

Abstract
Transthyretin (TTR) familial amyloid polyneuropathies (FAP) are autosomal dominant devastating afflictions, first described in Portugal, later in Japan and Sweden, now recognized worldwide. The TTR-Val30Met mutation is the most common, and depending on the geographic origin, a wide variation in age at onset of the disease is observed. In Europe, northern Sweden is the second most important area of the disease, and a late age of onset of 56 years has been reported.
The present work aims to estimate the penetrance in TTR-Val30Met Swedish families. Genealogical investigations, clinical data and genotyping were obtained in 77 TTR-Val30Met Swedish families. The penetrance in Val30Met carriers and variation within the endemic area, according to gender and transmitting parents was calculated by a newly developed bias free method.
The penetrance estimates were low i.e. 1.7% and 22% at age 30 and 60 years, respectively and far from complete (69%), by age 90 years. Differences between Piteå and Skellefteå regions were observed. Moreover, penetrance was significantly higher when the mutation was inherited from the mother than from the father.
The low penetrance observed in TTR-FAP kindreds and its variations are important information for the genetic counselling and treatment of Swedish FAP- patients and their families.
Keywords: Adult; Age of Onset; Aged; Aged, 80 and over; Amyloid Neuropathies, Familial; epidemiology; genetics; European Continental Ancestry Group; Female; Genetics, Population; Humans; Male; Middle Aged; Mutation; genetics; Pedigree; Penetrance; Prealbumin; genetics; Sweden; epidemiology
Keywords: tranthyretin, amyloidosis, genetics, penetrance, Swedish families
Introduction
Transthyretin (TTR) amyloid neuropathy is the most severe neuropathy of adulthood with autosomal dominant transmission. The disease is characterised by extracellular deposition of amyloid made up of mutated TTR, of which the most common mutation is the ATTR Val30Met. Its main clinical expression is a progressive sensory-motor neuropathy with autonomic manifestations frequently associated to cardiac dysfunction. Other manifestations include, ocular symptoms related to vitreous amyloidosis and eventually renal failure. A fatal outcome occurs after an average duration of 10 – 13 years[1, 2]. By removing the main source of mutated TTR, liver transplantation is the only therapeutic approach available, which can stabilize the disease when performed early in the course[3, 4].
Initially reported by Andrade (1952) in northern Portugal[5] in the area of Povoa de Varzim where the prevalence is 1/1000, the condition was subsequently recognized throughout the world including various European countries and two different foci in Japan [6, 7]. Apart from Portugal, the most important focus of the disease in Europe takes place in the north of Sweden where families are mainly clustered in limited areas around the towns of Skellefteå and Piteå[8].
Among 100 point mutations identified in the TTR gene, the Val30Met is by far the most common pathogenic variant and virtually the only one described in Portugal and Sweden[9]. Genotype phenotype correlations are unclear. Moreover, phenotypic variations and striking differences in the age of onset are observed between populations of Val30Met patients. Hence, the mean age of first symptoms is 33 years in Portugal and in the main endemic areas in Japan, [7,10] whereas in Sweden, the onset is rather late, at 56 years of age[11]. Screenings have not disclosed any protective variant of the TTR gene that could explain the higher age of onset in Sweden.
In Sweden, a population study had suggested that disease risk in carrier individuals could be rather low[11]. The age of onset is variable among pedigrees and is significantly higher with paternal inheritance compared with that of maternal inheritance[12]. This finding needs to be confirmed by estimating the penetrance (cumulative risk of being affected by a given age) in mutation carriers, i.e. the risk that a carrier individual be affected given (s)he has reached a given age.
Using a method to estimate penetrance that corrects for ascertainment bias toward affected individuals, significant differences in Portuguese and French families were noted[13].
The aim of the study was to assess the penetrance of Swedish TTR-Val30Met, using a bias-free method and to disclose differences in penetrance within the endemic area. In addition, to estimate the impact of transmitting parents’ gender on age at onset of the disease.
Patients and Methods
Patients
Among the 401 patients treated at the Department of Clinical Genetics Umeå University Hospital, since 1986 and identified with a Val30Met mutation, a complete genealogical investigation was available for 122 families. Since some families were related with each other pedigrees were merged, thus, ending with 85 pedigrees that could enter the analysis. Eight pedigrees contained 9 homozygous patients for the Val30Met mutation, and were excluded from the analysis as these patients were too few to allow a reliable estimation of risk associated with this genotype. Finally, 77 pedigrees were available for penetrance analysis.
In patients, the diagnosis of TTR amyloid neuropathy was based on the finding of amyloid deposits in biopsy specimens, however the deposits were not always immunohistochemically diagnoses as TTR-amyloid. The pathogenic TTR variant Val30Met mutation were identified by molecular genetic testing in all cases.
Genealogical investigation was carried out for all patients when possible since 1999 and data were entered using Cyrillic software (version 2.0).
A detailed history of the disease was taken from the patient and/or from medical records. The date of first symptoms was determined as accurately as possible.
For the present study, a total of 1353 subjects were recorded and clinical information was obtained on 235 affected individuals, including the probands. Overall, DNA analysis was performed in 215 individuals of the families. Among them 49 were asymptomatic carriers and 19 were non carriers. In addition, 35 asymptomatic individuals were found to be obligate carriers.
Genetic analysis
Over time different methods have been used to detect the TTR-Val30Met mutation. At first Southern blot were used to analyse the patients. Later they were diagnosed by polymerase chain reaction (PCR) based methods as previously described[11, 14].
Estimation of penetrance
a. Statistical method
We used the same method as Planté-Bordeneuve et al.[15] which allows a correction for ascertainment bias when using family data. The method is based on the maximum likelihood principle, using a survival analysis approach. Beside the correction for ascertainment, its main interest is to use phenotypic information on individuals whose genotype is unknown but whose probability of being a carrier is not negligible.
Briefly, the age-dependent penetrance function for mutation carriers was modelled by a Weibull distribution, defined by three parameters: λ (scale parameter), a (shape parameter) and κ, the fraction of individuals that would never be affected[16]. The penetrance function may be written:
Following survival analysis principles, the contribution of pedigree members to the likelihood depends on respectively the age at diagnosis for affected and the age at last news (or at death) for unaffected relatives.
The probability of unknown genotypes of untested individuals given observed genotypes in the families was computed from the frequency of the mutated allele in the general population assuming Hardy-Weinberg equilibrium for a founder (parents’ status unknown). Otherwise, this probability depends on parental genotypes assuming Mendelian transmission. In this analysis, disease allele frequency was estimated using the proportion of homozygotes among all affected individuals in the population, as explained in the appendix.
For sake of simplicity, the probability of being homozygous was neglected for the reasons given above.
To test for a difference according to gender, geographic origin or to the sex of the transmitting parent, we used a likelihood ratio test.
b. Family data used
All family data providing information on the risk of being affected for a relative were retained. Thus, at risk individuals or patients with reliable information on their date of birth, their age of onset or their date or age of death, if adapted, were included. In a minority of affected cases, the age of onset was extrapolated from the age of death, using the mean duration of the disease of 13 years in the Swedish population[2]. The branches of the families without information on the individual’s phenotype or date of birth or on the age at first manifestations were excluded.
On the basis of the known age of onset in previous reports of TTR amyloid neuropathy in Sweden, individuals younger than 18 years were not considered in the present study[8, 11, 12].
Ethics
The project has been approved by the Ethic Committee at Umea University
Results
Using the proportion of homozygotes for the Val30Met mutation among affected patients, the frequency of this mutation was estimated to be 0.04 (9/401) in the population and was fixed at this value in all subsequent analyses.
Figure 1 shows the penetrance curves for the whole sample and for males and females separately. Gender effect was not found significant (χ2 = 2.44, 2 df). In all the analyses performed on the different samples, there was no evidence for a plateau: the parameter κ converged to its bound 0, so that only the two parameters a and λ, were estimated. The risks and confidence intervals from age 30 to 90 years are shown in table 1. At age 30 and 60 years, the penetrance estimates were 1.7% and 22%, respectively. Even by age 90 years, penetrance was found to be far from complete (69%).
Figure 1.
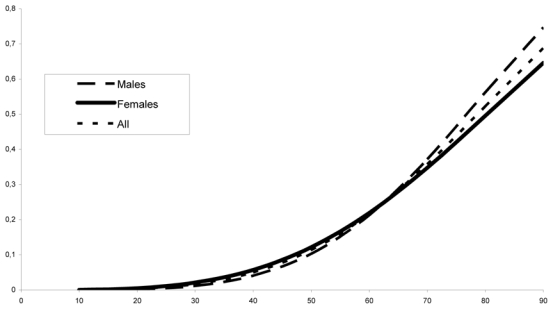
Estimated penetrance curve according to gender
Table 1.
Estimation of penetrance in the Swedish families
| Age (years) | Penetrance estimate | 95% confidence interval |
|---|---|---|
| 30 | 0.017 | 0.008–0.032 |
| 40 | 0.05 | 0.029–0.082 |
| 50 | 0.11 | 0.08–0.16 |
| 60 | 0.22 | 0.16–0.29 |
| 70 | 0.36 | 0.28–0.45 |
| 80 | 0.52 | 0.42–0.63 |
| 90 | 0.69 | 0.55–0.79 |
When subdividing the sample according to the three regions, the Lycksele was too small to allow a valid estimate (10 families), thus, only Piteå and Skellefteå were compared. As shown in figure 2, the penetrance was significantly lower for the Piteå region than for the Skellefteå region, with penetrance by age 80 of 32% and 65% respectively. The homogeneity test was highly significant (χ2 = 26.60, 2 df, p < 0.001).
Figure 2.
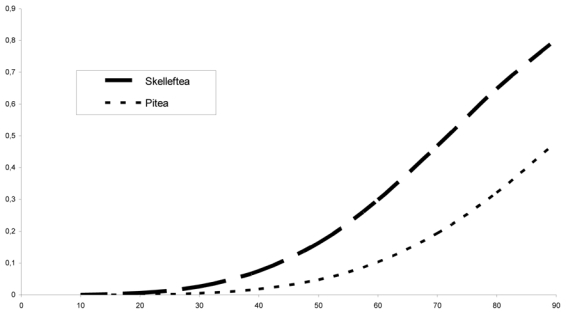
Estimated penetrance curve according to region
A clear difference according to the sex of the transmitting parent was found (figure 3). Among the 77 families, the gender of the transmitting parent was known for 762 individuals: for 435 the transmitting parent was the mother, and for 327 the transmitting parent was the father. The penetrance was found to be significantly higher when the mutation was inherited from the mother than from the father (χ2 = 7.84, 2 df, p < 0.02). Table 2 shows the penetrance estimates and confidence intervals according to the sex of the transmitting parent.
Figure 3.
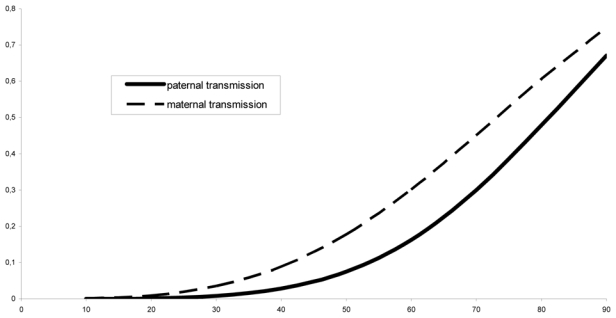
Estimated penetrance curve according to sex of the transmitting parent
Table 2.
penetrance estimates according to the sex of the transmitting parent
| Age (years) | Penetrance (95% confidence interval), when transmission by | |
|---|---|---|
| Father | Mother | |
| 30 | 0.008 (0.003–0.016) | 0.035 (0.013–0.059) |
| 40 | 0.028 (0.016–0.041) | 0.089 (0.043–0.14) |
| 50 | 0.075 (0.052–0.11) | 0.18 (0.10–0.27) |
| 60 | 0.16 (0.13–0.25) | 0.30 (0.21–0.44) |
| 70 | 0.30 (0.21–0.44) | 0.45 (0.34–0.61) |
| 80 | 0.48 (0.30–0.66) | 0.61 (0.47–0.76) |
| 90 | 0.67 (0.40–0.85) | 0.75 (0.57–0.89) |
Discussion
ATTR Val30Met mutation is by far the most frequent neuropathic form worldwide, and virtually the only one identified in endemic areas in Portugal, Japan, Sweden and Brazil. However, important differences in the disease expression, particularly regarding the age of onset and the penetrance seem to exist between countries. Significant differences between Portuguese and French populations have been noted[13], thus, cumulative disease risk in French carriers of the Val30Met mutation was estimated at 14% by age 50 and 50% by age 70 years, whereas the risk in Portuguese carriers was 80% by age 50 and 91% by age 70 years. Swedish carriers seem to be closer to French than Portuguese carriers with an even lower risk of 11 % by age 50 and 36% by age 70 years. The penetrance increases with age and does not display any plateau, a similar finding was noted in the French population, whereas a plateau was noted for the Portuguese. These results are consistent with the late age at onset averaging 56 years, previously reported in the Swedish population[11]. The reasons for the increase of penetrance with age can only be speculated on. The aging process involves a decreased ability to handle oxidative stress and also increases in the deposition of advanced glycation end products, of which both factors have been implicated in amyloid formation[17, 18].
The patients were all recruited from the Department of Medicine, Umeå University Hospital. This is a referral centre for FAP in Sweden. However, patients with onset at high age are generally not submitted for evaluation, especially not from hospitals with long experience in the disease (such as Skellefteå Hospital). It is therefore clear, that the patient material has a bias against younger patients, and to some degree patients with rapidly progressing disease. Moreover, marked differences in penetrance of the trait were noted within the endemic area. Such differences might be due to genetic factors (other than the TTR mutation) but also to environmental factors. In particular, it is noteworthy that Skellefteå is an area with different industries than those of Piteå and Lycksele. In Skellefteå, a large metallurgic industry and previously also textile manufactures are located and the later turned out to be a risk factor for developing the disease in a study of occupation and disease in northern Sweden[19]. Discordant penetrance of FAP in two pairs of monozygotic twins have been reported[20]. These observations along with the observed variation of penetrance could support the role of environmental factors to explain differences in expression of the mutation.
The other remarkable finding is the marked differences of penetrance according to the gender of the transmitting parent. Indeed, the penetrance of the trait was significantly higher when inherited from the mother than from the father. These data are in line with a previous genealogical analysis of Swedish pedigrees underlying that the age of onset was significantly higher when the mutation was inherited from the father and that anticipation (higher penetrance in younger generations) was more marked in descendants of affected mothers[12]. Differences of age of onset according to the gender of the transmitting parent have also been mentioned in Japanese and Portuguese families but such variation of penetrance has not been precisely analysed in other populations[10, 21]. It is tempting to link these differences to a parental imprinting phenomenon. In this setting, the occurrence of regions containing differential allele specific DMA methylation localised near the TTR gene might be interesting to explore. However, there is no evidence of other imprinted gene in the 18q11.2-12 region, so far (www.geneimprint.com). Another clue could be a possible role of the mitochondrial genome interacting with the expression or the pathogenesis of the TTR-Val30Met mutation.
The penetrance estimates are higher than previously reported. However, the previously reported frequency of symptomatic carriers at 2 % was estimated from a study of healthy volunteers from the endemic area and the known number of affected individuals[11]. The difference between the penetrance estimates cannot be due to an ascertainment bias, since our method takes into account that families without any affected individuals will not be ascertained. Neither can it be attributed to the high gene frequency in the Swedish population, comparatively with other populations such as the Portuguese,[10] as the method has been shown to be robust to a variation in this parameter[13]. Differences in reported prevalence could in part be due to an underestimation of affected individuals as probably is the case in the previous Swedish study, since it did not adjust for age. Indeed, the affected individuals were mostly older than 60 years, whereas those in the reference population were much younger.
In summary, different values of penetrance according to the geographic origin of the families within the endemic area were noted and substantiated the occurrence of anticipation related to female gender of the transmitting parent. The rather low penetrance observed in Swedish TTR-FAP kindreds and its variations are important information for the genetic counselling and treatment of Swedish FAP-patients and their families.
Abbreviations
- FAP
familial amyloidotic polyneuropathy
- TTR
transthyretin
Appendix
Computation of allele frequency from the proportion of homozygous individuals in a dominantly inherited disease
Let us consider a disease where the deleterious allele A is strictly dominant on the normal allele a (affected individuals may be either homozygous AA or heterozygous Aa).
Let q be the frequency of the A allele and P the proportion of homozygotes among all affected individuals in the population.
Assuming that Hardy-Weinberg proportions are valid, the theoretical frequency of homozygotes and heterozygotes in the population are respectively q2 and 2q(1−q).
Thus,
And finally,
References
- 1.Plante-Bordeneuve V, Said G. Transthyretin related familial amyloid polyneuropathy. Curr Opin Neurol. 2000 Oct;13(5):569–573. doi: 10.1097/00019052-200010000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235(5):479–485. doi: 10.1111/j.1365-2796.1994.tb01106.x. [DOI] [PubMed] [Google Scholar]
- 3.Suhr OB, Holmgren G, Steen L, Wikstrom L, Norden G, Friman S, Duraj FF, Groth CG, Ericzon BG. Liver transplantation in familial amyloidotic polyneuropathy. Follow-up of the first 20 Swedish patients. Transplantation. 1995;60(9):933–938. [PubMed] [Google Scholar]
- 4.Adams D, Samuel D, Goulon-Goeau C, Nakazato M, Costa PM, Feray C, Plante V, Ducot B, Ichai P, Lacroix C, Metral S, Bismuth H, Said G. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000 Jul;123 (Pt 7):1495–1504. doi: 10.1093/brain/123.7.1495. [DOI] [PubMed] [Google Scholar]
- 5.Andrade C. A peculiar form of peripheral neuropathy. Familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 6.Reilly MM, Adams D, Booth DR, Davis MB, Said G, Laubriat-Bianchin M, Pepys MB, Thomas PK, Harding AE. Transthyretin gene analysis in European patients with suspected familial amyloid polyneuropathy. Brain. 1995;118(Pt4):849–856. doi: 10.1093/brain/118.4.849. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002 Apr 9;58(7):1001–1007. doi: 10.1212/wnl.58.7.1001. [DOI] [PubMed] [Google Scholar]
- 8.Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O. Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered. 1993;43(5):288–294. doi: 10.1159/000154146. [DOI] [PubMed] [Google Scholar]
- 9.Saraiva MJ. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17(6):493–503. doi: 10.1002/humu.1132. [DOI] [PubMed] [Google Scholar]
- 10.Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Povoa do Varzim and Vila do Conde (north of Portugal) Am J Med Genet. 1995;60(6):512–521. doi: 10.1002/ajmg.1320600606. [DOI] [PubMed] [Google Scholar]
- 11.Holmgren G, Costa PM, Andersson C, Asplund K, Steen L, Beckman L, Nylander PO, Teixeira A, Saraiva MJ, Costa PP. Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet. 1994 May;31(5):351–354. doi: 10.1136/jmg.31.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drugge U, Andersson R, Chizari F, Danielsson M, Holmgren G, Sandgren O, Sousa A. Familial amyloidotic polyneuropathy in Sweden: a pedigree analysis. J Med Genet. 1993;30(5):388–392. doi: 10.1136/jmg.30.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plante-Bordeneuve V, Carayol J, Ferreira A, Adams D, Clerget-Darpoux F, Misrahi M, Said G, Bonaiti-Pellie C. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003 Nov;40(11):e120. doi: 10.1136/jmg.40.11.e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmgren G, Bergstrom S, Drugge U, Lundgren E, Nording-Sikstrom C, Sandgren O, Steen L. Homozygosity for the transthyretin-Met30-gene in seven individuals with familial amyloidosis with polyneuropathy detected by restriction enzyme analysis of amplified genomic DNA sequences. Clin Genet. 1992;41(1):39–41. doi: 10.1111/j.1399-0004.1992.tb03627.x. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs U, Zittermann A, Suhr O, Holmgren G, Tenderich G, Minami1 K, Koerfer R. Heart transplantation in a patient with senile cardiomyopathy. Am J Transplant. 2005;5:1159–1162. doi: 10.1111/j.1600-6143.2005.00805.x. [DOI] [PubMed] [Google Scholar]
- 16.Sposto R. Cure model analysis in cancer: an application to data from the Children’s Cancer Group. Stat Med. 2002 Jan 30;21(2):293–312. doi: 10.1002/sim.987. [DOI] [PubMed] [Google Scholar]
- 17.Ando Y, Nyhlin N, Suhr O, Holmgren G, Uchida K, el Sahly M, Yamashita T, Terasaki H, Nakamura M, Uchino M, Ando M. Oxidative stress is found in amyloid deposits in systemic amyloidosis. Biochem Biophys Res Commun. 1997;232(2):497–502. doi: 10.1006/bbrc.1996.5997. [DOI] [PubMed] [Google Scholar]
- 18.Nyhlin N, Ando Y, Nagai R, Suhr O, El Sahly M, Terazaki H, Yamashita T, Ando M, Horiuchi S. Advanced glycation end product in familial amyloidotic polyneuropathy (FAP) J Intern Med. 2000;247(4):485–492. doi: 10.1046/j.1365-2796.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 19.Hardell L, Holmgren G, Steen L, Fredrikson M, Axelson O. Occupational and other risk factors for clinically overt familial amyloid polyneuropathy. Epidemiology. 1995;6(6):598–601. doi: 10.1097/00001648-199511000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Holmgren G, Wikstrom L, Lundgren HE, Suhr OB. Discordant penetrance of the trait for familial amyloidotic polyneuropathy in two pairs of monozygotic twins. J Intern Med. 2004 Nov;256(5):453–456. doi: 10.1111/j.1365-2796.2004.01399.x. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto K, Ikeda S, Hanyu N, Takeda S, Yanagisawa N. A pedigree analysis with minimised ascertainment bias shows anticipation in Met30-transthyretin related familial amyloid polyneuropathy. J Med Genet. 1998;35(1):23–30. doi: 10.1136/jmg.35.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]