flow
- CNS
central nervous system
- CGRP
calcitonin-gene related peptide
- CVR
cerebrovascular resistance
- i.p
intraperitoneal
- i.v
intravenous
- MAP
mean arterial pressure
- OMAG
optical microangiography
- RVLM
rostral ventrolateral medulla
- RVM
rostral ventromedial medulla
- SAH
subarachnoid hemorrhage
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansar S, Edvinsson L. Subtype activation and interaction of protein kinase C and mitogen-activated protein kinase controlling receptor expression in cerebral arteries and microvessels after subarachnoid hemorrhage. Stroke. 2008;39:185–190. doi: 10.1161/STROKEAHA.107.487827. [DOI] [PubMed] [Google Scholar]
- Ansar S, Vikman P, Nielsen M, Edvinsson L. Cerebrovascular ETB, 5-HT1B, and AT1 receptor upregulation correlates with reduction in regional CBF after subarachnoid hemorrhage. Am J Physiol Heart Circ Physiol. 2007;293:H3750–3758. doi: 10.1152/ajpheart.00857.2007. [DOI] [PubMed] [Google Scholar]
- Arienta C, Balbi S, Caroli M, Fumagalli G. Depletion of calcitonin gene-related peptide in perivascular nerves during acute phase of posthemorrhagic vasospasm in the rabbit. Brain Res Bull. 1991;27:605–609. doi: 10.1016/0361-9230(91)90034-h. [DOI] [PubMed] [Google Scholar]
- Babic T, de Oliveira CV, Ciriello J. Collateral axonal projections from rostral ventromedial medullary nitric oxide synthase containing neurons to brainstem autonomic sites. Brain Res. 2008;1211:44–56. doi: 10.1016/j.brainres.2007.10.104. [DOI] [PubMed] [Google Scholar]
- Barbaro NM, Heinricher MM, Fields HL. Putative nociceptive modulatory neurons in the rostral ventromedial medulla of the rat display highly correlated firing patterns. Somatosens Mot Res. 1989;6:413–425. doi: 10.3109/08990228909144684. [DOI] [PubMed] [Google Scholar]
- Blessing WW. The lower brainstem and bodily homeostasis. New York: Oxford University Press; 1997. [Google Scholar]
- Blessing WW, Nalivaiko E. Regional blood flow and nociceptive stimuli in rabbits: patterning by medullary raphe, not ventrolateral medulla. J Physiol. 2000;524(Pt 1):279–292. doi: 10.1111/j.1469-7793.2000.t01-2-00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessing WW, Yu YH, Nalivaiko E. Raphe pallidus and parapyramidal neurons regulate ear pinna vascular conductance in the rabbit. Neurosci Lett. 1999;270:33–36. doi: 10.1016/s0304-3940(99)00459-0. [DOI] [PubMed] [Google Scholar]
- Bowker RM. The relationship between descending serotonin projections and ascending projections in the nucleus raphe magnus: a double labeling study. Neurosci Lett. 1986;70:348–353. doi: 10.1016/0304-3940(86)90577-x. [DOI] [PubMed] [Google Scholar]
- Boysen NC, Dragon DN, Talman WT. Parasympathetic tonic dilatory influences on cerebral vessels. Auton Neurosci. 2009;147:101–104. doi: 10.1016/j.autneu.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill J, Zhang JH. Subarachnoid Hemorrhage. Is It Time for a New Direction? Stroke. 2008 doi: 10.1161/STROKEAHA.108.533315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JD, Maire JJ, Martenson ME, Heinricher MM. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. J Neurosci. 2007;27:13222–13231. doi: 10.1523/JNEUROSCI.3715-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros A, Boukobza M, Porcher R, Sarov M, Valade D, Bousser MG. The clinical and radiological spectrum of reversible cerebral vasoconstriction syndrome. A prospective series of 67 patients. Brain. 2007;130:3091–3101. doi: 10.1093/brain/awm256. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Juul R, Jansen I. Perivascular neuropeptides (NPY, VIP, CGRP and SP) in human brain vessels after subarachnoid haemorrhage. Acta Neurol Scand. 1994;90:324–330. doi: 10.1111/j.1600-0404.1994.tb02732.x. [DOI] [PubMed] [Google Scholar]
- Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5:565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Fields HL, Bry J, Hentall I, Zorman G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983;3:2545–2552. doi: 10.1523/JNEUROSCI.03-12-02545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM. Anatomy and physiology of a nociceptive modulatory system. Philos Trans of the R Soc Lond B Biol Sci. 1985;308:361–374. doi: 10.1098/rstb.1985.0037. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6:1–9. doi: 10.1227/00006123-198001000-00001. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Sphenopalatine ganglion stimulation increases regional cerebral blood flow independent of glucose utilization in the cat. Brain Res. 1990;506:145–148. doi: 10.1016/0006-8993(90)91211-x. [DOI] [PubMed] [Google Scholar]
- Golanov EV, Reis DJ. Nitric oxide and prostanoids participate in cerebral vasodilation elicited by electrical stimulation of the rostral ventrolateral medulla. J Cereb Blood Flow Metab. 1994;14:492–502. doi: 10.1038/jcbfm.1994.61. [DOI] [PubMed] [Google Scholar]
- Golanov EV, Reis DJ. Contribution of oxygen-sensitive neurons of the rostral ventrolateral medulla to hypoxic cerebral vasodilatation in the rat. J Physiol. 1996;495 (Pt 1):201–216. doi: 10.1113/jphysiol.1996.sp021585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golanov EV, Ruggiero DA, Reis DJ. A brainstem area mediating cerebrovascular and EEG responses to hypoxic excitation of rostral ventrolateral medulla in rat. J Physiol. 2000;529(Pt 2):413–429. doi: 10.1111/j.1469-7793.2000.00413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA, Mulkey DK, Stornetta RL, Moreira TS, Takakura AT. The retrotrapezoid nucleus and central chemoreception. Adv Exp Med Biol. 2008;605:327–332. doi: 10.1007/978-0-387-73693-8_57. [DOI] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Hansen-Schwartz J, Hoel NL, Xu CB, Svendgaard NA, Edvinsson L. Subarachnoid hemorrhage-induced upregulation of the 5-HT1B receptor in cerebral arteries in rats. J Neurosurg. 2003a;99:115–120. doi: 10.3171/jns.2003.99.1.0115. [DOI] [PubMed] [Google Scholar]
- Hansen-Schwartz J, Hoel NL, Zhou M, Xu CB, Svendgaard NA, Edvinsson L. Subarachnoid hemorrhage enhances endothelin receptor expression and function in rat cerebral arteries. Neurosurgery. 2003b;52:1188–1194. 1194 1185. [PubMed] [Google Scholar]
- Heinricher MM, Ingram SL. The brainstem and nociceptive modulation. In: Bushnell MC, Basbaum AI, editors. The Senses, A Comprehensive Reference, Vol 5, Pain. San Diego: Academic Press; 2008. pp. 593–626. [Google Scholar]
- Heinricher MM, Kaplan HJ. GABA-mediated inhibition in rostral ventromedial medulla: role in nociceptive modulation in the lightly anesthetized rat. Pain. 1991;47:105–113. doi: 10.1016/0304-3959(91)90017-R. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Tortorici V, Fields HL. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63:279–288. doi: 10.1016/0306-4522(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. 2009;60:214–225. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM, Tortorici V. Interference with GABA transmission in the rostral ventromedial medulla: disinhibition of off-cells as a central mechanism in nociceptive modulation. Neuroscience. 1994;63:533–546. doi: 10.1016/0306-4522(94)90548-7. [DOI] [PubMed] [Google Scholar]
- Henninger N, Fisher M. Stimulating circle of Willis nerve fibers preserves the diffusion- perfusion mismatch in experimental stroke. Stroke. 2007;38:2779–2786. doi: 10.1161/STROKEAHA.107.485581. [DOI] [PubMed] [Google Scholar]
- Hermann DM, Luppi PH, Peyron C, Hinckel P, Jouvet M. Forebrain projections of the rostral nucleus raphe magnus shown by iontophoretic application of choleratoxin b in rats. Neurosci Lett. 1996;216:151–154. doi: 10.1016/0304-3940(96)13013-5. [DOI] [PubMed] [Google Scholar]
- Horiuchi J, McAllen RM, Allen AM, Killinger S, Fontes MAP, Dampney RAL. Descending vasomotor pathways from the dorsomedial hypothalamic nucleus: role of medullary raphe and RVLM. Am J Physiol Regul Integr Comp Physiol. 2004;287:R824–832. doi: 10.1152/ajpregu.00221.2004. [DOI] [PubMed] [Google Scholar]
- Juul R, Hara H, Gisvold SE, Brubakk AO, Fredriksen TA, Waldemar G, Schmidt JF, Ekman R, Edvinsson L. Alterations in perivascular dilatory neuropeptides (CGRP, SP, VIP) in the external jugular vein and in the cerebrospinal fluid following subarachnoid haemorrhage in man. Acta Neurochir (Wien) 1995;132:32–41. doi: 10.1007/BF01404845. [DOI] [PubMed] [Google Scholar]
- Kerman IA. Organization of brain somatomotor-sympathetic circuits. Exp Brain Res. 2008;187:1–16. doi: 10.1007/s00221-008-1337-5. [DOI] [PubMed] [Google Scholar]
- Kerman IA, Enquist LW, Watson SJ, Yates BJ. Brainstem substrates of sympatho-motor circuitry identified using trans-synaptic tracing with pseudorabies virus recombinants. J Neurosci. 2003;23:4657–4666. doi: 10.1523/JNEUROSCI.23-11-04657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman IA, Shabrang C, Taylor L, Akil H, Watson SJ. Relationship of presympathetic-premotor neurons to the serotonergic transmitter system in the rat brainstem. J Comp Neurol. 2006;499:882–896. doi: 10.1002/cne.21129. [DOI] [PubMed] [Google Scholar]
- Korsak A, Gilbey MP. Rostral ventromedial medulla and the control of cutaneous vasoconstrictor activity following i.c.v. prostaglandin E1. Neuroscience. 2004;124:709–717. doi: 10.1016/j.neuroscience.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Lambert G, Naredi S, Eden E, Rydenhag B, Friberg P. Sympathetic nervous activation following subarachnoid hemorrhage: Influence of intravenous clonidine. Acta Anaesthesiol Scand. 2002;46:160–165. doi: 10.1034/j.1399-6576.2002.460206.x. [DOI] [PubMed] [Google Scholar]
- Lohr M, Tzouras G, Molcanyi M, Ernestus RI, Hampl JA, Fischer JH, Sahin K, Arendt T, Hartig W. Degeneration of cholinergic rat Basal forebrain neurons after experimental subarachnoid hemorrhage. Neurosurgery. 2008;63:336–344. doi: 10.1227/01.NEU.0000320422.54985.6D. [DOI] [PubMed] [Google Scholar]
- Lombard F, Borel C. Pathogenesis of cerebral vasospasm. In: Bhardwaj A, Alkayed NJ, Kirsch JR, Traystman RJ, editors. Acute stroke: bench to bedside. New York: Informa Healthcare; 2007. pp. 29–44. [Google Scholar]
- Losiniecki A, Zuccarello M. Subarachnoid hemorrhage: effect on cerebral blood flow and cerebral metabolism. Front Biosci. 2008;13:1845–1856. doi: 10.2741/2804. [DOI] [PubMed] [Google Scholar]
- Lovick TA. The medullary raphe nuclei: a system for integration and gain control in autonomic and somatomotor responsiveness? Exp Physiol. 1997;82:31–41. doi: 10.1113/expphysiol.1997.sp004013. [DOI] [PubMed] [Google Scholar]
- Lovick TA, Robinson JP. Bulbar raphe neurones with projections to the trigeminal nucleus caudalis and the lumbar cord in the rat: a fluorescence double-labelling study. Exp Brain Res. 1983;50:299–308. doi: 10.1007/BF00239194. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256–263. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- Martenson ME, Cetas JS, Heinricher MM. A possible neural basis for stress-induced hyperalgesia. Pain. 2009;142:236–244. doi: 10.1016/j.pain.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki K, Zhou QQ, Guo W, Guan Y, Terayama R, Dubner R, Ren K. Changes in gene expression and neuronal phenotype in brain stem pain modulatory circuitry after inflammation. J Neurophysiol. 2002;87:750–760. doi: 10.1152/jn.00534.2001. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp Physiol. 2008;93:773–797. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Rosin DL, West G, Takakura AC, Moreira TS, Bayliss DA, Guyenet PG. Serotonergic Neurons Activate Chemosensitive Retrotrapezoid Nucleus Neurons by a pH-Independent Mechanism. J Neurosci. 2007;27:14128–14138. doi: 10.1523/JNEUROSCI.4167-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Matsumura K, Hubschle T, Nakamura Y, Hioki H, Fujiyama F, Boldogkoi Z, Konig M, Thiel HJ, Gerstberger R, Kobayashi S, Kaneko T. Identification of sympathetic premotor neurons in medullary raphe regions mediating fever and other thermoregulatory functions. J Neurosci. 2004;24:5370–5380. doi: 10.1523/JNEUROSCI.1219-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Li A. CO2 dialysis in the medullary raphe of the rat increases ventilation in sleep. J Appl Physiol. 2001;90:1247–1257. doi: 10.1152/jappl.2001.90.4.1247. [DOI] [PubMed] [Google Scholar]
- Ootsuka Y, Blessing WW. Inhibition of medullary raphe/parapyramidal neurons prevents cutaneous vasoconstriction elicited by alerting stimuli and by cold exposure in conscious rabbits. Brain Res. 2005 doi: 10.1016/j.brainres.2005.05.062. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, Porreca F. Antinociceptive and nociceptive actions of opioids. J Neurobiol. 2004;61:126–148. doi: 10.1002/neu.20091. [DOI] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Diemer NH, Svendgaard NA. Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery. 2003;52:165–175. doi: 10.1097/00006123-200301000-00022. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Svendgaard NA. A new experimental model in rats for study of the pathophysiology of subarachnoid hemorrhage. Neuroreport. 2002;13:2553–2556. doi: 10.1097/00001756-200212200-00034. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Svendgaard NA. Experimental subarachnoid hemorrhage: cerebral blood flow and brain metabolism during the acute phase in three different models in the rat. Neurosurgery. 2004;54:426–436. doi: 10.1227/01.neu.0000103670.09687.7a. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J Neurosurg. 2005a;102:1046–1054. doi: 10.3171/jns.2005.102.6.1046. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery. 2005b;56:1082–1092. [PubMed] [Google Scholar]
- Rathner JA, Madden CJ, Morrison SF. Central pathway for spontaneous and prostaglandin E2-evoked cutaneous vasoconstriction. Am J Physiol Regul Integr Comp Physiol. 2008;295:R343–354. doi: 10.1152/ajpregu.00115.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathner JA, McAllen RM. Differential control of sympathetic drive to the rat tail artery and kidney by medullary premotor cell groups. Brain Res. 1999;834:196–199. doi: 10.1016/s0006-8993(99)01568-1. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Golanov EV, Galea E, Feinstein DL. Central neurogenic neuroprotection: central neural systems that protect the brain from hypoxia and ischemia. Ann N Y Acad Sci. 1997;835:168–186. doi: 10.1111/j.1749-6632.1997.tb48628.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- Salo LM, Nalivaiko E, Anderson CR, McAllen RM. Control of cardiac rate, contractility, and atrioventricular conduction by medullary raphe neurons in anesthetized rats. Am J Physiol Heart Circ Physiol. 2009;296:H318–324. doi: 10.1152/ajpheart.00951.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JM, Rincon F, Fernandez A, Resor C, Kowalski RG, Claassen J, Connolly ES, Fitzsimmons BF, Mayer SA. Cerebral infarction associated with acute subarachnoid hemorrhage. Neurocrit Care. 2007;7:10–17. doi: 10.1007/s12028-007-0003-2. [DOI] [PubMed] [Google Scholar]
- Schubert GA, Thome C. Cerebral blood flow changes in acute subarachnoid hemorrhage. Front Biosci. 2008;13:1594–1603. doi: 10.2741/2783. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- Seylaz J, Hara H, Pinard E, Mraovitch S, MacKenzie ET, Edvinsson L. Effect of stimulation of the sphenopalatine ganglion on cortical blood flow in the rat. J Cereb Blood Flow Metab. 1988;8:875–878. doi: 10.1038/jcbfm.1988.145. [DOI] [PubMed] [Google Scholar]
- Shiokawa Y, Svendgaard NA. Cerebrovascular sensory innervation involved in the development of cerebral vasospasm following a subarachnoid hemorrhage. J Auton Nerv Syst. 1994;49(Suppl):S167–170. doi: 10.1016/0165-1838(94)90107-4. [DOI] [PubMed] [Google Scholar]
- Spencer SE, Sawyer WB, Wada H, Platt KB, Loewy AD. CNS projections to the pterygopalatine parasympathetic preganglionic neurons in the rat: a retrograde transneuronal viral cell body labeling study. Brain Res. 1990;534:149–169. doi: 10.1016/0006-8993(90)90125-u. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Platt KB, Loewy AD. CNS cell groups regulating the sympathetic outflow to adrenal gland as revealed by transneuronal cell body labeling with pseudorabies virus. Brain Res. 1989;491:274–296. doi: 10.1016/0006-8993(89)90063-2. [DOI] [PubMed] [Google Scholar]
- Talman WT, Corr J, Nitschke Dragon D, Wang D. Parasympathetic stimulation elicits cerebral vasodilatation in rat. Auton Neurosci. 2007;133:153–157. doi: 10.1016/j.autneu.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Horst GJ, Hautvast RW, De Jongste MJ, Korf J. Neuroanatomy of cardiac activity-regulating circuitry: a transneuronal retrograde viral labelling study in the rat. Eur J Neurosci. 1996;8:2029–2041. doi: 10.1111/j.1460-9568.1996.tb00723.x. [DOI] [PubMed] [Google Scholar]
- Titova E, Ostrowski RP, Zhang JH, Tang J. Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol Res. doi: 10.1179/174313209X382412. (in press) [DOI] [PubMed] [Google Scholar]
- Toda N, Tanaka T, Ayajiki K, Okamura T. Cerebral vasodilatation induced by stimulation of the pterygopalatine ganglion and greater petrosal nerve in anesthetized monkeys. Neuroscience. 2000;96:393–398. doi: 10.1016/s0306-4522(99)00557-6. [DOI] [PubMed] [Google Scholar]
- Treggiari MM, Romand JA, Martin JB, Reverdin A, Rufenacht DA, de Tribolet N. Cervical sympathetic block to reverse delayed ischemic neurological deficits after aneurysmal subarachnoid hemorrhage. Stroke. 2003;34:961–967. doi: 10.1161/01.STR.0000060893.72098.80. [DOI] [PubMed] [Google Scholar]
- Underwood MD, Iadecola C, Sved A, Reis DJ. Stimulation of C1 area neurons globally increases regional cerebral blood flow but not metabolism. J Cereb Blood Flow Metab. 1992;12:844–855. doi: 10.1038/jcbfm.1992.116. [DOI] [PubMed] [Google Scholar]
- Wang RK. Three-dimensional optical micro-angiography maps directional blood perfusion deep within microcirculation tissue beds in vivo. Phys Med Biol. 2007;52:N531–537. doi: 10.1088/0031-9155/52/23/N01. [DOI] [PubMed] [Google Scholar]
- Wang RK, Jacques S, Ma Z, Hurst S, Gruber A. Three-dimensional optical angiography. Optics Express. 2007;15:4083–4097. doi: 10.1364/oe.15.004083. [DOI] [PubMed] [Google Scholar]
- Wilkins RH. Attempts at treatment of intracranial arterial spasm in animals and human beings. Surg Neurol. 1973;1:148–159. [PubMed] [Google Scholar]
- Wise RA, Hoffman DC. Localization of drug reward mechanisms by intracranial injections. Synapse. 1992;10:247–263. doi: 10.1002/syn.890100307. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Narcotic analgestics: CNS sites and mechanisms of action as revealed by intracerebral injection techniques. Pain. 1978;4:299–359. doi: 10.1016/0304-3959(77)90145-2. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Nishizawa S, Tsukada H, Kakiuchi T, Yokoyama T, Ryu H, Uemura K. Cerebral blood flow autoregulation following subarachnoid hemorrhage in rats: chronic vasospasm shifts the upper and lower limits of the autoregulatory range toward higher blood pressures. Brain Res. 1998;782:194–201. doi: 10.1016/s0006-8993(97)01278-x. [DOI] [PubMed] [Google Scholar]
- Yarnitsky D, Lorian A, Shalev A, Zhang ZD, Takahashi M, Agbaje-Williams M, Macdonald RL. Reversal of cerebral vasospasm by sphenopalatine ganglion stimulation in a dog model of subarachnoid hemorrhage. Surg Neurol. 2005;64:5–11. doi: 10.1016/j.surneu.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Zagon A. Innervation of serotonergic medullary raphe neurons from cells of the rostral ventrolateral medulla in rats. Neuroscience. 1993;55:849–867. doi: 10.1016/0306-4522(93)90446-m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Dynamic OMAG blood flow imaging in vivo after microinjection of bicuculline into the RVM with a temporal time resolution of 1 minute. The changes in flow can readily be seen over the course of 30 minutes. (See video). (B) Measured changes of the diameter of the vessel indicated in (A) over 30 minutes after activation of the RVM. Scale bar = 500μm.