

Abstract
Previous studies have demonstrated that the JC virus (JCV) late regulatory protein agnoprotein is phosphorylated by the serine/threonine-specific protein kinase-C (PKC) and mutants of this protein at the PKC phosphorylation sites exhibit defects in the viral replication cycle. We have now investigated whether agnoprotein phosphorylation is regulated by PP2A, a serine/threonine-specific protein phosphatase and whether JCV small t antigen (Sm t-Ag) is involved in this regulation. Protein–protein interaction studies demonstrated that PP2A associates with agnoprotein and dephosphorylates it at PKC-specific sites. Sm t-Ag was also found to interact with PP2A and this interaction inhibited the dephosphorylation of agnoprotein by PP2A. The interaction domains of Sm t-Ag and agnoprotein with PP2A were mapped, as were the interaction domains of Sm t-Ag with agnoprotein. The middle portion of Sm t-Ag (aa 82–124) was found to be critical for the interaction with both agnoprotein and PP2A and the N-terminal region of agnoprotein for interaction with Sm t-Ag. To further understand the role of Sm t-Ag in JCV regulation, a stop codon was introduced at Ser90 immediately after splice donor site of the JCV early gene and the functional consequences of this mutation were investigated. The ability of this mutant virus to replicate was substantially reduced compared to WT. Next, the functional significance of PP2A in JCV replication was examined by siRNA targeting. Downregulation of PP2A caused a significant reduction in the level of JCV replication. Moreover, the impact of Sm t-Ag on agnoprotein phosphorylation was investigated by creating a double mutant of JCV, where Sm t-Ag stop codon mutant was combined with an agnoprotein triple phosphorylation mutant (Ser7, Ser11 and Thr21 to Ala). Results showed that double mutant behaves much like the triple phosphorylation mutant of agnoprotein during viral replication cycle, which suggests that agnoprotein might be an important target of Sm t-Ag with respect to the regulation of its phosphorylation. Collectively, these results suggest that there is an interplay between agnoprotein, Sm t-Ag and PP2A with respect to the regulation of JCV life cycle and this could be important for the progression of the JCV-induced disease, PML.
Keywords: JC virus, Agnoprotein, Dephosphorylation, Gene expression, Replication, Small t antigen, PP2A
Introduction
One of the interesting characteristics of viruses is to use host machinery for their own purpose in that they modify the host system in such a way as to optimize viral replication. Reciprocally, some viral regulatory proteins are modified by the host system and thereby function differently at different stages of the infection cycle. One of the prime examples of polyomavirus-mediated alteration of the host system is the inactivation of the cellular protein phosphatase 2A (PP2A) by SV40 Sm t-Ag (Sontag et al., 1993). PP2A targets a variety of viral (Prives, 1990) and cellular proteins (Arroyo and Hahn, 2005; Garcia et al., 2000; Howe et al., 1998; Lin et al., 2006; Sontag et al., 1997; Ugi et al., 2002); and plays critical roles in their regulation. PP2A is an abundant serine/threonine-specific protein phosphatase in mammalian cells and is critical for many biological processes, including development, differentiation and growth control (Janssens and Goris, 2001). This enzyme exists as a complex composed of a core and a variable unit. The core enzyme consists of a catalytic C subunit (∼36 kDa) and a regulatory A subunit (∼65 kDa). The variable unit exists as different isoforms but generally fall into three families designated B, B′ and B″ (Kremmer et al., 1997). This large number of variable subunits allows the formation of numerous, but functionally distinct, substrate-specific isoforms of PP2A. SV40 Sm t-Ag has been reported to replace the B subunit in the PP2A complex and may thereby modify the function of the enzyme complex (Hahn et al., 2002).
JCV is a small human polyomavirus and is the etiological agent of a fatal demyelinating disease, progressive multifocal leukoencephalopathy (PML) (Berger and Concha, 1995; Raj and Khalili, 1995; Safak et al., 2005). It shows a high degree of sequence homology to the human BK virus (BKV) and the rhesus macaque simian virus-40 (SV40) polyomaviruses in coding regions (∼70%), however, the non-coding regulatory region of each virus is highly divergent between these viruses. This divergence of the non-coding region plays a critical role in the unique pattern of gene expression of each virus in different cell types and tissues (Frisque and White, 1992; Major and Ault, 1995).
The late coding region of JCV encodes a small and highly basic regulatory protein, agnoprotein, whose functions in the JCV infection cycle are beginning to be investigated. Recent findings suggest that it may play an important role in the JCV replication cycle (Sariyer et al., 2006). In this regard, analysis of phosphorylation site mutants, where Ser7, Ser11 and Thr21 were mutated into Ala, showed that mutant viruses were unable to continue the viral replication cycle (Sariyer et al., 2006). In addition, we have previously demonstrated that agnoprotein associates with JCV large T antigen (LT-Ag) and the cellular transcription factor YB-1 and negatively affect their role in JCV transcription (Safak et al., 2001, 2002). We have also analyzed the effect of agnoprotein on cell cycle progression and found that cells stably expressing agnoprotein largely accumulate at the G2/M phase of the cell cycle (Darbinyan et al., 2002). Moreover, it was recently reported that agnoprotein interacts with the cellular proteins FEZ1 and HP1α and may promote the transport of virions from the nucleus to the cytoplasm (Suzuki et al., 2005) and thereby facilitates the propagation of JCV (Okada et al., 2005).
The JCV early coding region encodes the regulatory proteins, LT-Ag and Sm t-Ag (Frisque et al., 1984) and several spliced variants of the early proteins, called T's (Frisque, 2001; Frisque et al., 2003). LT-Ag is known to play critical roles in both viral DNA replication (Lynch and Frisque, 1990, 1991; Lynch et al., 1994; Tavis and Frisque, 1991) and viral gene transcription (Khalili et al., 1987; Lashgari et al., 1989; Tyagarajan and Frisque, 2006). This protein was also shown to have an oncogenic potential and its expression can lead to the induction of tumors of neuronal origin in experimental animals (Krynska et al., 1999; Small et al., 1986; Varakis et al., 1978). T′ proteins were recently found to differentially interact with the retinoblastoma family of tumor suppressor proteins (Frisque et al., 2006; Trowbridge and Frisque, 1995; Tyagarajan and Frisque, 2006). Although the function of JCV Sm t-Ag in JCV regulation and cellular transformation is unknown, functional studies with SV40 Sm t-Ag showed an important role in permissive and nonpermissive infections. Sm t-Ag enhances the ability of SV40 LT-Ag to transform cells (Chen and Hahn, 2003; Gaillard et al., 2001; Nunbhakdi-Craig et al., 2003; Porras et al., 1996, 1999; Zhao et al., 2003). It is also believed that Sm t-Ag antagonizes LT-Ag-induced cellular apoptosis and thereby contributes to more efficient transformation of rat embryo fibroblasts (Kolzau et al., 1999). In addition, transgenic animals created with a Sm t-Ag deletion mutant of SV40 genome developed tumors almost exclusively in highly mitotic tissues such as lymphoid organs (Carbone et al., 1989; Choi et al., 1988) suggesting that Sm t-Ag may play a significant role in tumor induction in nonproliferative tissues. The tumorigenic potential of Sm t-Ag was linked to the inhibition of PP2A by this protein (Ali and DeCaprio, 2001; Rundell and Parakati, 2001; Sontag et al., 1993). Recent reports also indicate that Sm t-Ag may function as a transfer factor to specify PP2A targeting in cells and thereby modulate the transcriptional activity of cellular genes including the androgen receptor (Yang et al., 2005).
In this communication, we have investigated the regulatory role of PP2A on agnoprotein dephosphorylation as well as the effect of Sm t-Ag on this process. We provide evidence that agnoprotein is targeted by PP2A for dephosphorylation and this process can be inhibited by Sm t-Ag, suggesting the presence of interplay between these three proteins.
Results
Agnoprotein coimmunoprecipitates with PP2A subunits
We have recently demonstrated that JCV agnoprotein is phosphorylated by PKC and that phosphorylation site mutants are unable to continue the viral replication cycle (Sariyer et al., 2006). Since most phosphorylated proteins are subjected to regulation by phosphatases (Arroyo and Hahn, 2005; Boutros et al., 2007; Gallego et al., 2006; Gallego and Virshup, 2005, 2007; Jakubiec and Jupin, 2007; Jakubiec et al., 2006; Miller et al., 1982; Wang and Liu, 2007) we sought to investigate whether the phosphatase, PP2A (Heriche et al., 1997; Zhou et al., 2003) targets JCV agnoprotein. To test this possibility, SVG-A cells were transfected/infected with JCV Mad-1 strain and whole cell extracts were prepared. Extracts were subsequently examined for the presence of an agnoprotein:PP2A complex by immunoprecipitation followed by Western blotting. As shown in Fig. 1A, the immunocomplexes obtained upon incubation of the protein extracts with α-PP2A C antibody contained agnoprotein (lane 6), while those immunoprecipitated with pre-immune sera (control) did not (lane 4), which indicates the specificity of the coimmunoprecipitation. In parallel, we also investigated whether agnoprotein coprecipitates with both A and B subunits of PP2A (Fig. 1B). Results showed that in fact it does but the level of band intensity for agnoprotein resulting from coprecipitation with both anti-A and anti-B antibodies of PP2A is not as strong as for that of C subunit case (compare lane 4 to 5 and 6 respectively).
Fig. 1.
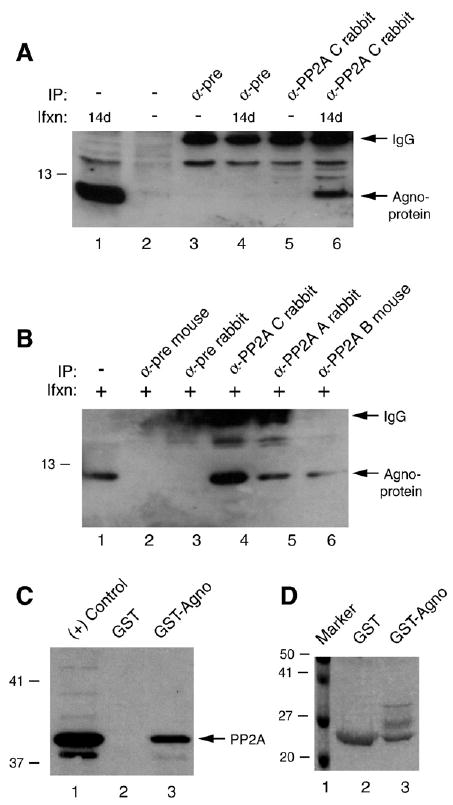
Agnoprotein interacts with PP2A. (A) Coimmunoprecipitation of agnoprotein with PP2A C subunit. Whole cell extracts (500 μg) were prepared from SVG-A cells [uninfected (−) or infected with JCV Mad-1] at the indicated days (d) post infection, subjected to immunoprecipitation (IP) using α-pre or α-PP2A C antibody (2 μg) as indicated. Immunocomplexes were then analyzed by Western blotting using α-agnoprotein antibody as described in Materials and methods. In lane 1, whole cell extract from SVG-A cells (20 μg) was loaded as a positive control for agnoprotein. IgG-labeled arrow points the light chain of the antibodies that were used for IP and detected by the secondary antibody. Ifxn denotes infection. (B) In parallel to the immunoprecipitation studies described in panel B, protein complexes were also precipitated (IP) with antibodies that are directed against A (α-PP2A A rabbit polyclonal, B (α-PP2A B mouse monoclonal) and C (α-PP2A C rabbit polyclonal) subunits of PP2A or either with normal mouse serum (α-pre M) or normal rabbit serum (α-pre R) as indicated using whole cell extracts from SVG-A cells prepared at 14 days post infection. Immunocomplexes were then analyzed by Western blotting using an anti-agno rabbit antibody. (C) GST pulldown assay. Whole cell extracts prepared from SVG-A cells were incubated either with GST or GST–Agnoprotein (Agno) (2 μg each), immobilized on GST–Sepharose beads. Columns were extensively washed with binding buffer and retained proteins were analyzed by Western blotting using α-PP2A C antibody as described in Materials and methods. In lane 1, whole cell extract from SVG-A cells (20 μg) was loaded as a positive control. (D) SDS-PAGE analysis of GST and GST–agnoprotein followed by coomassie staining.
To further investigate the interaction between PP2A and agnoprotein, in vitro GST pull-down experiments were performed. Whole cell extracts were prepared from glial cells (U-87MG), which endogenously express a high level of PP2A, and incubated with bacterially produced GST and GST–Agno fusion proteins immobilized on glutathione–Sepharose beads. After washing, proteins retained on the columns were analyzed by immunoblotting using an antibody raised against agnoprotein. As shown in Fig. 1C, incubation of whole cell extracts with Sepharose columns containing GST or GST–Agno resulted in the specific retention of PP2A on a column that contains GST–Agno (lane 3) but not on a column that contains GST alone (lane 2). Fig. 1D illustrates the Coomassie blue-stained SDS-PAGE gel of the full-length agnoprotein fused to GST, which was used in this study. Of note, GST–agnoprotein is not produced as a single band in bacteria as previously reported (Safak et al., 2001). Altogether, results from in vivo and in vitro protein–protein interaction studies strongly suggest that agnoprotein and PP2A interact and form heterocomplexes.
Eighteen to thirty-six amino acid region of agnoprotein interacts with PP2A
In the next series of experiments, we mapped the region of agnoprotein which interacts with PP2A. A series of bacterially expressed agnoprotein deletion mutants fused to GST were incubated with protein extracts from glial cells (U-87MG) as described in Materials and methods. After washing the columns, protein complexes were resolved by SDS-PAGE and analyzed by Western blotting using an anti-PP2A C antibody. As shown in Fig. 2A, removal of the regions of agnoprotein positioned between residues 1 and 36 (lane 7); and 1 and 54 (lane 8) completely inhibited association of agnoprotein with PP2A. We observed a reduced binding activity of a clone, which retains the residues 1–54 (lane 4) compared to WT (lane 3). In addition, a deletion mutant that retained the residues 1–36 showed a comparably high level of binding activity to PP2A (lane 5). Furthermore, two deletion mutants that contained residues 18 to 36 (lane 6 and 9) showed slightly higher binding efficiency compared to WT (lane 3), which indicates that the residues located between amino acid 18 to 36 of agnoprotein are critical for binding to PP2A. Fig. 2B illustrates the Coomassie blue-stained SDS-PAGE gel analysis of the full-length and mutant agnoproteins, which were used in the GST pull-down assay. A schematic of the GST pull-down assay depicting the regions of agnoprotein, which bind to PP2A is presented in Fig. 2C.
Fig. 2.
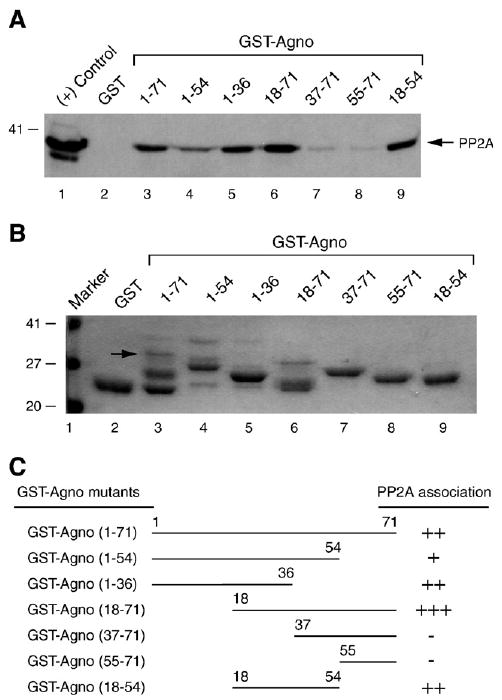
Eighteen to thirty-six amino acid region of agnoprotein is critical for interaction with PP2A. (A) Bacterially produced GST or GST–agno fusion protein or agnoprotein deletion mutants that were fused to GST were immobilized on GST–Sepharose beads (2 μg each) and incubated with whole cell extracts (300 μg) prepared from SVG-A cells overnight at 4 ° by racking. Unbound proteins were washed and bound proteins were analyzed by Western blotting using α-PP2A C antibody. In lane 1, whole cell extract was directly loaded on the gel as a positive control (20 μg). (B) SDS-PAGE analysis of GST, GST–Agno full-length and GST–Agno deletion mutants. Position of the GST–Agno full length was indicated by an arrow. (C) Summary of the results obtained from in vitro mapping assays. The relative binding activity of agnoprotein and its deletion mutants is represented by + or − signs. +++, very strong binding; ++, strong binding; +, binding; and −, no binding.
Agnoprotein is dephosphorylated by PP2A
Above, we showed that PP2A targets JCV agnoprotein. In addition, our recent studies demonstrated that agnoprotein is a substrate for PKC (Sariyer et al., 2006). Since PP2A is known to be a serine/threonine phosphatase, we next determined whether PP2A could dephosphorylate PKC-phosphorylated agnoprotein. Agnoprotein was then produced as a fusion protein (GST–Agno) in bacteria, purified to its homogeneity and phosphorylated by PKC (Sariyer et al., 2006). Dephosphorylation assay was carried out using PP2A enzyme obtained from two different sources, commercially available and immunoprecipitated one. However, before addressing this question, we first determined the activity of commercially available pure PP2A towards its substrate. As shown in Fig. 3A, pure PP2A enzyme is active and successfully dephospharylates its substrate (compare lane 1 to lane 2). We then carried out PP2A-mediated dephosphorylation of agnoprotein using either this commercially available pure enzyme or using the one that is immunoprecipitated from whole cell extracts, prepared from U-87MG cells. As demonstrated in Fig. 3B, commercially obtained pure PP2A successfully dephosphorylates agnoprotein in a time course study (compare lane 1 to lanes 2–6) so does an immunoprecipitated PP2A (Fig. 3C, lanes 2 and 3). However, the complexes that were coimmunoprecipitated with pre-immune serum did not have a significant effect on the level of agnoprotein dephosphorylation (lanes 4 and 5), suggesting that dephosphorylation of agnoprotein by PP2A is specific. In order to further demonstrate the specificity of the PP2A-mediated dephosphorylation of agnoprotein, a PP2A-specific inhibitor, okadaic acid, was included in the reaction. In the presence of this inhibitor, we observed a complete inhibition of the PP2A-mediated dephosphorylation of agnoprotein (lane 6). Taken together, these studies demonstrate that PP2A specifically dephosphorylates PKC-phosphorylated agnoprotein.
Fig. 3.
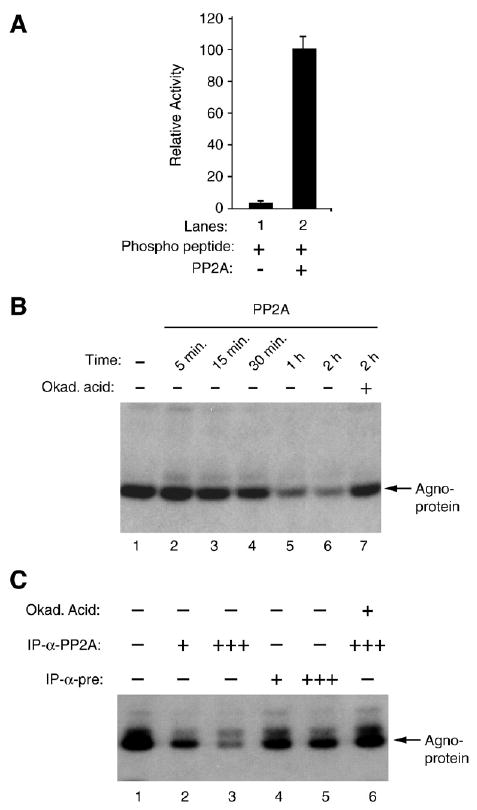
PP2A dephosphorylates agnoprotein. (A) Enzymatic activity of purified PP2A on authentic substrate was measured according to manufacturer's recommendations (Upstate) and expressed as relative activity compared to without enzyme in the reaction. (B) Time course activity of PP2A on phosphorylated agnoprotein. PKC-phosphorylated GST–agnoprotein was equally distributed into reaction tubes, subjected to dephosphorylation by purified PP2A at different time points and analyzed by autoradiography. Okadaic acid was used as a specific inhibitor of PP2A in the reaction (lane 7). (C) PKC-phosphorylated GST–agnoprotein was also subjected to dephosphorylation by immunoprecipitated-PP2A and analyzed by autoradiography as described in Materials and methods. PP2A was immunoprecipitated (IP) from whole cell extracts prepared from SVG-A cells (500 μg) using either α-pre (2 μg) or α-PP2A C antibody (2 μg) prior to dephosphorylation reaction. Immunoprecipitated PP2A was split into 4 equal portions. One (+) or three (+++) portions of immunoprecipitated PP2A were used in the reactions as indicated. In lane 6, okadaic acid (Okad. Acid) was used to inhibit the dephosphorylation reaction.
Sm t-Ag associates with PP2A
JCV Sm t-Ag shows about 70% sequence identity at the amino acid level to its counterpart SV40 Sm t-Ag. SV40 Sm t-Ag was previously shown to inhibit the regulatory function of PP2A (Sontag et al., 1993). To determine whether JCV Sm t-Ag might also interact with cellular PP2A, we performed in vitro GST pull-down experiments. Bacterially expressed GST or full-length Sm t-Ag fused to GST was immobilized on glutathione–Sepharose beads and incubated with whole cell extracts prepared from U-87MG cells. After washing the beads with incubation buffer, bound proteins were resolved by SDS-10% PAGE and analyzed by Western blotting using an anti-PP2A C antibody. As demonstrated in Fig. 4A, PP2A was retained on the Sepharose column containing GST–Sm t-Ag full length (lane 3) but an interaction of this type between PP2A and GST alone was not observed (lane 2), indicating the specificity of association between Sm t-Ag and PP2A. In addition, we identified the protein domains within Sm t-Ag, which confers the interaction with PP2A using a series of Sm t-Ag deletion mutants fused with GST which were prepared and incubated with whole cell extracts prepared from U-87MG cells. A Sm t-Ag carboxy-terminal deletion mutant retaining residues 1 to 124 showed a strong binding activity (lane 4) but less than full-length (lane 3). Another carboxy-terminal deletion mutant retaining residues 1 to 81, however, lost its binding activity to PP2A (lane 5). Removal of the amino-terminal region of Sm t-Ag positioned between residues 1 to 81 did not severely affect its interaction with PP2A (lane 6, aa 82–172). The magnitude of interaction of this clone with PP2A appears to be closer to that was observed for full-length (compare lane 6 with lane 3). As the deletion of the residues between 1 to 124 severely affected the ability of Sm t-Ag to interact with PP2A (lane 7), retaining the residues between 82 to 124 showed strong binding activity to PP2A (lane 8) indicating that the core interaction domain of Sm t-Ag with PP2A is located between residues 82 to 124 toward the central portion of the protein.
Fig. 4.
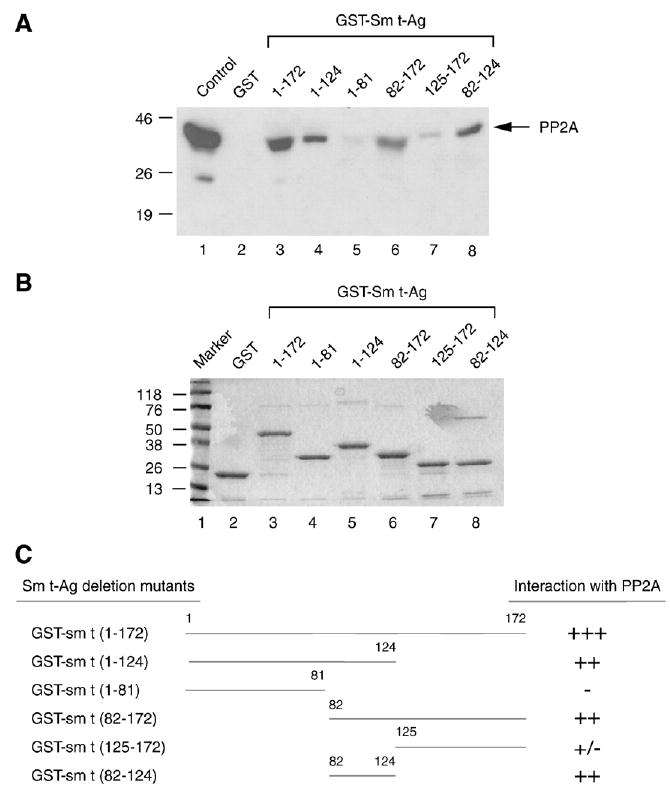
The PP2A interaction domain of the Sm t-Ag localizes to its middle portion. (A) Sm t-Ag and its deletion mutants were expressed in bacteria as fusion proteins and immobilized on glutathione-S-transferase (GST) beads. Whole cell lysates prepared from U-87MG cells were incubated with either GST–Sm t-Ag or its deletion mutants for 2 h. The columns were extensively washed with binding buffer as described in Materials and methods and the proteins retained in the column were fractionated by SDS-10% PAGE and analyzed by Western blotting with an antibody directed against PP2A. (B) Analysis of GST, GST–Sm t-Ag and the deletion mutants of Sm t-Ag by SDS-12% PAGE. (C) Schematic representation of Sm t-Ag and its deletion mutants. The relative binding activity of Sm t-Ag and its deletion mutants is represented by + or − signs. +++, very strong binding; ++, strong binding; +, binding; +/−, weak binding and −, no binding.
Late agnoprotein interacts with early protein Sm t-Ag
Our previous studies have shown that agnoprotein can physically and functionally interact with the JCV early regulatory protein, LT-Ag. We have now analyzed if agnoprotein can interact with another JCV early regulatory protein, Sm t-Ag by coimmunoprecipitation and GST pull-down assays. Whole cell extracts were prepared from SVG-A cells infected or uninfected with JCV Mad-1 strain as described in Materials and methods, and incubated with either pre-immune serum (control) or anti-Sm t-Ag antibody. Immunocomplexes were analyzed by Western blotting for agnoprotein. As shown in Fig. 5A, immunocomplexes obtained upon incubation of the protein extracts with an anti-Sm t-Ag antibody contained agnoprotein (lane 5). However, we did not detect agnoprotein immunoprecipitated with the control pre-immune serum (lane 4), which indicates specificity.
Fig. 5.
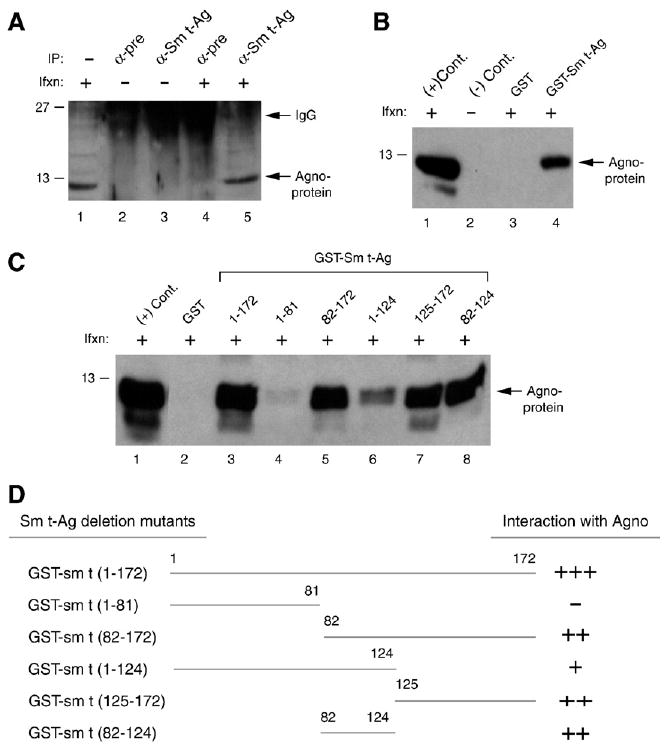
The C-terminal portion of Sm t-Ag is important for agnoprotein interaction. (A) Agnoprotein coimmunoprecipitates with Sm t-Ag. Whole cell extracts (500 μg) prepared from SVG-A cells infected (Ifxn) or uninfected with JCV Mad-1 were subjected to immunoprecipitation (IP) using α-pre rabbit and α-Sm t-Ag antibody and analyzed by Western blotting using α-agno antibody. In lane 1, whole cell extracts from infected cells were loaded as a positive control. (B) GST pulldown assay. Whole cell extracts, prepared from SVG-A cells, infected with JCV Mad-1 were incubated with either GST or GST–Sm t-Ag, immobilized on Glutathione–Sepharose beads. Bound proteins were analyzed by Western blotting using an α-agno antibody as described for Fig. 2. In lane 1, whole cell extract (20 μg) from infected cells was loaded as a positive (+) control. In lane 2, whole cell extract (20 μg) from uninfected cells was loaded as a negative (−) control. (C) The agnoprotein interaction domain of Sm t-Ag maps to the C-terminal portion of the protein. GST pulldown assay was carried out as described for panel B. (D) Summary of the results from in vitro mapping assays as described for Fig. 4C.
To investigate further the interaction between agnoprotein and Sm t-Ag in vitro, GST pull-down experiments were performed. As shown in Fig. 5B, incubation of Sepharose columns containing GST or GST–Agno with whole cell extracts prepared from SVG-A cell infected with JCV Mad-1 resulted in the specific retention of agnoprotein on the column containing GST–Sm t-Ag (lane 4) but not on that containing GST alone (lane 3). Of note, no retention of agnoprotein by GST column alone indicates the specificity of the interaction between agnoprotein and Sm t-Ag. Next, we mapped the interaction domain(s) of Sm t-Ag with agnoprotein performing GST pull-down assays using deletion mutants of Sm t-Ag (Fig. 5C). The C-terminal region of the protein containing residues 82–172 is critical for the interaction between Sm t-Ag and agnoprotein (lane 5). In addition, a similar binding activity of 82–124 and 125–172 regions suggested that C-terminal region of Sm t-Ag may contain more than one region responsible for interaction of agnoprotein with Sm t-Ag.
Sm t-Ag inhibits PP2A-mediated dephosphorylation of agnoprotein
Previous studies have shown that Sm t-Ag of SV40 interacts with the regulatory A subunit of PP2A and this interaction results in the replacement of regulatory B subunit. Since PKC-phosphorylated agnoprotein is a substrate for PP2A (Fig. 3B and C), we reasoned that JCV Sm t-Ag may inhibit PP2A-mediated dephosphorylation of agnoprotein. In order to investigate this possibility, we performed in vitro inhibition studies. Agnoprotein was produced in bacteria as a fusion protein (GST–Agno), purified and phosphorylated by PKC (Sariyer et al., 2006). PP2A was immunoprecipitated from whole cell extracts (U-87MG cells) using anti-PP2A C antibody and was used as a dephosphorylation enzyme in the reaction. As shown in Fig. 6A, PKC-phosphorylated agnoprotein was incubated either with increasing concentration of GST–Sm t-Ag (lanes 7–9) or GST (lanes 10–12) and results showed that PP2A dephosphorylated agnoprotein (lane 6) in a similar extent as was observed for Fig. 3C. However, addition of Sm t-Ag to the dephosphorylation reaction significantly inhibited the PP2A-mediated dephosphorylation of agnoprotein (lanes 7–9) compared to the control (lane 6). Under identical reaction conditions, addition of GST protein did not alter significantly the ability of PP2A to dephosphorylate agnoprotein (lanes 10–12). Alkaline phosphatase (AP) was, however, used as a negative control in the reaction to remove the phosphate groups from agnoprotein (lane 2) as previously demonstrated (Sadowska et al., 2003). Additionally, addition of GST Sm t-Ag into the AP reaction in increasing amounts did not have an effect (lanes 3–5) on the reaction, further suggesting that inhibition of PP2A by Sm t-Ag is specific. Taken together, our in vitro dephosphorylation studies showed that Sm t-Ag has the ability to inhibit the PP2A-mediated dephosphorylation of agnoprotein.
Fig. 6.
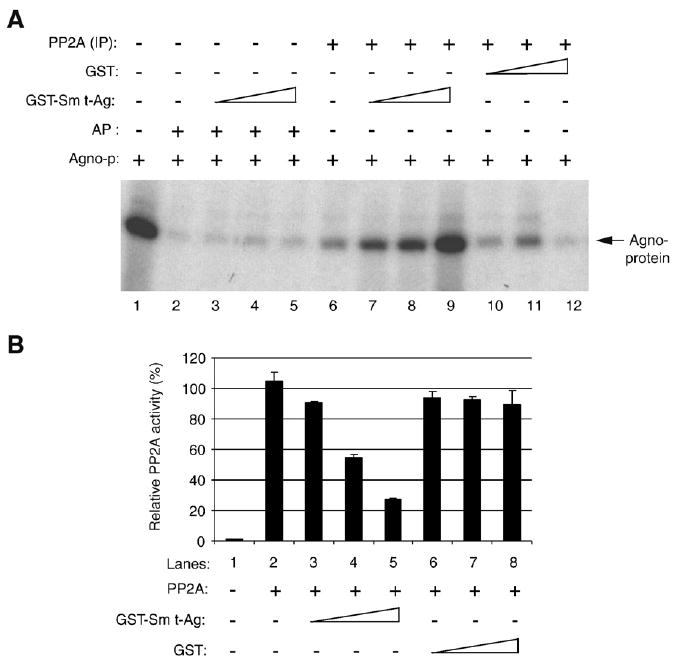
Sm t-Ag inhibits agnoprotein dephosphorylation by PP2A. (B) Full-length agnoprotein was expressed in bacteria as a GST fusion protein (GST–Agno) and purified its homogeneity and phosphorylated by PKC as described previously (Sariyer et al., 2006). PKC-phosphorylated agnoprotein was subjected to dephosphorylation by PP2A, fractionated on SDS-PAGE and analyzed by autoradiography as described in Materials and methods. In lane 2, alkaline phosphatase (AP) was used as a control to dephosphorylate agnoprotein (Sadowska et al., 2003). In lanes 3–5, an increasing amount of GST–Sm t-Ag (0.5, 1.0 and 1.5 μg/lane respectively) was added to the reaction. Similarly, in lanes 7–9, GST–Sm t-Ag was also added into the reaction in increasing amount as described for lanes 3–5. In lane 10–12, an increasing amount of GST (0.5, 1.0 and 1.5 μg/lane respectively) alone was used. In lane 1, phosphorylated GST–agnoprotein was loaded as a positive control. (B) JCV Sm t-Ag inhibits PP2A activity. Inhibition reaction is described in Materials and methods in detail. Briefly, GST and GST Sm t-Ag was expressed in bacteria and purified to their homogeneity. Both proteins were added into reaction in increasing amounts (0.5. 1.0 and 1.5 μg/lane) as indicated and the amount of phosphate released from substrate was colorimetrically determined at 600 nm wavelengths. The enzyme activity was expressed in “relative PP2A activity” in percent.
Moreover, we addressed the question whether Sm t-Ag directly inhibits PP2A activity or Sm t-Ag binds to agnoprotein and obscures the substrate availability, leading to indirect inhibition of PP2A activity. Two differentiate between these two possibilities; we purchased a PP2A phosphatase kit from Upstate, which contains pure PP2A and an authentic substrate for PP2A; and performed phosphatase assay in the presence of either increasing amounts of GST–Sm t-Ag (Fig. 6B, lanes 3–5) or that of GST (Fig. 6B, lanes 6–8). Our results show that Sm t-Ag significantly inhibited PP2A activity when directly added to the reaction (Fig. 6, lanes 3–5), but GST alone did not (Fig. 6, lanes 6–7) suggesting that Sm t-Ag exhibits the ability, at least in part, to directly inhibit PP2A in vitro.
C-terminal portion of Sm t-Ag is critical for its function
The N-terminal 82-amino acid region of Sm t-Ag is shared with LT-Ag. Therefore, any unique function associated with Sm t-Ag most likely lies within its unique C-terminal region. Comparison of JCV and SV40 Sm t-Ags at the amino acid level indicates that both proteins exhibit closer to 70% homology although more divergent sequences are clustered in the middle portion of the each molecule (Fig. 7A). Functional domains such as J domain, PP2A binding domain and cystein clusters are positioned relatively similar locations within each protein (Fig. 7B). Moreover, our results demonstrated that the C-terminal region of Sm t-Ag is critical for interaction with both agnoprotein (Fig. 5C) and PP2A (Fig. 4A). To understand the role of Sm t-Ag in JCV regulation, we introduced a stop codon at amino acid Ser90 immediately after splice donor site of the gene so that the unique C-terminal portion of the protein was not produced. We then evaluated the impact of this stop codon on viral replication and gene regulation. For this purpose, primary human fetal glial cells (PHFG) were either transfected/infected with JCV Mad-1 WT strain or JCV Mad-1 Sm t-Ag mutant genome. Low molecular weight DNA was then isolated at indicated time points and analyzed by Dpn I digestion and Southern blotting. As shown in Fig. 7D, the replication efficiency of WT genome increased substantially by 21d posttransfection (lanes 2–4), but that of Mut was significantly inhibited (lanes 5–7), suggesting that the unique C-terminal region of Sm t-Ag plays a critical role in JCV replication. In parallel, we also analyzed the expression level of LT-Ag, VP-1 and agnoprotein from both WT and mutant backgrounds by Western blotting. We obtained consistent results as observed for the replication studies (Fig. 7D) in that the expression level of LT-Ag, VP-1 and agnoprotein from Mut virus is significantly reduced compared to WT (Fig. 7E, compare lanes 2–4 to 5–7 in respective panels). In Fig. 7E, bottom panel, the absence of Sm t-Ag expression was demonstrated in cells that were transfected/infected with mutant virus genome. In addition, we also subcloned the early coding region of WT and mutant separately into an expression vector to demonstrate the efficient splicing of the early transcripts regardless of the presence of the stop codon in the genome. Our results showed that the expression level of LT-Ag for both WT and mutant genomes was relatively equal by Western blotting (data not shown), suggesting that the reduced level of replication of mutant genome versus WT was not due to an inefficient splicing of early transcripts of the mutant, but rather due to the lack of expression of the C-terminal region of Sm t-Ag. These results suggest that the unique region of Sm t-Ag plays an important role in the JCV replication cycle.
Fig. 7.
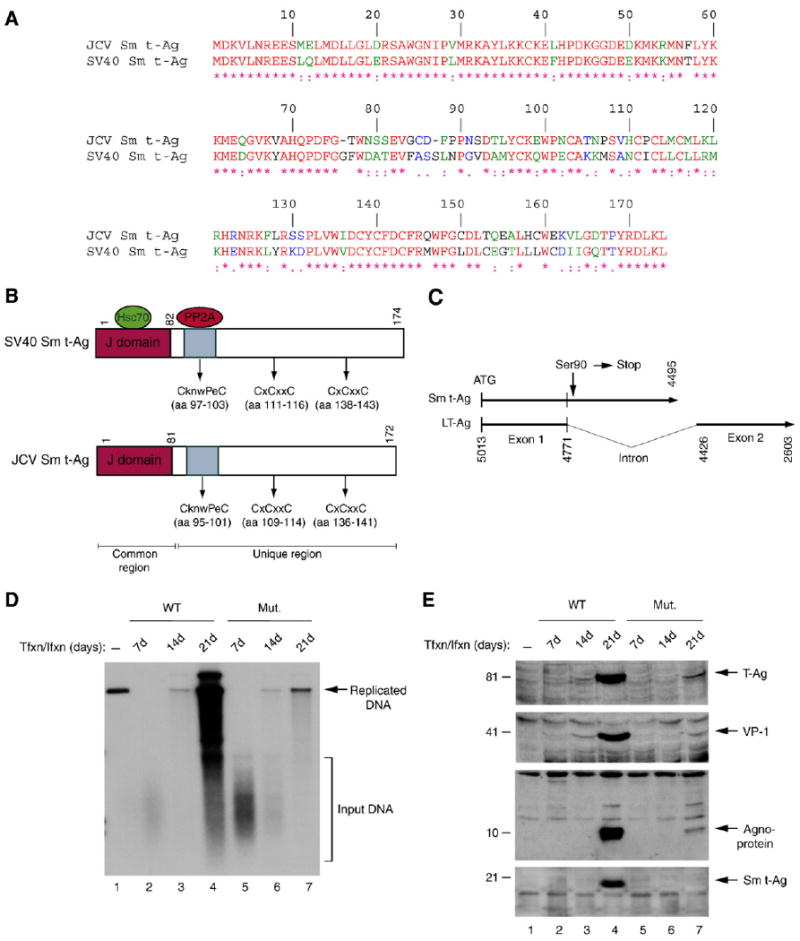
Viral replication and gene expression were substantially down-regulated in the absence of full-length Sm t-Ag expression. (A) Amino acid alignment of JCV Sm t-Ag and SV40 Sm t-Ag. (B) Schematic comparison of SV40 Sm t-Ag regions with that of JCV Sm t-Ag. The position of J domains, PP2A binding domains and cystein clusters are indicated. (C) Schematic representation of JCV Mad-1 early coding region, where Ser90 (TCT) of Sm t-Ag was converted into a stop codon as described in Materials and methods. Numbering is according to JCV Mad-1 strain (GenBank # NC_001699, formerly J02226). (D) Dpn I assay. Primary human fetal glial cells (PHFG) (4 million cells/75 cm2 flask) were transfected/infected either with WT (5 μg) or Mut genome (5 μg) by lipofectin method. At 7d, 14d and 21d posttransfection, low molecular weight DNA was isolated (Ziegler et al., 2004) and analyzed by Dpn I assay (Hirt, 1976). (E) In parallel, either nuclear or cytoplasmic extracts were prepared from transfectants and analyzed by Western blotting using anti-LT-Ag (PAb-2000), anti-VP-1 (PAB597, a gift from Dr. W. Atwood), anti-agnoprotein and anti-Sm t-Ag antibodies as indicated.
Effect of PP2A inhibition on JCV replication
Next, we investigated the effect of PP2A inhibition on JCV replication by RNA interference. SVG-A cells were transfected/infected with JCV genome in the presence and absence of a non-specific siRNA or siRNA specific for PP2A. Two days post-transfection, the replication efficiency of the samples was determined by Dpn I assay. Results showed that the replication of the JCV genome was significantly inhibited (approximately 5-fold, Figs. 8A and B, lane 5) compared to untreated samples (lane 3) or samples, which were treated with non-specific siRNA (lane 4) suggesting that PP2A plays an important role in JCV replication cycle. This reduced replication in siRNA-treated samples (lane 5) cannot be simply attributed to the inefficient transfection efficiency of JCV genome into the cells, because the level of input DNA in three experimental groups appears to be equal (compare lane 3 to 4 and 5). In addition, since we did not observe a significant level of difference between untreated and treated samples with respect to input DNA levels, the observed effect on JCV DNA replication appears to be mostly due to a specific inhibition of PP2A by siRNA rather than a non-specific toxicity of the treatment.
Fig. 8.
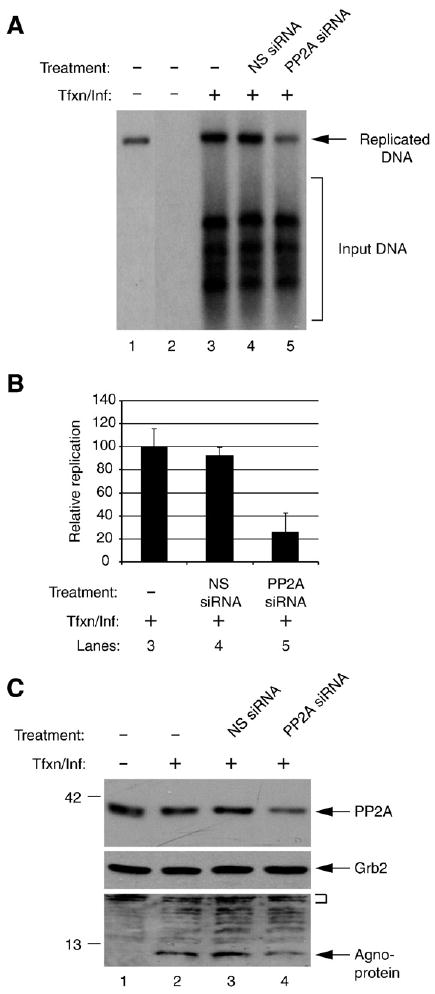
Effect of PP2A inhibition on JCV replication and gene expression. (A) Inhibition of PP2A negatively affects JCV replication. SVG-A cells. 2 × 105 cells per 35-mm tissue culture dish were transfected/infected with JCV genome (2 μg/plate) in the presence and absence of 50 pmol of non-targeting siRNA or Smartpool PP2AC siRNA (PP2AC siRNA, Dharmacon, Lafayette, CO). At the 48 h post infection, cells were trypsinized and split into two equal portions. One half of the samples were used to prepare low molecular weight DNA for Dpn I assay and the other portion was used to prepare whole cell extracts for Western blotting. DNA was digested with both Bam HI and Dpn I and analyzed by Southern blotting using labeled JCV genomic DNA as probe. Dpn I enzyme digests only transfected (input) DNA and leaves intact the newly replicated DNA. In lane 1, plasmid containing the Mad-1 genome was digested with Bam HI only and 0.1 ng of it was run on the gel as a positive control. In lane 2, the DNA sample from untransfected/uninfected cells were processed as in lanes 3–5 and was loaded as a negative control. The arrow points to the replicated DNA and the bracket indicates the input DNA. Tfxn/Inf denotes transfection/infection, i.e., introduction of the Mad-1 JCV genome into cells by transfection. (B) Quantitative representation of the results from panel A. The bands corresponding to the replicated DNA was quantitated in each lane (lanes 3–5) by densitometry and normalized relative to the band intensity on lane 3. The standard deviations of the data are presented as error bars (C) Western blot analysis of the whole cell extracts prepared in parallel to the replication assay in panel A, using specific antibodies against agnoprotein, PP2AC and Grb2. In lane 1, sample from untransfected/uninfected cells was loaded as a negative control. The small bracket denotes the non-specific bands that indicate the equal protein loading for agnoprotein. Grb2 was probed as a loading control for PP2AC.
In parallel, the expression levels of both PP2A and agnoprotein were also analyzed by Western blotting. The expression levels of both proteins were considerably lower in samples treated with the specific siRNA (Fig. 8C, lane 4) compared to either non-treated (lane 2) or treated with non-specific siRNA (lane 3). These findings suggest that PP2A plays a critical role in the JCV replication cycle.
Effect of a double mutation on Sm t-Ag and agnoprotein on viral replication cycle
Our results demonstrate that Sm t-Ag may influence the level of agnoprotein dephosphorylation through the inhibition of PP2A. We then reason that a triple phosphorylation mutant of agnoprotein should be resistant to mutations in Sm t-Ag. To address this question, we have created a double mutant of JCV Mad-1 strain on Sm t-Ag and agnoprotein as described in Materials and methods, and analyzed the impact of these mutations on the replication properties of JCV by Dpn I assay. WT and mutant genomes were transfected into SVG-A cells and low molecular weight DNA was isolated at 5, 10 and 15 day posttransfection and analyzed by Southern blotting. As shown in Fig. 9C, the double mutant behaves very much like agnoprotein triple phosphorylation mutant alone during the replication cycle as previously reported (Sariyer et al., 2006). That is, this double mutant is also unable to continue to its replication after first round of infection cycle (compare lanes 2–4 with 5–7). These findings confirms our assumption that agnoprotein might be an important target of Sm t-Ag with respect to the regulation of its dephosphorylation, and therefore it appears that the triple phosphorylation mutant of agnoprotein is resistant to a mutation on Sm t-Ag.
Fig. 9.
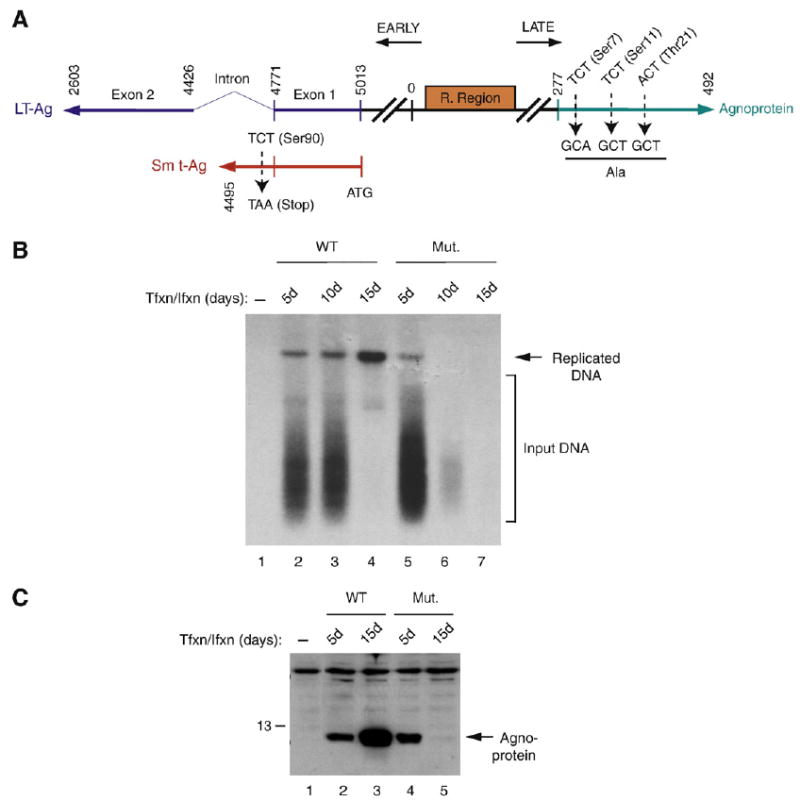
Agnoprotein triple phosphorylation mutant is resistant to a mutation in Sm t-Ag. (A) Schematic representation of a double mutant of JCV Mad-1 strain on Sm t-Ag and agnoprotein. On agnoprotein, Ser7, Ser11 and Thr21 were converted into Ala. On Sm t-Ag, Ser90 was converted into a stop codon. (B). Analysis of the growth properties of JCV WT Mad-1 and double mutant by Dpn I assay. SVG-A cells (5 million cells/75 cm2 flask) were transfected/infected either with WT (5 μg) or Mut genome (5 μg) by lipofectin method. At 5d, 10d and 15d posttransfection, low molecular weight DNA was isolated (Ziegler et al., 2004) and analyzed by Dpn I assay (Hirt, 1976). (C) In parallel, cytoplasmic extracts were prepared from transfectants and analyzed by Western blotting using anti-agnoprotein antibody.
Discussion
The function of regulatory proteins is often modulated by posttranslational modifications, including phosphorylation. This modification is known to affect the activity of many target molecules in a positive and negative manner depending on the nature of the target. For instance, previous studies have shown that the initiation of viral DNA replication is regulated by the phosphorylation state of SV40 LT-Ag (Cegielska et al., 1994b; Scheidtmann et al., 1991b), and PP2A is the primary phosphatase, involved in this regulation. PP2A removes the phosphate moieties from Ser120, Ser123 and Thr124 on SV40 LT-Ag and stimulates the origin unwinding activity of this protein (Cegielska et al., 1994a,b; Cegielska and Virshup, 1993). It is also possible that PP2A plays regulatory roles in the function of agnoprotein through the regulation of the phosphorylation state of this protein during the infection cycle.
We have recently showed that agnoprotein is subject to phosphorylation by PKC and further analysis of agnoprotein phosphorylation site mutants, showed that they are defective for viral replication (Sariyer et al., 2006). Since agnoprotein is phosphorylated by a serine/threonine kinase, PKC, we reasoned that agnoprotein might be a substrate for the PP2A. In the present study, we demonstrate that PP2A dephosphorylates agnoprotein (Fig. 3B and C) and that the N-terminal half of agnoprotein, in which the PKC phosphorylation sites are localized (Fig. 3D), is critical for interaction with PP2A (Fig. 2A). It should be mentioned here that the interaction observed between PP2A and agnoprotein could be both stoichiometric and enzymatic, because there are a number of cases reported in the literature supporting this case. For instance, SV40 Sm t-Ag was reported to have a stable interaction with PP2A and inhibits the activity of this enzyme (Yang et al., 1991). Similarly, PP2A was also reported to form a stable complex with Shc and regulates the dephosphorylation of this protein (Ugi et al., 2002). As such, the interaction that we observed between agnoprotein and PP2A could be in both levels, stoichiometric and enzymatic.
In addition, we showed that Sm t-Ag interacts with the viral late agnoprotein, through its C-terminal region (Fig. 5C). Additional mapping studies revealed that the interaction domain of Sm t-Ag with agnoprotein and PP2A overlaps (Fig. 4A), suggesting that agnoprotein and PP2A may compete with one another for association with Sm t-Ag. The importance of interactions among these three proteins was demonstrated by the observation that Sm t-Ag inhibits agnoprotein dephosphorylation by PP2A (Fig. 6A), which may be an important regulatory mechanism for JCV replication.
Since the interaction domain of Sm t-Ag with agnoprotein and PP2A is located within its unique region, we analyzed the effect of this region on the JCV life cycle by mutational analysis. We introduced a stop codon at Ser90 (Fig. 7C) immediately after the splice donor site of JCV early genome so that the C-terminal unique region of Sm t-Ag is not produced during the infection cycle (Fig. 7E). Results showed that replication of JCV without the unique region of Sm t-Ag is severely impaired (Fig. 7D). This suggests that Sm t-Ag, although not directly involved in JCV replication, appears to play an important role in JCV life cycle, perhaps through interaction with agnoprotein and PP2A. Similarly, we investigated the functional significance of PP2A in JCV replication cycle by siRNA targeting, and observed that that inhibition of PP2A expression results in a considerable reduction in viral replication (Fig. 8A), which also emphasizes the critical role of PP2A in JCV life cycle. Mutational studies with SV40 Sm t-Ag supports our findings and such studies demonstrated that Sm t-Ag significantly contributes to the viral replication cycle (Cicala et al., 1994; Jog et al., 1990; Shenk et al., 1976; Topp, 1980). In particular, studies conducted by Cicala et al (Cicala et al., 1994) demonstrated that mutant virus that lacks the Sm t-Ag replicates less efficiently than the wild-type virus. The role of Sm t-Ag in viral DNA replication was even further investigated by microinjection studies, where SV40 DNA was injected into CV1 cells with and without purified Sm t-Ag and was analyzed by Southern blotting. It was found that the replication of either wild-type or Sm t-Ag deletion mutant DNA increased three-to fivefold in cells coinjected with purified Sm t-Ag, suggesting that SV40 Sm t-Ag has a stimulatory effect on viral DNA replication in vivo.
Addition of a phosphate moiety to the target molecules is catalyzed by specific kinases and removal of the same group is carried out by specific phosphatases. As a result, there is a well-balanced network of kinases and phosphatases in a cell and this equilibrium is crucial for a cell to accurately respond to the environmental cues. Outside invaders such as viruses shift this balance toward one side by modifying the cell environment in such a way that the host machinery is exploited by the virus for the successful completion of its life cycle. As we demonstrate in this work for JCV Sm t-Ag of SV40 was also previously shown to target PP2A in host cells (Sontag et al., 1993, 1997). Mapping and point mutation analysis of the interaction domain of SV40 Sm t-Ag with PP2A revealed that central portion of the protein, specifically residues including Cys97, Pro101 and Cys103 are critical in this interaction (Mungre et al., 1994; Porras et al., 1996; Sontag et al., 1993). These critical residues are also located within the interaction domain of JCV Sm t-Ag with PP2A. The functional significance of PP2A targeting by SV40 Sm t-Ag is not fully understood for SV40 infection cycle. However, it appears that at least in the absence of viral infection cycle, this targeting may facilitate cell cycle progression and therefore play a critical role in the onset of cell transformation (Hahn et al., 2002).
Recent crystallography studies have shown that not only does the unique region of SV40 Sm t-Ag interact with PP2A but also the J domain (Cho et al., 2007). The previous biochemical interaction studies between SV40 Sm t-Ag and PP2A appear to be consistent with the these recent crystallographic findings, because although the J domain is not essential for the interaction between SV40 Sm t-Ag and PP2A, the deletion of this domain significantly decreased the inhibitory activity of Sm t-Ag on the PP2A core dimer (Mateer et al., 1998), suggesting that the J domain enhances the binding of Sm t-Ag to the PP2A subunit. Our binding studies (Fig. 4) is also consistent with the these crystallographic and previous biochemical studies (Cho et al., 2007; Mateer et al., 1998), in that although the core interaction domain of JCV Sm t-Ag with PP2A is located within the unique region of the protein, the J domain appears to significantly contribute to this interaction, because the deletion of the first 81 amino acid region of JCV Sm t-Ag negatively affects this interaction (Fig. 4A).
Our in vitro inhibition studies demonstrated that JCV Sm t-Ag is able to inhibit PP2A-mediated agnoprotein dephosphorylation (Fig. 6A). These results are similar to the previously reported studies with SV40 Sm t-Ag. It was demonstrated that SV40 Sm t-Ag can inhibit the PP2A-mediated dephosphorylation SV40 large T antigen and cellular transcription factor, p53 (Scheidtmann et al., 1991a,b) in vitro. In infection cases, however we think that a partial inactivation of PP2A by Sm t-Ag is more likely, because a total inactivation would lead to deleterious consequences, such as an early cell death and therefore an abortive infection. A recent report by Yang et al. (Yang et al., 2005) supports such a partial inactivation case, which suggests that Sm t-Ag can orient PP2A towards specific substrates rather than totally inhibiting its enzymatic activity.
In light of the data presented in this work with respect to functional interactions among the three proteins, combined with the previous analysis of agnoprotein phosphorylation mutants (Sariyer et al., 2006), one can speculate that Sm t-Ag may directly contribute to the maturation process of infectious viral particles through the regulation of the timing of agnoprotein dephosphorylation by PP2A. In other words, Sm t-Ag may regulate the timing of the dephosphorylation of agnoprotein by PP2A during the infection cycle. It is possible that during the early phases of the infection cycle, Sm t-Ag may inhibit the dephosphorylation of agnoprotein by PP2A and may prevent the process of the formation of immature capsids (i.e. empty capsids). In fact, characterization of a double mutant of JCV (Fig. 9B) suggested that in deed Sm t-Ag may be involved in regulation of agnoprotein dephosphorylation, because this double mutant behaves much like the triple phosphorylation mutant of agnoprotein during the infection cycle. That is, this mutant cannot continue its replication cycle as the case observed for the triple phosphorylation mutant of agnoprotein, suggesting that Sm t-Ag may in fact target agnoprotein for the regulation of its phosphorylation during the infection cycle and therefore may contribute the maturation of viral capsids. It is also possible that the dephosphorylated form of agnoprotein is critical for the efficient release of viral particles from infected cells toward the late stages of the infection cycle. In this case, PP2A may regain its full activity by being released from Sm t-Ag. Agnoprotein may play a critical role in the release of PP2A from Sm t-Ag by competing for the same binding site on Sm t-Ag. Our mapping studies support this possibility, since we showed that the Sm t-Ag interaction domains for PP2A and agnoprotein overlap (Figs. 4A and 5C).
Viruses have developed interesting strategies to deregulate cells through PP2A. Cytomegalovirus (CMV), for example, contains a cellular PP2A holoenzyme, which is probably involved in the dephosphorylation of cellular proteins during the early infection cycle (Michelson et al., 1996). DNA tumor viruses, including adenoviruses and polyomaviruses also encode proteins that specifically target PP2A. E4orf4 from adenovirus interacts with PP2A and down-regulates JunB transcription, which may play a role in the viral infection cycle by regulating the apoptotic response of the infected cells (Kleinberger and Shenk, 1993). In this regard, it was found that the interaction of E4orf4 with PP2A leads to the apoptosis of transformed cells (Shtrichman and Kleinberger, 1998). Sm t-Ags of SV40 and polyomavirus as well as polyomavirus middle T were also found to target PP2A (Cegielska et al., 1994a; Pallas et al., 1990; Virshup et al., 1989, 1992), and this targeting appears to be required for viral-induced transformation. It may also be important for the viral replication cycle, perhaps through the modification of PP2A-dependent signaling pathways by viral regulatory proteins in infected cells. Taken together, studies with the regulatory functions of JCV proteins will provide new insight into the mechanism of the regulatory pathways of JCV infection cycle, which may in turn help us to develop effective therapeutic strategies against PML.
Materials and methods
Cell culture
SVGA is a clonal population of a human glial cell line that was established by transformation of fetal glial cell line with an origin defective SV40 mutant and has been described previously (Major et al., 1985). U-87MG (ATCC HTB14) is a human glioblastoma cell line. Both SVGA and U87MG cell lines were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin, 100 μg/ml). They were maintained at 37 °C in a humidified atmosphere with 7% CO2. Primary Human Fetal Glial (PHFG) cells were prepared and maintained as previously described (Daniel and Frisque, 1993; Swenson et al., 1996).
Plasmids
Sm t-Ag full-length and its deletion mutants were cloned into a prokaryotic expression vector (pGEX2T) at Bam HI/Eco RI site by a PCR amplification method using specific primers.
Forward primers were
Sm t-Ag 5′-Bam HI (5′-ACCTTCCAGGATCCATGGACAAAGTGCTGAATAGGGAG-3′),
FP aa 82 (5′-ACCTTCCA GGATCCATGGTTGGTTGTGATTTTCCTCCTAAT-3′),
FP aa 125 (5′-ACCTTCAGGATCCATGTTTTTAAGAAGCAGCCCACTT-3′).
Reverse primers were
Sm t-Ag 3′-Eco RI (5′-ACCTCCAGAATTCTTAAAGCTTTAGATCCCTGTA -3′),
RP aa 124 (5′-ACCTCCAGAATTCTTATTTTCAGTTTCTATGCCTTAA-3′),
PR aa 81 (5′-ACCTCCAGAATTCCTCTGAACTATTCCATGTACCAAA-3)
The resulting PCR fragments were digested with Bam HI/Eco RI and subcloned into the Bam HI/Eco RI sites of the pGEX2T vector. GST–Agnoprotein (1–71) and deletion mutants of agnoprotein fused to GST protein (1–54, 1–36, 18–37, 37–71, 55–71 and 18–54) were described previously (Safak et al., 2001).
Site-directed mutagenesis
A stop codon was introduced into Sm t-Ag coding region right after splice donor site converting Ser90 codon to a stop (TAA) codon utilizing a ExSite-PCR based Kit (Stratagene). The following primers were used in the PCR-mediated mutagenesis as described previously (Akan et al., 2006; Sariyer et al., 2006).
Primer 1. 5′-TCA GAG GTT GGT TGT GAT TAA CCT CCT AAT TCT GAT ACC C-3′ Primer 2. 5′-G GGT ATC AGA ATT AGG AGG TTA ATC ACA ACC AAC CTC TGA -3′. This plasmid is called Bluescript KS Mad-1 Sm t-Ag Mut (Ser90 to Stop). We also created a double mutant of JCV Mad-1 strain on agnoprotein and Sm t-Ag as follows. Creation of a triple phosphorylation mutant of agnoprotein was previously described (Sariyer et al., 2006) and the plasmid was called Bluescript KS (BstXI−) Mad-1 Mut3(Agno Ser7, Ser11 and Thr21 to Ala). Sm t-Ag mutant described above was also subloned into Bluscript KS (BstXI−) background. By exchanging the BstXI/EcoNI fragment on Sm t-Ag mutant with the same fragment form triple phosphorylation mutant, we have created a double mutant of JCV Mad-1 and called Bluescript (BstXI−) Sm t-Ag (Ser90 to stop) +Mut3(Agno Ser7, Ser11 and Thr21 to Ala).
Immunoprecipitation and Western blotting
Five hundred micrograms of whole cell lysates were prepared from SVG-A cells infected with JCV Mad-1 or from uninfected cells in lysis buffer {25 mM Tris–HCl (pH 7.4), 150 mM NaCl, 10 mM HEPES pH 7.0, 1 mM DTT, 10% Glycerol, 1 mM EDTA, 0.5 NP-40 and complete protease inhibitors (Sigma)}. Cell lysates (0.5 mg) were incubated with either normal mouse or rabbit serum (2 μg) or with an anti-PP2A C antibody directed against the C (2 μg, Santa Cruz, Cat #: SC-14020) or the A subunit (2 μg, Santa Cruz, Cat #: SC-15355) or the B (2 μg, Upstate, Cat #: 05-592) subunit by rocking at 4 °C for 2 h. Immunocomplexes were washed three times with lysis buffer and analyzed by Western blotting using a polyclonal antibody raised against agnoprotein (Safak et al., 2002). A reciprocal immunoprecipitation and Western blot analysis was also performed on the same extracts to detect agnoprotein–Sm t-Ag interaction as described above. Five hundred micrograms of whole cell extract prepared from infected cells were immunoprecipitated with either normal rabbit serum (2 μg) or rabbit polyclonal antibody raised against the C-terminal unique region of Sm t-Ag (PAb 2081, 2 μg, we raised this antibody ourselves in rabbit) and immunocomplexes were analyzed by Western blotting using polyclonal anti-agnoprotein antibody (Safak et al., 2002).
GST affinity chromatography assay (GST-pull down)
Expression and purification of GST and GST fusion proteins were previously described (Safak et al., 1999a,b). Interaction of agnoprotein with PP2A was tested by GST-pull down assay as described previously (Safak et al., 2001, 2002). Two micrograms of either GST or GST–Agno immobilized on Sepharose beads were incubated with whole cell extracts prepared from U-87MG cells for 2 h at 4 °C in lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.5% Nonidet P-40. Columns were then washed extensively with lysis buffer and retained protein complexes were resolved by SDS-10% PAGE followed by Western blot analysis using an anti-PP2A C antibody. Interaction of Sm t-Ag with PP2A or that of agnoprotein was also tested as described above.
In vitro kinase assay
Kinase assays were performed as previously described (Sariyer et al., 2006). Briefly, GST and GST-agnoprotein full-length (1–71) were expressed in bacteria and were purified as described previously (Kim et al., 2003; Sadowska et al., 2003). One microgram of bacterially expressed GST or GST-agnoprotein (1–71) was incubated with 10 U of PKC (Promega) in kinase reaction mixture containing 25 mM Tris–HCl (pH 7.4), 20 mM MgCl2, 5 mM DTT, 2.5 μCi of [γ-32P]-ATP, and 10% glycerol at 30 °C for 1 h. After the kinase reaction, phosphorylated proteins were used for inhibition studies and were fractionated by SDS-12% PAGE and visualized by autoradiography.
Inhibition of PP2A by JCV Sm t-Ag
The purified PP2A enzyme (Cat. #: 14-111), phosphatase assay kit (Cat. #: 17-313) and a substrate for PP2A (K-R-pT-R-R, Cat. #: 12-219) were all purchased from Upstate Company. GST and GST–Sm t-Ag proteins were expressed in bacteria and purified to their homogeneity. The release of phosphate from the substrate by PP2A was colorimetrically measured by Malachite green phosphatase assay according to the manufacturer's recommendations. Either GST alone (control) or GST Sm t-Ag was added to the reaction as described under the related figure legend and the amount of the released phosphate was determined by extrapolating the calorimetrically measured data points to the standard curves according to the manufacturer's recommendations.
Inhibition of dephosphorylation assay
One microgram of bacterially expressed GST–agnoprotein was phosphorylated by PKC as described above (see in vitro kinase assay). The kinase reaction was stopped by addition of 1 μg of Protein Kinase C Inhibitor Peptide (Calbiochem, Cat. 05-23-4904) into the reaction mixture. Phosphorylated GST–agnoprotein was desphosphorylated both with purified and immunoprecipitated PP2A. The purified PP2A enzyme, phosphatase assay kit (Cat. #: 17-313) and an authentic substrate for PP2A (Cat. #: 12-219) were both purchased from Upstate. Phosphorylated GST–agnoprotein was equally distributed into reaction tubes. Immunoprecipitated PP2A was prepared from whole cell extracts from SVGA cells (500 μg) using a monoclonal antibody against PP2A (3 μl, Upstate, Cat. 16-125D). Immunocomplexes were washed three times with pNPP Ser/Thr assay buffer (Upstate, Cat. 20-179) and was resuspended in the same buffer. Immunoprecipitated PP2A (IP-PP2A) enzyme was subsequently used for dephosphorylation assay as described in the related figure legend. Normal serum was also used for immunoprecipitation as a negative control above. In some reactions, a specific inhibitor of PP2A (okadaic acid, 2 ng/ml) (Sigma, Cat. 1355544) was used to inhibit PP2A activity. All reactions were performed in pNPP Ser/Thr assay buffer (50 μl) at 30 °C for various durations of time as indicated under related figure legends. Dephosphorylated proteins were fractionated by 12% SDS-PAGE and visualized by autoradiography.
Whole cell and nuclear extract preparation
Whole cell and nuclear extracts were prepared as described previously (Dignam et al., 1983; Sadowska et al., 2003; Safak et al., 2002).
Targeting of PP2A by siRNA and JCV replication
The replication efficiency of JCV was assayed by employing Dpn I assay as described previously (Akan et al., 2006; Sariyer et al., 2006). SVG-A cells (2×105 cells per 35-mm tissue culture dish) were transfected/infected with JCV genome (2 μg/plate) in the presence and absence of 200 μmol of Smartpool PP2Ac siRNA or non-targeting siRNA (NS siRNA, Dharmacon, Lafayette, CO). At the 48 h post infection, low molecular weight DNA containing both input and replicated viral DNA was isolated by Qiagen spin columns (Ziegler et al., 2004), digested with Bam HI and Dpn I enzymes, resolved on 1% agarose gel and analyzed by Southern blotting using labeled probe prepared from whole Mad-1 genome (Ziegler et al., 2004). Dpn I enzyme digests only transfected (input) DNA and leaves replicated DNA intact.
In addition, to analyze the effect of Sm t-Ag stop mutant on viral replication, primary human fetal glial cells (PHFG) (4 million cells/75 cm2 flask) were transfected/infected either with WT (5 μg) or Mut genome [where Ser90 (TCT) codon of Sm t-Ag is replaced with a stop codon (TAA), 5 μg] employing lipofectin according to manufacturer's recommendations (Invitrogen, Carlsbad, CA). At 7d, 14d and 21d posttransfection, low molecular weight DNA was isolated and processed for Dpn I assay as described above. Moreover, in order to analyze the effect of double mutant on viral replication cycle, SVG-A cells (2×105 cells/T75 cm2 tissue culture flask) were transfected/infected with JCV genome Mad-1 WT or double mutant (a combination of Sm t-Ag stop mutant and a triple phosphorylation mutant of agnoprotein) (10 μg/flask), as described above. Samples were then processed for either Dpn I assay or Western blotting as indicated under related figure legends.
Acknowledgments
We would like to thank past and present members of the Center for Neurovirology for sharing of ideas and reagents, particularly to Dr. M. White for insightful discussion. This work was made possible by grants awarded by NIH to KK and MS.
References
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349(1):66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Ali SH, DeCaprio JA. Cellular transformation by SV40 large Tantigen: interaction with host proteins. Semin Cancer Biol. 2001;11(1):15–23. doi: 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24(52):7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- Berger JR, Concha M. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J Neurovirology. 1995;1(1):5–18. doi: 10.3109/13550289509111006. [DOI] [PubMed] [Google Scholar]
- Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7(7):495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- Carbone M, Lewis AM, Jr, Matthews BJ, Levine AS, Dixon K. Characterization of hamster tumors induced by simian virus 40 small t deletion mutants as true histiocytic lymphomas. Cancer Res. 1989;49(6):1565–1571. [PubMed] [Google Scholar]
- Cegielska A, Moarefi I, Fanning E, Virshup DM. T-antigen kinase inhibits simian virus 40 DNA replication by phosphorylation of intact T antigen on serines 120 and 123. J Virol. 1994a;68(1):269–275. doi: 10.1128/jvi.68.1.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A, Shaffer S, Derua R, Goris J, Virshup DM. Different oligomeric forms of protein phosphatase 2A activate and inhibit simian virus 40 DNA replication. Mol Cell Biol. 1994b;14(7):4616–4623. doi: 10.1128/mcb.14.7.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A, Virshup DM. Control of simian virus 40 DNA replication by the HeLa cell nuclear kinase casein kinase I. Mol Cell Biol. 1993;13(2):1202–1211. doi: 10.1128/mcb.13.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hahn WC. SV40 early region oncoproteins and human cell transformation. Histol Histopathol. 2003;18(2):541–550. doi: 10.14670/HH-18.541. [DOI] [PubMed] [Google Scholar]
- Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. Structural basis of PP2A inhibition by small t antigen. BLos Biol. 2007;5:1–10. doi: 10.1371/journal.pbio.0050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YW, Lee IC, Ross SR. Requirement for the simian virus 40 small tumor antigen in tumorigenesis in transgenic mice. Mol Cell Biol. 1988;8(8):3382–3390. doi: 10.1128/mcb.8.8.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicala C, Avantaggiati ML, Graessmann A, Rundell K, Levine AS, Carbone M. Simian virus 40 small-t antigen stimulates viral DNA replication in permissive monkey cells. J Virol. 1994;68(5):3138–3144. doi: 10.1128/jvi.68.5.3138-3144.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel AM, Frisque RJ. Transcription initiation sites of prototype and variant JC virus early and late messenger RNAs. Virology. 1993;194(1):97–109. doi: 10.1006/viro.1993.1239. [DOI] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21(36):5574–5581. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11(5):1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ. Structure and function of JC virus T′ proteins. J Neurovirology. 2001;7(4):293–297. doi: 10.1080/13550280152537120. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, White FA. In: The molecular biology of JC virus, causative agent of progressive multifocal leukoencephalopathy. R ERP, editor. Humana Press Inc.; Totowa, NJ: 1992. [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51(2):458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ, Bollag B, Tyagarajan SK, Kilpatrick LH. T′ proteins influence JC virus biology. J Neurovirology. 2003;9(Suppl 1):15–20. doi: 10.1080/13550280390195270. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, Hofstetter C, Tyagarajan SK. Transforming activities of JC virus early proteins. Adv Exp Med Biol. 2006;577:288–309. doi: 10.1007/0-387-32957-9_21. [DOI] [PubMed] [Google Scholar]
- Gaillard S, Fahrbach KM, Parkati R, Rundell K. Overexpression of simian virus 40 small-T antigen blocks centrosome function and mitotic progression in human fibroblasts. J Virol. 2001;75(20):9799–9807. doi: 10.1128/JVI.75.20.9799-9807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, Kang H, Virshup DM. Protein phosphatase 1 regulates the stability of the circadian protein PER2. Biochem J. 2006;399(1):169–175. doi: 10.1042/BJ20060678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, Virshup DM. Protein serine/threonine phosphatases: life, death, and sleeping. Curr Opin Cell Biol. 2005;17(2):197–202. doi: 10.1016/j.ceb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8(2):139–148. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- Garcia A, Cereghini S, Sontag E. Protein phosphatase 2A and phosphatidylinositol 3-kinase regulate the activity of Sp1-responsive promoters. J Biol Chem. 2000;275(13):9385–9389. doi: 10.1074/jbc.275.13.9385. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22(7):2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heriche JK, Lebrin F, Rabilloud T, Leroy D, Chambaz EM, Goldberg Y. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science. 1997;276(5314):952–955. doi: 10.1126/science.276.5314.952. [DOI] [PubMed] [Google Scholar]
- Hirt BJ. Selective extraction of polyoma DNA from infected mouse cell culture. J Mol Biol. 1976;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Howe AK, Gaillard S, Bennett JS, Rundell K. Cell cycle progression in monkey cells expressing simian virus 40 small t antigen from adenovirus vectors. J Virol. 1998;72(12):9637–9644. doi: 10.1128/jvi.72.12.9637-9644.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubiec A, Jupin I. Regulation of positive-strand RNA virus replication: the emerging role of phosphorylation. Virus Res. 2007;129(2):73–79. doi: 10.1016/j.virusres.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubiec A, Tournier V, Drugeon G, Pflieger S, Camborde L, Vinh J, Hericourt F, Redeker V, Jupin I. Phosphorylation of viral RNA-dependent RNA polymerase and its role in replication of a plus-strand RNA virus. J Biol Chem. 2006;281(30):21236–21249. doi: 10.1074/jbc.M600052200. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353(Pt 3):417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jog P, Joshi B, Dhamankar V, Imperiale MJ, Rutila J, Rundell K. Mutational analysis of simian virus 40 small-t antigen. J Virol. 1990;64(6):2895–2900. doi: 10.1128/jvi.64.6.2895-2900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili K, Feigenbaum L, Khoury G. Evidence for a shift in 5′-termini of early viral RNA during the lytic cycle of JC virus. Virology. 1987;158(2):469–472. doi: 10.1016/0042-6822(87)90224-8. [DOI] [PubMed] [Google Scholar]
- Kim J, Woolridge S, Biffi R, Borghi E, Lassak A, Ferrante P, Amini S, Khalili K, Safak M. Members of the AP-1 Family, c-Jun and c-Fos, functionally interact with JC virus early regulatory protein large T antigen. J Virol. 2003;77(9):5241–5252. doi: 10.1128/JVI.77.9.5241-5252.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger T, Shenk T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J Virol. 1993;67(12):7556–7560. doi: 10.1128/jvi.67.12.7556-7560.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolzau T, Hansen RS, Zahra D, Reddel RR, Braithwaite AW. Inhibition of SV40 large T antigen induced apoptosis by small T antigen. Oncogene. 1999;18(40):5598–5603. doi: 10.1038/sj.onc.1202942. [DOI] [PubMed] [Google Scholar]
- Kremmer E, Ohst K, Kiefer J, Brewis N, Walter G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol Cell Biol. 1997;17(3):1692–1701. doi: 10.1128/mcb.17.3.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krynska B, Otte J, Franks R, Khalili K, Croul S. Human ubiquitous JCV(CY) T-antigen gene induces brain tumors in experimental animals. Oncogene. 1999;18(1):39–46. doi: 10.1038/sj.onc.1202278. [DOI] [PubMed] [Google Scholar]
- Lashgari MS, Tada H, Amini S, Khalili K. Regulation of JCVL promoter function: transactivation of JCVL promoter by JCV and SV40 early proteins. Virology. 1989;170(1):292–295. doi: 10.1016/0042-6822(89)90381-4. [DOI] [PubMed] [Google Scholar]
- Lin SS, Bassik MC, Suh H, Nishino M, Arroyo JD, Hahn WC, Korsmeyer SJ, Roberts TM. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. J Biol Chem. 2006;281:23003–23012. doi: 10.1074/jbc.M602648200. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. J Virol. 1990;64(12):5812–5822. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Factors contributing to the restricted DNA replicating activity of JC virus. Virology. 1991;180(1):306–317. doi: 10.1016/0042-6822(91)90035-a. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Haggerty S, Frisque RJ. DNA replication of chimeric JC virus-simian virus 40 genomes. Virology. 1994;204(2):819–822. doi: 10.1006/viro.1994.1600. [DOI] [PubMed] [Google Scholar]
- Major EO, Ault GS. Progressive multifocal leukoencephalopathy: clinical and laboratory observations on a viral induced demyelinating disease in the immunodeficient patient. Curr Opin Neurol. 1995;8(3):184–190. [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A. 1985;82(4):1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateer SC, Fedorov SA, Mumby MC. Identification of structural elements involved in the interaction of simian virus 40 small tumor antigen with protein phosphatase 2A. J Biol Chem. 1998;273(52):35339–35346. doi: 10.1074/jbc.273.52.35339. [DOI] [PubMed] [Google Scholar]
- Michelson S, Turowski P, Picard L, Goris J, Landini MP, Topilko A, Hemmings B, Bessia C, Garcia A, Virelizier JL. Human cytomegalovirus carries serine/threonine protein phosphatases PP1 and a host-cell derived PP2A. J Virol. 1996;70(3):1415–1423. doi: 10.1128/jvi.70.3.1415-1423.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Barrett RE, Britton CB, Tapper ML, Bahr GS, Bruno PJ, Marquardt MD, Hays AP, McMurtry JG, 3rd, Weissman JB, Bruno MS. Progressive multifocal leukoencephalopathy in a male homosexual with T-cell immune deficiency. N Engl J Med. 1982;307(23):1436–1438. doi: 10.1056/NEJM198212023072307. [DOI] [PubMed] [Google Scholar]
- Mungre S, Enderle K, Turk B, Porras A, Wu YQ, Mumby MC, Rundell K. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-t antigen in vitro decrease viral transformation. J Virol. 1994;68(3):1675–1681. doi: 10.1128/jvi.68.3.1675-1681.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Craig L, Machleidt T, Sontag E. Simian virus 40 small tumor antigen induces deregulation of the actin cytoskeleton and tight junctions in kidney epithelial cells. J Virol. 2003;77(5):2807–2818. doi: 10.1128/JVI.77.5.2807-2818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 2005;6(5):452–457. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60(1):167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- Porras A, Bennett J, Howe A, Tokos K, Bouck N, Henglein B, Sathyamangalam S, Thimmapaya B, Rundell K. A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small-t antigen and is required for transformation in small-t antigen-dependent assays. J Virol. 1996;70(10):6902–6908. doi: 10.1128/jvi.70.10.6902-6908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras A, Gaillard S, Rundell K. The simian virus 40 small-t and large-T antigens jointly regulate cell cycle reentry in human fibroblasts. J Virol. 1999;73(4):3102–3107. doi: 10.1128/jvi.73.4.3102-3107.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C. The replication functions of SV40 T antigen are regulated by phosphorylation. Cell. 1990;61(5):735–738. doi: 10.1016/0092-8674(90)90179-i. [DOI] [PubMed] [Google Scholar]
- Raj GV, Khalili K. Transcriptional regulation: lessons from the human neurotropic polyomavirus, JCV. Virology. 1995;213(2):283–291. doi: 10.1006/viro.1995.0001. [DOI] [PubMed] [Google Scholar]
- Rundell K, Parakati R. The role of the SV40 ST antigen in cell growth promotion and transformation. Semin Cancer Biol. 2001;11(1):5–13. doi: 10.1006/scbi.2000.0341. [DOI] [PubMed] [Google Scholar]
- Sadowska B, Barrucco R, Khalili K, Safak M. Regulation of human polyomavirus JC virus gene transcription by AP-1 in glial cells. J Virol. 2003;77(1):665–672. doi: 10.1128/JVI.77.1.665-672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Gallia GL, Ansari SA, Khalili K. Physical and functional interaction between the Y-box binding protein YB-1 and human polyomavirus JC virus large T antigen. J Virol. 1999a;73(12):10146–10157. doi: 10.1128/jvi.73.12.10146-10157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Gallia GL, Khalili K. Reciprocal interaction between two cellular proteins, Puralpha and YB- 1, modulates transcriptional activity of JCVCY in glial cells. Mol Cell Biol. 1999b;19(4):2712–2723. doi: 10.1128/mcb.19.4.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75(3):1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Sadowska B, Barrucco R, Khalili K. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol. 2002;76(8):3828–3838. doi: 10.1128/JVI.76.8.3828-3838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Major E, Khalili K. Human polyomavirus, JC virus, and progressive multifocal encephalopathy. In: Howard IG, Gendelman E, Everall Ian Paul, Lipton Stuart A, Swindells Susan, editors. The Neurology of AIDS. Oxford University Press; New York: 2005. pp. 461–474. [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80(8):3893–3903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidtmann KH, Mumby MC, Rundell K, Walter G. Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: inhibition by small-t antigen. Mol Cell Biol. 1991a;11(4):1996–2003. doi: 10.1128/mcb.11.4.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidtmann KH, Virshup DM, Kelly TJ. Protein phosphatase 2A dephosphorylates simian virus 40 large T antigen specifically at residues involved in regulation of DNA-binding activity. J Virol. 1991b;65(4):2098–2101. doi: 10.1128/jvi.65.4.2098-2101.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenk T, Carbon J, Berg P. Construction and analysis of viable deletion mutants of simian virus 40. J Virol. 1976;18:664–671. doi: 10.1128/jvi.18.2.664-671.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R, Kleinberger T. Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J Virol. 1998;72(4):2975–2982. doi: 10.1128/jvi.72.4.2975-2982.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JA, Khoury G, Jay G, Howley PM, Scangos GA. Early regions of JC virus and BK virus induce distinct and tissue-specific tumors in transgenic mice. Proc Natl Acad Sci U S A. 1986;83(21):8288–8292. doi: 10.1073/pnas.83.21.8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell. 1993;75(5):887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- Sontag E, Sontag JM, Garcia A. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-kappaB activation. EMBO J. 1997;16(18):5662–5671. doi: 10.1093/emboj/16.18.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Okada Y, Semba S, Orba Y, Yamanouchi S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, Sawa H. Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J Biol Chem. 2005;280(26):24948–24956. doi: 10.1074/jbc.M411499200. [DOI] [PubMed] [Google Scholar]
- Swenson JJ, Trowbridge PW, Frisque RJ. Replication activity of JC virus large T antigen phosphorylation and zinc finger domain mutants. J Neurovirology. 1996;2(2):78–86. doi: 10.3109/13550289609146541. [DOI] [PubMed] [Google Scholar]
- Tavis JE, Frisque RJ. Altered DNA binding and replication activities of JC virus T-antigen mutants. Virology. 1991;183(1):239–250. doi: 10.1016/0042-6822(91)90136-y. [DOI] [PubMed] [Google Scholar]
- Topp WC. Variable defectiveness for lytic growth of the dl 54/59 mutants of simian virus 40. J Virol. 1980;33(3):1208–1210. doi: 10.1128/jvi.33.3.1208-1210.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge PW, Frisque RJ. Identification of three new JC virus proteins generated by alternative splicing of the early viral mRNA. J Neurovirology. 1995;1(2):195–206. doi: 10.3109/13550289509113966. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Frisque RJ. Stability and function of JC virus large T antigen and T′ proteins are altered by mutation of their phosphorylated threonine 125 residues. J Virol. 2006;80(5):2083–2091. doi: 10.1128/JVI.80.5.2083-2091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugi S, Imamura T, Ricketts W, Olefsky JM. Protein phosphatase 2A forms a molecular complex with Shc and regulates Shc tyrosine phosphorylation and downstream mitogenic signaling. Mol Cell Biol. 2002;22(7):2375–2387. doi: 10.1128/MCB.22.7.2375-2387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varakis J, ZuRhein GM, Padgett BL, Walker DL. Induction of peripheral neuroblastomas in Syrian hamsters after injection as neonates with JC virus, a human polyoma virus. Cancer Res. 1978;38(6):1718–1722. [PubMed] [Google Scholar]
- Virshup DM, Kauffman MG, Kelly TJ. Activation of SV40 DNA replication in vitro by cellular protein phosphatase 2A. EMBO J. 1989;8(12):3891–3898. doi: 10.1002/j.1460-2075.1989.tb08568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virshup DM, Russo AA, Kelly TJ. Mechanism of activation of simian virus 40 DNA replication by protein phosphatase 2A. Mol Cell Biol. 1992;12(11):4883–4895. doi: 10.1128/mcb.12.11.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu Y. Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal. 2007;19(7):1372–1382. doi: 10.1016/j.cellsig.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11(4):1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Vitto MJ, Busby SA, Garcia BA, Kesler CT, Gioeli D, Shabanowitz J, Hunt DF, Rundell K, Brautigan DL, Paschal BM. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2A onto the androgen receptor. Mol Cell Biol. 2005;25(4):1298–1308. doi: 10.1128/MCB.25.4.1298-1308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cells. 2003;3(5):483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Zhou J, Pham HT, Ruediger R, Walter G. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem J. 2003;369(Pt 2):387–398. doi: 10.1042/BJ20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler K, Bui T, Frisque RJ, Grandinetti A, Nerurkar VR. A rapid in vitro polyomavirus DNA replication assay. J Virol Methods. 2004;122(1):123–127. doi: 10.1016/j.jviromet.2004.08.012. [DOI] [PubMed] [Google Scholar]