

The phthalate dioxygenase system (PDS), a two-component enzyme system in Burkholderia cepacia DB01, initiates the aerobic breakdown of phthalate by forming cis-4,5-dihydro-4,5-dihydroxyphthalate. PDS comprises the monomeric phthalate dioxygenase reductase (PDR), an enzyme that contains both FMN and a plant-type [2Fe-2S] ferredoxin (Fdx), and a dioxygenase (PDO), a hexamer of α subunits, each containing a Rieske [2Fe-2S] center and a ferrous mononuclear center [1]. FMN in PDR accepts a hydride from NADH and rapidly (≥1000 s−1 [2]) shuttles an electron to the [2Fe-2S] ferredoxin center. Fully reduced PDR reduces the Rieske centers in PDO that, in turn, transfer the electrons to the iron mononuclear centers during the course of the oxygenation reaction (Figure 1).
Figure 1.
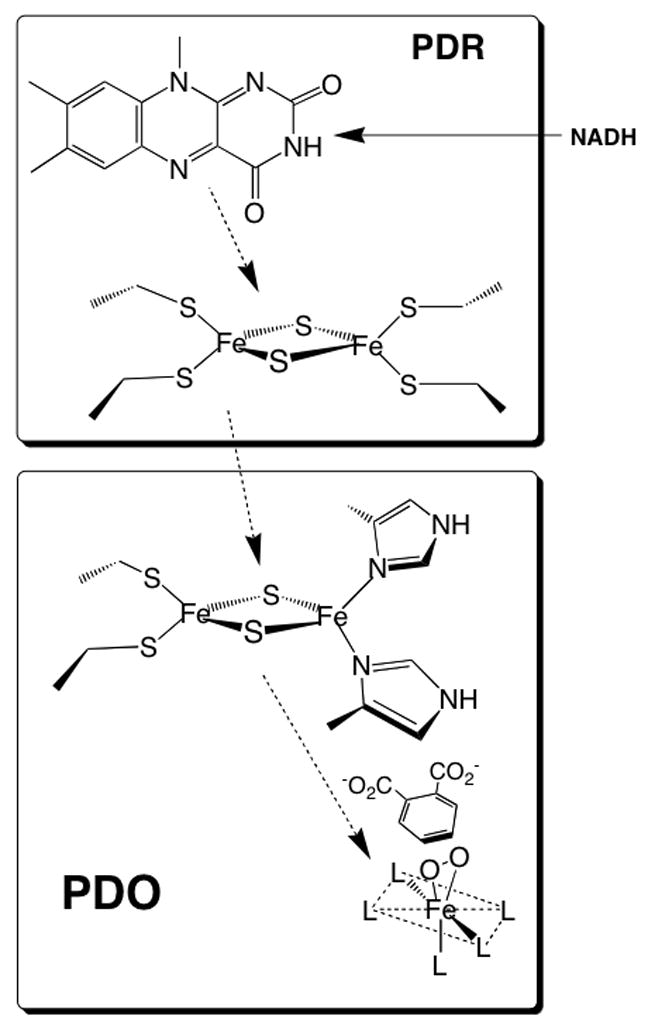
Phthalate dioxygenase system. This figure shows the various redox cofactors and the general pathway for electron transfer. The ligands of the iron mononuclear center are represented by “L”. The figure also shows the substrate (phthalate) and an oxygen molecule bound to the iron side-on, a binding arrangement demonstrated for the NDO system [25]. Because no X-ray structure of PDO is available, assignment of ligands and geometry is not specified. However, the X-ray structures of related dioxygenases (see below) shows that the mononuclear site can be described as a 2-His 1-carboxylate “facial triad” [26]. Likely ligands in PDO are the well conserved His-181, His-186, and Asp-358. Other studies have indicated that one or two waters are also ligated in various states of PDO[27].
It was shown previously that unlike most Rieske dioxygenases (e.g., naphthalene (NDO) [3], carbazole (CarDO)) [4], cumene [5], biphenyl [6] and many others), which are either α3β3 or α3 multimers, PDO is an α6 hexamer with two stacked α3 trimers positioned one on top the other [1]. However, the arrangements of monomers within these two trimers is most likely similar to the head-to-tail arrangement of subunits observed for the α3 subunits in other Rieske dioxygenases like NDO [3]. Published structures of Rieske dioxygenases show the Rieske center to be positioned in proximity of the Fe-mononuclear center of the adjacent subunit, with an interaction between the centers mediated by the aspartate located at the interface [7; 8; 9; 10; 11; 12]. Such proximity is enabled by the Rieske center of one subunit being imbedded within a cavity of the adjacent subunit. While such a structural arrangement facilitates efficient electron transfer from the Rieske center to the Fe-mononuclear center, it allows limited opportunities for the Fdx domain in PDR to come close to the Rieske center in PDO to effect efficient electron transfer. One such approach can be through the loop 105–125 in PDO that is positioned on top of the adjacent subunit. Located within 15 Å from the Rieske center, this loop is exposed to solvent and thus can serve as an anchoring point for PDR. The distal end of the loop seems to be rich in positively charged residues that could provide electrostatic protein-protein interactions between PDO and PDR (figure 2). In PDO, in particular, the distal end of the loop contains a string of amino acids (Lys117, Lys119, His120, Lys121, and Tyr123) that are candidates for participation in PDR binding.
Figure 2.

Sequence alignment of PDO with other Rieske oxygenases. Residues that are highly conserved relative to PDO are marked in yellow and residues that are similar to those in PDO are marked in green. Other residues conserved among several Rieske oxygenases are marked in blue. Cysteine (A) and histidine (B), which are [2Fe-2S] Rieske center ligands, and tryptophan W94 (C) are very well conserved among the Rieske dioxygenases. Also marked is a 105–125 putative PDR binding domain. Residues mutated in this study are shown in red.
It has been reported previously that for dioxygenase systems containing a reductase component and a Fdx component as two separate proteins, the specificity of oxygenase:Fdx interaction is much higher than that of the reductase:Fdx interaction. Thus, while it is often possible to substitute a ferredoxin reductase from an unrelated system without a significant loss of dioxygenase system activity, the respective ferredoxins cannot be easily replaced [13; 14; 15]. It is not surprising, therefore, that the loop thought to provide this specificity is not conserved, but is specific for each of the dioxygenases. Nevertheless, sequence analysis shows significant homology between the distal end of the loop in PDO and that of carbazole 1,9-dioxygenase (CarDO). The recently published crystal structure of the oxygenase-Fdx complex of carbazole 1,9 oxygenase [16] shows that the [2Fe-2S] center of its Fdx is located about 15 Å from the Rieske center in the dioxygenase. The Fdx is positioned in proximity to the distal end of the loop (115–125 in CarDO), which is rich in positively charged amino acids, similar to the loop in PDO. In particular, Lys120 and Lys122 of CarDO align with Lys119 and Lys121 in PDO, and the positively charged Arg118 in CarDO corresponds to Lys117 in PDO (figure 2). Tyrosine 124 in CarDO is also conserved (Tyr123 in PDO). We note that despite the fact that this region is well aligned between the sequences of PDO and CarDO, it is not well-conserved among all the Rieske oxygenases, including NDO. While the residues of the 115–125 domain in CarDO are clearly involved in oxygenase-Fdx interaction, it is also possible that the observed alignment with 105–125 loop in PDO is fortuitous and does not serve the same functional role as in CarDO.
Located directly adjacent to the Rieske center of CarDO is Trp95 (Trp94 in PDO, Trp106 in NDO), which is very well conserved among the Rieske dioxygenases (figure 2). This tryptophan is in van der Waals contact with the iron of the Rieske center. In CarDO it is also within 5 Å of the two glutamines (Gln115 and Gln119) of the 105–125 loop, and in van der Waals contact with residues (Ile354 and Phe256) of the 353–358 domain located on the adjacent CarDO subunit (see figures 3A and 3B). Residues in both of these domains are in van der Waals contact with residues surrounding the [2Fe-2S] center on the Fdx (including His48 and His68 that ligate the iron-sulfur center). A similar arrangement seems to exist in NDO where Trp106 is in van der Waals contact with Leu133, Leu128, and Tyr124, and with Leu384 from the adjacent NDO subunit. A crystal structure of NDO in complex with the relevant Fdx is not yet available, so it is not possible to verify whether these domains are similarly involved in NDO-Fdx interactions. If an analogous arrangement exists in PDO; Trp94 seems to be well-positioned to facilitate electron transfer from the Fdx in PDR to the Rieske center of PDO.
Figure 3.
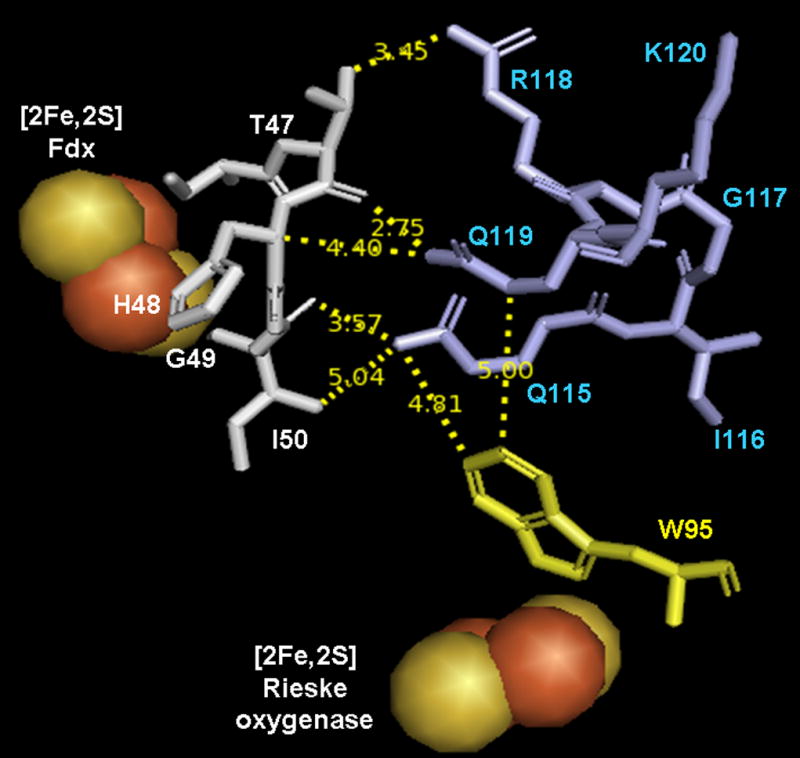
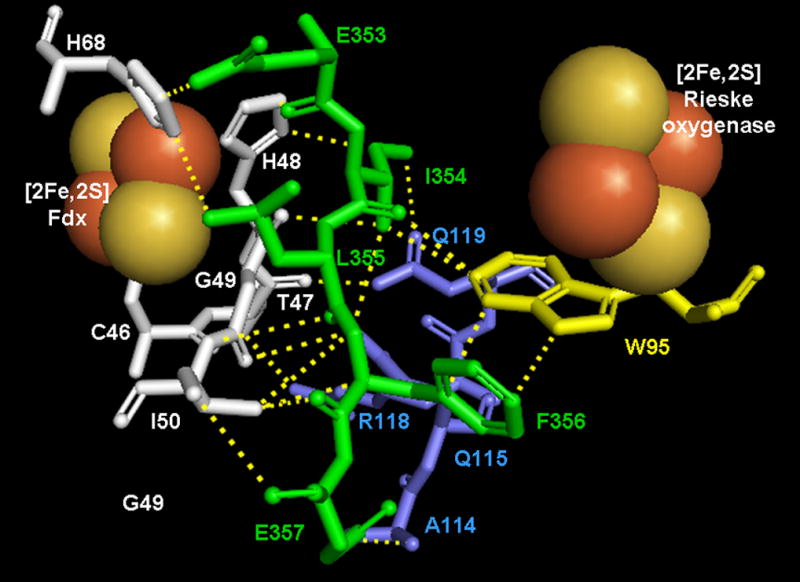
Amino acid arrangement in the vicinity of the [2Fe-2S] Rieske center of CarDO in its complex with Fdx (based on 2DE5). Fdx residues are shown in white, residues of CarDO subunits are shown in light blue if belonging to the same monomer as the Rieske center shown and in green if belonging to an adjacent oxygenase subunit. Conserved Trp 95 (Trp94 in PDO) is shown in yellow. A and B represent different views of the amino acids arrangement with A highlighting the interaction of Trp95 with the same subunit residues and B highlighting Trp95 interactions with residues from the adjacent oxygenase subunit. Dotted lines on B represent short range interactions (<4 Å) between selected residues. Figures were built in PyMOL 2006 from DeLano Scientific Inc., which incorporated Open-Source PyMOL 099rc6.
The work presented herein provides insight into the roles that the positively charged residues of the distal end of the 105–125 loop of PDO play in dioxygenase-reductase interactions. This work also elucidates the potential role of the conserved tryptophan (Trp94) in electron transfer from the Fdx center of PDR to the Rieske center in PDO.
MATERIALS AND METHODS
Construction of Plasmids
Genomic DNA that encodes PDO was inserted into the pET11A vector as described elsewhere [12]. Substitutions of target amino acids were carried out using the QuikChange site-direction mutagenesis kit (Stratagene) following the recommended protocol. For single amino acid substitutions (Lys117Ala, Lys119Ala, His120Ala, Lys121Ala, Tyr123Ala or Trp94Ala, Trp94Tyr, and Trp93Phe) the pET11a plasmid containing the nucleotide sequence encoding PDO from Burkholderia cepacia DB01 was used as a template with the complementary oligonucleotide primers given in Table 1. Substituted nucleotides are underlined in the sequence; all primers are shown as a 5′ to 3′ sequence). For the construction of the five-point substituted mutant (5-variant; substitutions Lys117Ala, Lys119Ala, His120Ala, Lys121Ala, and Tyr123Ala) a previously created plasmid with the Tyr123Ala point mutation was used as a template. The other four substitutions were done simultaneously using the primer shown in Table 1. Substituted nucleotides are underlined in the sequence by a solid line; substitutions done previously (mutation Tyr123Ala) are underlined by a dotted line. All primers were purchased from Integrated DNA Technologies, Inc. Successful substitutions were verified by nucleotide sequencing of the entire PDO gene. PDO was expressed in E. coli C41(DE3) cells purchased from Avidis. Protein expression and purification were performed as described previously [12]. Expression and purification of WT PDO and the variant PDOs were carried out in parallel to ensure maximum possible reproducibility of expression and purification conditions.
Table 1.
Primers used for the construction of PDO mutants.
| Mutation | Primers |
|---|---|
| PDO: Lys117Ala | GTAGCATGACCGACGCGGTCAAGCACAAGGCC |
| GGCCTTGTGCTTGACCGCGTCGGTCATGCTAC | |
| PDO: Lys119Ala | GACCGACAAGGTCGCGCACAAGGCCTATCCC |
| GGGATAGGCCTTGTGCGCGACCTTGTCGGTC | |
| PDO: His120Ala | CGACAAGGTCAAGGCCAAGGCCTATCCCGTGC |
| GCACGGGATAGGCCTTGGCCTTGACCTTGTCG | |
| PDO: Lys121Ala | ACCGACAAGGTCAAGCACGCGGCCTATCCCG |
| CGGGATAGGCCGCGTGCTTGACCTTGTCGGT | |
| PDO: Tyr123Ala | TCAAGCACAAGGCCGCTCCCGTGCAGGAATGG |
| CCATTCCTGCACGGGAGCGGCCTTGTGCTTGA | |
| PDO: 5 Point Mutation | GCATGACCGACGCGGTCGCGGCCGCGGCCGCTCCCGTGCAGGAA |
| TTCCTGCACGGGAGCGGCCGCGGCCGCGACCGCGTCGGTCATGC | |
| PDO: Trp94Tyr | CTGCGCTGCCTGTATCACGGCTATAAGTTTGACGTCG |
| CGACGTCAAACTTATAGCCGTGATACAGGCAGCGCAG | |
| PDO: Trp94Phe | GCGCTGCCTGTATCACGGCTTCAAGTTTGACGTC |
| GACGTCAAACTTGAAGCCGTGATACAGGCAGCGC | |
| PDO: Trp94Ala | GCTGCCTGTATCACGGCGCGAAGTTTGACGTC |
| GACGTCAAACTTCGCGCCGTGATACAGGCAGC | |
| PDR: 6 HIS insertion | 5′-CCGCAGAGCTGGTTCTCGACCTTCACCACCACCACCACCACTAAGTATCGCGGAGCAAAC-3′ |
| 5′-GTTTGCTCCGCGATACTTAGTGGTGGTGGTGGTGGTGAAGGTCGAGAACCAGCTCTGCGC-3′ |
PDR that contained a tag of six histidines on its C-terminus was constructed using the QuikChange II XL site-direction mutagenesis kit (Stratagene) following the recommended protocol and using the GenElute HP plasmid midiprep kit in a vacuum format (Sigma) for the isolation of plasmid DNA. Plasmid pET11a containing the nucleotide sequence that encodes for PDR from Burkholderia cepacia DB01 [12] was used as a template. The nucleotides coding for the six histidine residues were inserted using the complementary oligonucleotide primers listed in Table 1 (inserted histidine codons are underlined). Successful mutation of the codons was verified by nucleotide sequencing of the entire PDO gene. Protein was expressed in C41 E. coli (Avidis) and cell lysis was performed as described previously for PDO [12; 17]. After ultracentrifugation (1h, 105,000 g), clear lysate was collected and applied to a DEAE column equilibrated with 20 mM KPi, pH 7.8, and washed with 2–3 column volumes of the same buffer. Protein was eluted by a gradient of 20–150 mM KPi, with PDR eluting at about 80 mM KPi. Fractions showing a characteristic PDR absorbance spectrum (absorbance maxima at 345, 410, and 465 nm) were pooled and applied to a Ni-NTA Agarose (QIAGEN) column equilibrated with 50 mM KPi, pH 7.8, containing 5 mM imidazole. The protein was washed with 2 column volumes of the same buffer containing 20 mM imidazole, and then with 2 column volumes of the buffer containing 50 mM imidazole. His-tagged protein was eluted with the same buffer, but containing 500 mM imidazole. Hexa-his tagged PDR exhibited strong binding/precipitation on regenerated cellulose filters from Amicon Ultracentrifugal concentrators (Millipore). Similar binding to the filters was also observed in the absence of imidazole. Therefore, to eliminate imidazole, to effect a buffer exchange, and to concentrate the enzyme, fractions containing PDR were pooled, and the enzyme was fractionated by precipitation with 40 and 75 percent ammonium sulfate. The 40–75 percent fraction was re-suspended in 0.1M HEPES, pH 7.8, 5% glycerol and frozen in liquid nitrogen. Enzyme obtained by this protocol had activity of about 200 units/mg1 [18; 19] and was determined to be homogeneous by SDS-PAGE gel.
Concentrations of enzymes were determined spectrophotometrically using Δε575 = 2.38 mM−1cm−1 and Δε466= 17.54 mM−1cm−1 for the extinction coefficient difference between oxidized and reduced PDO and PDR, respectively. PDO activity was determined in steady-state assays by monitoring the change in absorbance at 340 nm due to consumption of NADH. Reaction mixtures contained 0.2 μM PDO, 3–5 mM phthalate, 105–250 μM NADH in 0.1 M HEPES, pH 7.8, at 22 °C. Reactions were initiated by the addition of 0.2 μM PDR. Ferrous ammonium sulfate (FAS), prepared as an anaerobic solution of known concentration, was added to the assays when necessary. The activity measured in this manner does not represent the maximum PDO activity, but rather the activity at 1:1 PDO:PDR stoichiometry under these specific conditions. These conditions were chosen because they are practical and reproducible. In addition, they are close to the physiological ratio between PDO and PDR in Burkholderia cepacia and also have been used previously in determination of the rates of electron transfer between PDO and PDR. Iron content in PDO was determined by Inductively Coupled Plasma High Resolution Mass Spectrometry (ICP) using a Finnegan Element Instrument from Thermo Finnegan Co.
PDO and PDR samples used for stopped-flow studies (20–40 μM before mixing) were made anaerobic by vacuum/gas exchange (Ar) 10 times over about 25 minutes, and the samples were finally overlaid with about 2 psi of purified argon. Reduction of PDR was achieved by anaerobic titration with 10 mM of anaerobically prepared sodium dithionite solution. PDO was reduced by photoreduction 5-deazaflavin as described previously [20; 21] using 10 mM glycine as an ultimate electron donor. The oxidation state of the enzymes was monitored through the changes in their optical spectra using a Shimadzu UV 2051PC spectrophotometer. Experiments were performed in 0.1 M HEPES, pH 7.8, 23 °C in the presence of 3 mM phthalate.
Kinetic data were acquired using a Kinetic Instruments, Inc., stopped-flow spectrophotometer in single-wavelength mode. Oxidation of the Rieske centers in PDO and their reduction by dithionite was observed at 575 nm, a wavelength that corresponds to the secondary maximum in the difference absorption spectrum between oxidized and reduced PDO. Reduction of the Rieske centers in PDO by PDRre was monitored by observing the absorbance changes at 610 nm or 640 nm, the wavelengths within the absorbance band of the flavin semiquinone form of PDR. Data collection and analysis were performed using program A, which employs the Marquardt-Levenberg algorithm [22], and was developed in our laboratory by Chung-Jen Chiu, Rong Chang, Joel Dinverno, and David P. Ballou, University of Michigan. Rates obtained from curve fitting (mean values from at least 5 independent experiments) were within 15% of each other unless specified otherwise.
Product analysis was performed using a Waters HPLC system equipped with a Waters 441 detector (λ=250 nm). An Aminex HPX-87H column running isocratically in 9 mM H2SO4 at 65 °C with 0.75 mL/min flow rate was used for product separation. Data was recorded and analyzed with the Empower software package (Waters). Samples for HPLC analysis were prepared by filtering out the enzyme with Microcon-YM30 concentrators (Amicon, Co.). Filtrate solutions were collected and stored at −20 °C until analysis was performed. Control experiments using previously extracted DHD verified that no DHD deterioration occurs during the freeze/thaw steps.
Bacterial growth media components were from Fisher Scientific and Difco, carbenicillin was from Apollo Scientific, IPTG was from Research Products International; other reagents were from Fisher Scientific and Sigma.
RESULTS AND DISCUSSION
Iron content and Steady-State Catalytic Activity of WT and mutant forms of PDO
Recombinant WT PDO as purified contained 2.6 ± 0.2 Fe/monomer and exhibited a steady-state turnover activity of 3.8 s−1 (Table 2) as measured by the steady-state assays described in Materials and Methods, similar to that reported previously [12].
Table 2.
Iron content and steady-state turnover activity of PDO WT and the mutants
| Fe/Rieske (± σ) | TN (± σ), s−1 | Coupling*, % | |
|---|---|---|---|
| WT | 2.5 ± 0.2 | 3.8 ± 0.3 | 100.0 ± 0.5 |
| K117A | 2.5 ± 0.2 | 2.7 ± 0.2 | 100.7 ± 1.0 |
| K119A | 2.9 ± 0.2 | 3.4 ± 0.2 | 100.1 ± 1.0 |
| H120A | 2.6 ± 0.2 | 3.0 ± 0.4 | 100.3 ± 1.0 |
| K121A | 2.6 ± 0.2 | 2.3 ± 0.4 | 100.3 ± 1.0 |
| Y123A | 3.2 ± 0.3 | 1.4 ± 0.2 | 100.1 ± 1.0 |
| 5 point mutation | 2.8 ± 0.3 | 0.35 ± 0.09 | 100.6 ± 1.0 |
| W94Y | 2.6 ± 0.2 | 0.9 ± 0.2 | 100.2 ± 1.0 |
| W94F | 2.4 ± 0.1 | 1.4 ± 0.2 | 100.9 ± 1.0 |
| W94A | 2.4 ± 0.1 | 1.0 ± 0.2 | 100.1 ± 1.0 |
Coupling is defined as the ratio between the amount of DHD produced and the amount of NADH oxidized in the course of steady state reaction.
PDO Variants
Point mutations ((Lys117Ala, Lys119Ala, His120Ala, Lys121Ala, or Tyr123Ala, as well as Trp94Ala, Trp94Phe, and Trp94Tyr) did not result in any significant changes in the amount of Fe present in mutant PDO samples as purified (Table 2). In steady-state assays, the turnover activity of the mutants was less than that of the WT enzyme. Of the five variants substituted in the 105–125 domain, the Tyr123Ala mutant exhibited the greatest decrease in steady-state activity, about 63 percent. Overall, for these five mutants, the activity in steady-state reactions was decreased on average by ~30 percent, compared to that in the WT PDO. The steady-state turnover activity of Trp94 variants was 2.5 – 4-fold less than that of the WT enzyme (Table 2).
As purified, the 5-variant, which contained all five point mutations (Lys117Ala, Lys119Ala, His120Ala, Lys121Ala, and Tyr123Ala), also contained an amount of iron similar to that in the WT PDO. Its steady-state turnover activity was, however, decreased more than 10-fold compared to that in WT PDO (Table 2).
Product Formation and Coupling of the Steady-State Reaction
For WT PDO, the catalysis of DHD formation is tightly coupled to the electron delivery from NADH (through the reductase (PDR)) [12; 23]. Similarly tight coupling (100 ± 1 percent) was also observed with all the PDO variants investigated in this study (see Table 2). Thus, substitutions of the positively charged residues in the 105–125 domain of PDO and substitution of the linked tryptophan-94 did not affect product formation and the coupling of electron transfer from PDR to DHD formation. This result is consistent with those mutations having little effect on the mononuclear site, where the reaction with oxygen takes place.
Kinetics of the Reaction of Reduced Forms of PDO with O2
The absorption spectrum of oxidized PDO in the visible region is dominated by the absorption of the Rieske center, which in the oxidized form has maxima at 465 and 575 nm and in the reduced form has significantly diminished absorbance in the visible region. This feature allows for convenient determination of the Rieske center redox state. As was shown previously [17], reduced Rieske centers of WT PDO become oxidized during catalysis by fast electron transfer to the iron in the mononuclear center, which reacts with O2. In the absence of PDR and phthalate, the kinetics of the oxidation has two phases, with rates of ~ 1 (kox1) and ~ 0.1 (kox2) s−1. These were previously attributed to electron transfers from two Rieske centers to the same mononuclear Fe(II) [17; 23] (see Figure 4A and 4B). The oxidation characterized by kox1 is presumed to be due to the Rieske center that is linked to the mononuclear center by Asp178. These electron transfers are significantly accelerated in conditions more like those in catalysis. For example, in the presence of oxidized PDR approximately 50% of the oxidation of the Rieske center occurs at 4–5 s−1, while in the presence of phthalate, kox1 increases to 40–50 s−1. When both PDR and phthalate are present, phases at both 4–5 and 40–50 s−1 can be observed. By contrast, if iron is not present in the mononuclear center, reduced Rieske centers are oxidized very slowly (at 1–3 × 10−3 s−1), presumably in a direct reaction between the Rieske [2Fe-2S] center and O2 [17]. For WT PDO used in this study the fast oxidation of the Rieske center that occurs in the presence of phthalate (kox1) was 49 s−1, and the slower oxidation, which is characteristic of electron transfer with no PDR present (kox2) was about 0.2 s−1 (see Table 3 and Figure 5). A small contribution of an even slower rate of oxidation of the Rieske center (~ 10−4 s−1) was observed in the samples of recombinant PDO and was attributed to the presence of damaged PDO subunits [12].
Figure 4.
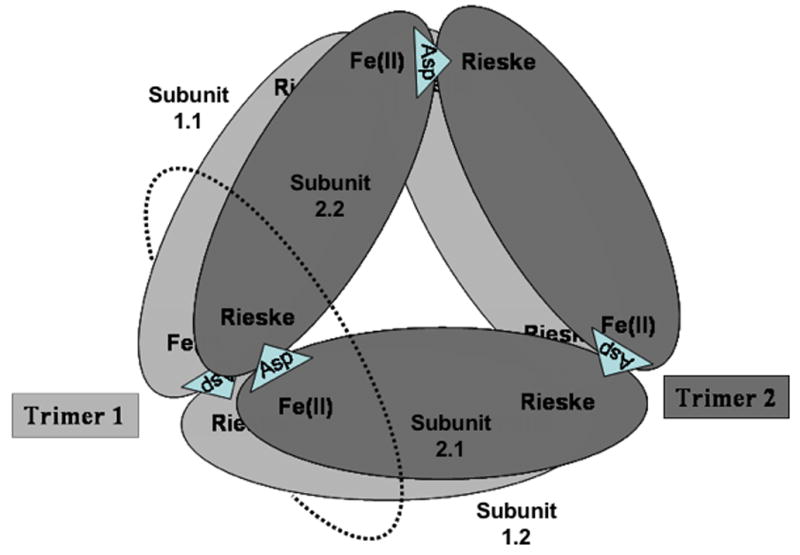
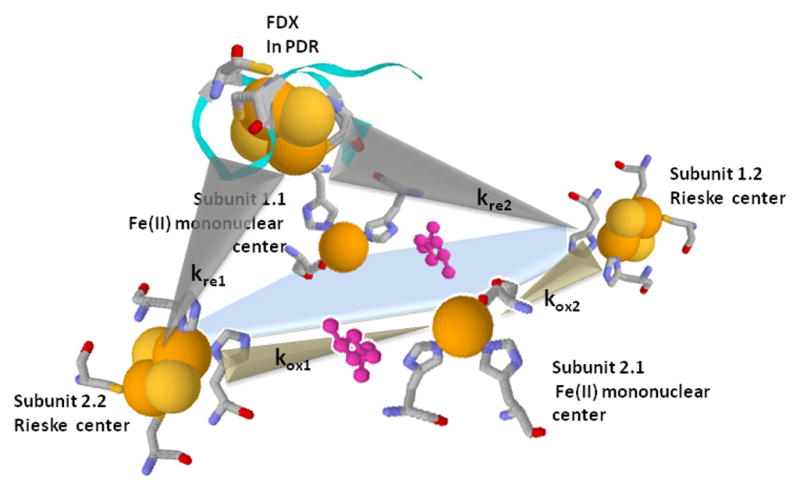
Electron transfers between the Rieske centers in PDO and the Fdx center in PDR. A. Proposed arrangement of subunits within the PDO hexamer. Bridging ASP178 is shown as a triangle on the appropriate subunit interfaces. Dotted circle indicates the plane of cross-section shown on figure 3B; B. Schematic of the cross-section of the PDO multimer in the plane defined by the dotted circle shown in 3A with PDR Fdx attached. Two Rieske centers and two Fe-mononuclear centers from four different PDO subunits are located proximally, forming a 2 Rieske - 2 Fe mononuclear active cluster. Each Rieske center is able to deliver electrons to either of the two mononuclear centers. However, only one of these electron transfers (kox1) is facilitated by a bridging aspartate (ASP178 in PDO, shown in rose), which is critical for rapid catalysis. The PDR Fdx domain that binds to PDO can potentially supply electrons to both Rieske centers within the active cluster; however, one center is “preferred”, with the electron transfer rate (kre1) about 20-fold higher than the other (kre2)
Table 3.
Rates of Rieske center reduction and oxidation in PDO WT and the mutants.
| Rieske center Reduction by PDR* | Rieske center Oxidation*** | |||||||
|---|---|---|---|---|---|---|---|---|
| kre1, s−1 | Are1, % | kre2, s−1 | Are2, % | kox1, s−1 | Aox1, % | kox2, s−1 | Aox1, % | |
| WT | 94 | 64 | 2.0 | 36 | 49 | 60 | 0.23 | 9 |
| K117A | 31 | 81 | 0.7 | 19 | 57 | 54 | 0.23 | 9 |
| K119A | 90 | 65 | 1.1 | 35 | 53 | 41 | 0.16 | 10 |
| H120A | 72 | 69 | 2.2 | 31 | 59 | 68 | 0.35 | 9 |
| K121A | 80 | 61 | 1.8 | 39 | 49 | 79 | 0.17 | 10 |
| Y123A | 50 | 69 | 2.9 | 31 | 49 | 60 | 0.18 | 5 |
| 5 point variant | 16 | 74 | 1.6 | 26 | 59 | 63 | 0.08 | 5 |
| W94Y | 22 | 100 | n.d. ** | n.d. | 35 | 52 | n.d. ** | n.d. |
| W94F | 25 | 100 | n.d. ** | n.d. | 32 | 40 | n.d. ** | n.d. |
| W94A | 74 | 72 | 1.4 | 28 | 19 | 31 | 0.11 | 11 |
Anaerobicallyreduced PDR (15 μM) was mixed with anaerobic oxidized PDO (15f μM) (concentration before mixing). Enzymes were in 0.1M HEPES, pH 7.8, containing 3 mM phthalate. Are1 and Are2 denote the fractions of the Rieske centers reduced based on the two fastest observed phases of semiquinone formation in PDR. In all samples the fast phase (kre1) accounted for reduction of only 35–40% of all available Rieske centers.
n.d. – phase was not detected. An increased contribution of a slower, 1–4 10−2 s−1 reduction phase (for Rieske center reduction by PDR) or 1–4 10−3 s−1 oxidation phase (for Rieske center oxidation) was observed but not evaluated in this study, because such phases are not physiologically significant.
Anaerobically reduced PDO (20 μM) in 0.1M HEPES, pH 7.8 in the presence of 3 mM phthalate was mixed with the same buffer containing 3 mM phthalate and 250 μM O2 (concentration before mixing). Aox1 and Aox2 denote the fraction of the Rieske centers oxidized during the respective oxidation phase. Total contribution of these two fast phases account to less than 100% of all available Rieske centers due to the contributing of slower (> 5 10−3 s−1) oxidation phases.
Figure 5.
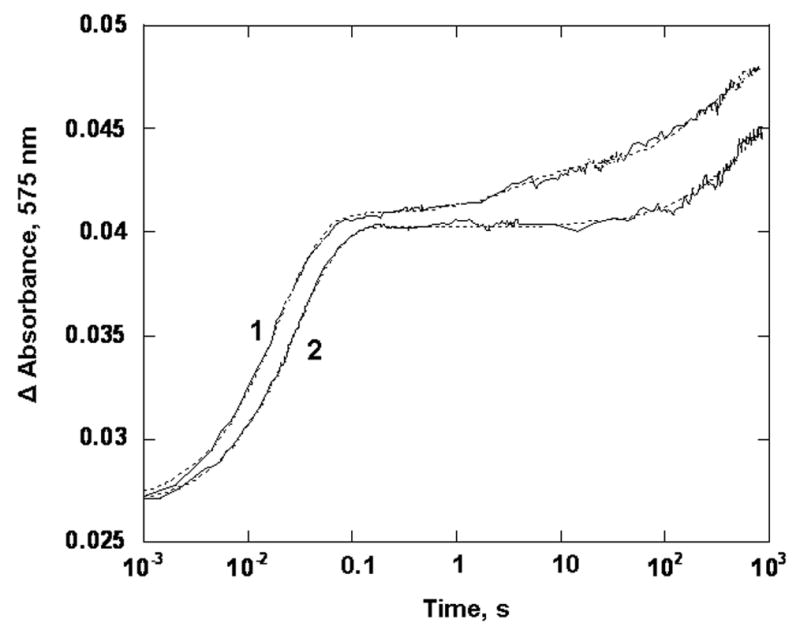
Oxidation of reduced PDO by oxygen. PDO (20 μM) was mixed with 250 μM O2 in 0.1M HEPES pH 7.8, in the presence of 3 mM phthalate at 22 °C. Presented are experimental trances (solid line) and the fits obtained with the parallel reaction model (dotted lines) for WT PDO (1) and the W94Y variant (2), All concentrations are those before mixing in the stopped-flow spectrophotometer. Traces were recorded at 575 nm and truncated at 900 s for presentation.
PDO Variants
Rieske centers of PDO with single point mutations (Lys117Ala, Lys119Ala, His120Ala, Lys121Ala, and Tyr123Ala) as well as the 5-variant enzyme reacted with oxygen at essentially the same rates as WT PDO (Table 3). Two fast phases, one with a rate of about 50 s−1 (kox1, Table 3), characteristic for the oxidation of the Rieske center in the presence of phthalate, and the second one with a rate of about 0.1–0.2 s−1 (kox2, Table 3), were present in all of the samples.
For Trp94 variants, the effect of substituting of Trp94 with Tyrosine or Phenylalanine on the oxidation properties of the Rieske center in PDO was somewhat different from that observed in the alanine substituted mutant. For both the W94Y and the W94F variants the fast rates of Rieske center oxidation (kox1, Table 3 and Figure 5) were not significantly affected by the substitution, decreasing only about 30% relative to the WT PDO. However, these mutations had a significant impact on the slower rates of Rieske center oxidation (kox2, Table 3 and Figure 5), resulting in a 10–20-fold decrease in the observed oxidation rates. Substitution of Trp94 with alanine had essentially opposite effects, with the fast rates of Rieske center oxidation affected by the mutation to a much greater extent (up to 4-fold decrease relative to WT PDO) than the slower rates (about a 2-fold decrease).
At saturating PDR concentrations WT PDO exhibits a steady-state TN of ~25 s−1. Thus, the slower rate of oxidation of the Rieske center (kox2) observed in single turnover experiments (even when it is accelerated to about 4.5–5 s−1 in the presence of PDR) seems too slow to be significant for steady-state catalysis. In steady-state catalysis both electrons required for the oxygenation reaction are delivered to the mononuclear center by a single Rieske center, most likely at rates kox1 of ~40 s−1. The Rieske center, after delivering the first electron, is re-reduced by the reductase very rapidly (kre1 of 100–200 s−1) so that this step is not observed. In single turnover (with no reductase present), the electron supply is limited to that available in the pre-reduced Rieske centers. Therefore, because the mononuclear iron in PDO is in the ferrous form at the completion of the reaction, the electrons required for dioxygenation must come from two Rieske centers, resulting in only 0.5 DHD being formed per Rieske center oxidized [23]. In single turnover experiments the slower electron transfer, which likely occurs between the Rieske and mononuclear centers that are not connected by a bridging aspartate (see Figure 4B), also contributes to DHD formation, albeit more slowly, and allows for full utilization of all electrons stored by the Rieske centers of PDO. When NADH is not abundant in the cell, a second electron from the reductase may be unavailable. If the mononuclear ferrous ion binds O2 and receives an electron from its Rieske center partner, a form of activated oxygen could develop and lead to deleterious reactive oxygen species. However, as shown in single turnover experiments, the second nearby reduced Rieske center can contribute its electron to permit the formation of product and thereby prevent the release of such reactive oxygen species. This second electron transfer (with the designated rate kox2, Figure 4B) was not observed in W94Y and W94F mutants, indicating that W94 plays an integral role in delivering the electrons from the Rieske center to mononuclear center by this “alternate” route.
On average, “as purified” PDO contains 2.6–2.7 atoms of Fe(II) per monomer (see Table 2), which indicates that in these enzymes up to 40% of the mononuclear centers are not populated with iron. Consistent with that data, a slower oxidation phase with the rate of 1–3 10−3 s−1 was also present in all the samples; this rate was indicative of the oxidation of that fraction of Rieske centers that did not have access to mononuclear centers containing Fe(II). A small, but somewhat variable very slow (~ 10−4 s−1) oxidation phase of the Rieske center, which is probably due to damaged PDO subunits, was also present in both WT and mutant PDO. The relative contributions of these slow phases were not evaluated.
PDO:PDR interaction
We used steady-state kinetics to determine the Km values for PDR. Fitting the data to the Michaelis-Menten equation gave values for the WT and mutant PDO shown in Table 4. The single residue substitution mutants and WT PDO used in this study exhibit Km values for PDR in the reaction with PDO that are 5–10 μM. The variant containing all 5 mutations in the putative PDR binding domain, however, exhibited a 5-fold higher Km value compared to that of WT PDO. Analogous constants for W94 variants were not determined.
Table 4.
Km values for PDR in the PDOS
| PDO sample | Km, μM |
|---|---|
| WT | 8.2 ± 0.8 |
| K117A | 6.7 ± 0.4 |
| K119A | 7.3 ± 0.5 |
| H120A | 5.6 ± 0.6 |
| K121A | 7.1 ± 0.8 |
| Y123A | 6.3 ± 0.4 |
| 5 point mutation | 40 ± 4 |
Kinetics of the reaction of reduced PDR with oxidized forms of PDO
Fully reduced PDR (3 electrons) transmits electrons from its ferredoxin [2Fe-2S] center to the Rieske [2Fe-2S] center of PDO. Intramolecular electron transfer in PDR is very fast (≥ 1050 s−1, [2]) so that after reducing the Rieske center, the ferredoxin center in PDR is immediately re-reduced by the FMNH−, forming the flavin semiquinone. This form of PDR form has distinct absorbance between 600–670 nm, an area of relatively small spectral contribution from PDO. This property allows for monitoring the rates of PDR flavin semiquinone formation, and by extension, the reduction of the Rieske centers in PDO. In WT PDO the steady-state turnover rate2 at 1:1 PDO (Rieske):PDR stoichiometry was about 4 s−1. Thus, in an anaerobic reaction of equimolar amounts of reduced PDR with oxidized PDO only kinetic phases of semiquinone formation with rates ≥ 4 s−1 would be relevant for catalysis. In WT PDO there are two kinetic phases related to Rieske center reduction, as assessed by corresponding FMN semiquinone formation in PDR: one with a rate of ~ 105 s−1 and a second one with a rate of ~ 2 s−1 (kre1 and kre2 in Table 3). Based on the kinetic data and on the stoichiometries of the PDO:PDR interaction, it was previously proposed that under these conditions only one PDR molecule can bind and effectively transfer an electron to one of the two closely positioned Rieske centers in the PDO multimer (located on two adjacent PDO monomers – Fig. 4) [12]. Because the mononuclear iron is already ferrous, no further electrons can be delivered to this active site pair (when no O2 is present). In the slower phase, additional semiquinone appears, indicating that a second PDR must be involved in reducing the second Rieske center. This step likely requires dissociation of the first PDR and association of the second PDR, thus potentially limiting the rate of reduction of the second Rieske center (kre2). The fast phase of flavin semiquinone formation observed in the reaction of reduced PDR with WT PDO in this study was about 2-fold less than that observed previously [12; 17]. This difference might be due to lower specific activity observed for the PDR sample used in the experiments (about 80 units/mg as compared to 200 units/mg reported previously [18; 19]). Nevertheless, the comparison can be made between the rates of observed electron transfer from the PDRred to PDOox in WT and in the variants.
Both fast and slow phases of FMN semiquinone formation were measured in the reaction of reduced PDR with oxidized PDO variants. In all cases, the fast phase rates (kre1) of reduction of the Rieske were less than that observed in the WT enzyme.
PDO Variants
Single point mutations had a limited impact on the rate of the fast phase of reduction of the Rieske center by PDR. The amplitude of the first phase compared to that in WT varied from about 96% for Lys119Ala (not a statistically significant effect) to about 50% for Tyr123Ala, and to about 35% for Lys117Ala (see Table 3 and Figure 6). In the 5-variant PDO an even larger effect was observed on kre1 (to 16 s−1), which is ~ 6-fold lower than that in the WT enzyme. This diminished reduction rate plays a major role in the 10-fold decrease of the steady-state turnover rate for this variant. However, the slow phase of Rieske center reduction (kre2) appears to be much less perturbed by the substitutions (see Table 3 and Figure 6). The faster pathway is most likely the primary route of electron transfer in catalysis (see Discussion).
Figure 6.
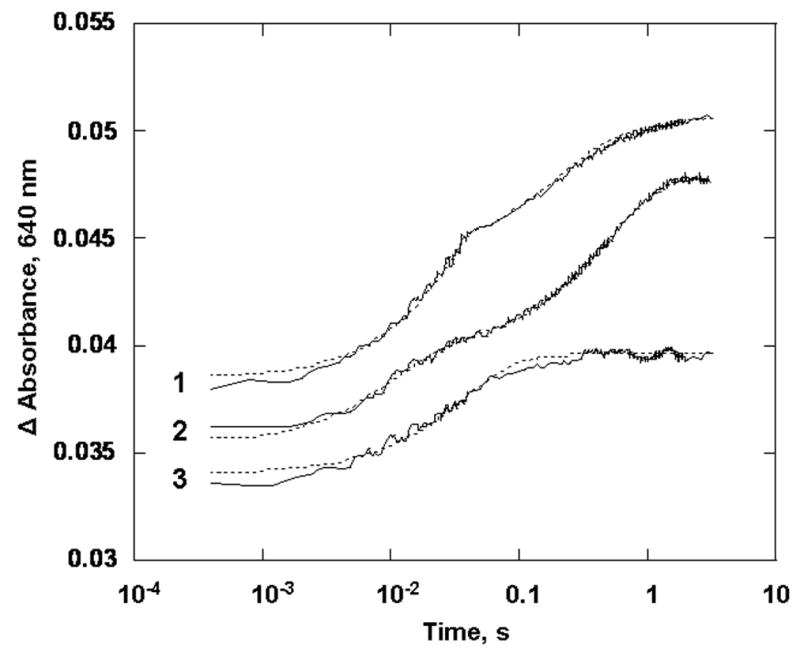
Reaction of oxidized PDO (15 μM) with a stoichiometric amount of reduced PDR ([PDO Rieske]:[PDR] = 1:1) in 0.1M HEPES pH 7.8, in the presence of 3 mM phthalate at 22 °C. Experimental traces (solid line) and fits obtained with the parallel reaction model (dotted lines) for K117A (1), WT (2) and W94F variant (3). Traces are offset from each other by 0.01 absorbance units for clarity. All concentrations are those before mixing in the stopped-flow spectrophotometer. Presented traced were recorded at 640 nm.
Substitution of Trp94 with Tyrosine or Phenylalanine affected both phases of the reduction of the Rieske center by the Fdx center in PDR. For W94F and W94Y there was ~ 4-fold decrease in electron transfer rates (kre1) for the faster phase. The slower phase of electron transfer (kre2) was not observed for these variants (see Figure 6, Table 3 and comments therein). In contrast, the W94A variant behaved similarly to WT PDO. We have no explanation for why substitution of tryptophan with tyrosine or phenylalanine had a greater effect on electron transfer rates than substitution with alanine. The slower electron transfer step in the reduction of the Rieske center (that leads to formation of the semiquinone form of PDR) is not likely to be important in steady-state catalysis, which under these conditions (1:1 PDO to PDR stoichiometry) is about 4 s−1 (Table 2). The fact that the slower electron transfer step (kre2) was not observed in W94Y and W94F mutants indicates that these substitutions resulted in a significant rearrangement of PDO subunits such that the Fdx domain in PDR does not have optimal access to the second Rieske center (e.g., the Rieske center on subunit 1.2 of Fig. 4B) of any of the PDO trimer interfaces.
Kinetics of PDO Rieske center reduction by sodium dithionite
The Rieske center in PDO can be reduced by dithionite; however, in WT PDO the reaction is extremely slow [11]. As was demonstrated previously, the rate of this reaction is determined by the conformational state of the PDO multimer. When the subunits are tightly packed, as in the WT PDO hexamer, dithionite does not have easy access to the Rieske center. However, disruption of the interface between the subunits (e.g. by Asp178 substitutions in PDO) increased the rate of Rieske center reduction by dithionite by several orders of magnitude [11], presumably by allowing better solvent (and dithionite) access to the center.
PDO variants
Substitutions of the residues in the distal end of the 105–125 domain in PDO did not have substantial effects on the rates of Rieske center reduction by sodium dithionite. More than 90 percent of the centers were reduced at rates of 1–4 10−3 s−1, with a small number of Rieske centers (less than 10% on average) reduced at a faster rate (0.1–0.4 s−1), similar to that observed in WT PDO (Table 5).
Table 5.
Rates of Rieske center reduction by sodium dithionite in PDO WT and the mutants.
| Rieske center reduction* | ||||
|---|---|---|---|---|
| k1, s−1 | A1 (%) | k2, s−1 | A2 (%) | |
| WT | 0.12 | 10 | 1.0 10−3 | 90 |
| K117A | 0.24 | 2 | 0.8 10−3 | 98 |
| K119A | 0.31 | 13 | 4.2 10−3 | 87 |
| H120A | 0.27 | 8 | 1.6 10−3 | 92 |
| K121A | n.d. ** | n.d. | 3.6 10−3 | 100 |
| Y123A | 0.48 | 9 | 2.2 10−3 | 91 |
| 5 point mutation | 0.32 | 6 | 1.6 10−3 | 94 |
| W94Y | 3.1 | 2 | 0.012 | 98 |
| W94F | n.d. | n.d. | 0.040 | 100 |
| W94A | 5.9 | 100 | n.d. | n.d. |
An anaerobic solution of PDO (30–40 μM) in 0.1M HEPES, pH 7.8 in the presence of 3 mM phthalate was mixed with anaerobically prepared 2 mM sodium dithionite in the same buffer. Concentrations are presented before mixing.
n.d. – phase not detected
Substitutions of Trp94 in PDO with aromatic amino acids (tyrosine and phenylalanine) brought about more than 10-fold increases in observed rates of reduction by dithionite (Table 5 and Figure 7). Substitution with alanine (Trp94Ala) resulted in about 6,000-fold faster reduction by dithionite than with WT PDO. This rapid rate of reduction was similar to that observed for the Asp178 variants of PDO, in which the substitutions of this residue resulted in increased solvent access to the Rieske center [11]. Overall, these results indicate that W94 substitutions bring about conformational changes in the PDO multimer that increase solvent access to the Rieske centers.
Figure 7.
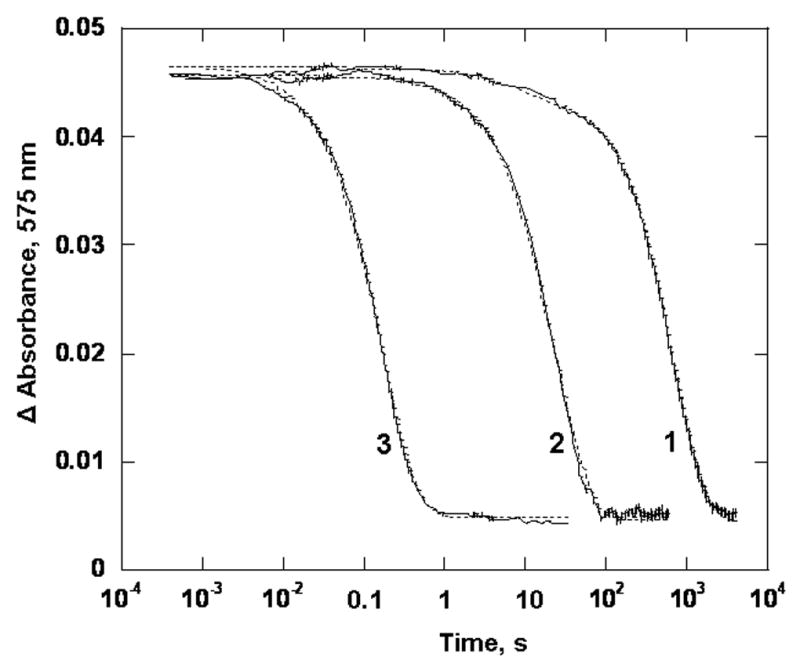
Reaction of anaerobic oxidized PDO (40 μM) with 1 mM anaerobically prepared sodium dithionite in 0.1 M HEPES pH 7.8, in the presence of 3 mM phthalate at 22 °C. Presented are experimental trances (solid line) and the fits obtained with the parallel reaction model (dotted lines) for WT PDO (1), variants W94F (2) and W94A (3), Traces were recorded at 575 nm.
CONCLUSIONS
As described above, the distal end of 105–125 loop in PDO appears to be at least part of the binding site for PDR. Results presented in this study clearly demonstrate that this region is important for dioxygenase-reductase interactions. Although no single residue among those investigated in this study is by itself solely responsible for the interaction, cumulatively, the residues in the 105–125 loop exert significant influence both on the binding of the reductase to its oxidative counterpart and on the rate of electron transfer from the Fdx center in the reductase to the Rieske center in PDO. However, the fact that even in the 5-variant PDO this electron transfer rate is decreased by only ~6-fold, correlating with a similar magnitude decline in the steady-state turnover rate, shows that electron transfer was not completely disrupted. Thus, additional residues must also be involved in electron transfer within the PDO-PDR complex.
Only about 35–40% of the Rieske centers are reduced in the fast phase (see comment to Table 3). As mentioned, at the concentrations of PDR used in these experiments it is likely that only one PDR binds at the site where two Rieske and two mononuclear centers interact (Fig. 4b). Thus, while this PDR is most likely to interact preferentially with one of the Rieske centers, resulting in electrons being transferred at fast (kre1) rate, a second reductase molecule is necessary to provide electrons for the reduction of the second Rieske center. Such an electron transfer, with a slower rate of kre2 is likely to be rate limited by dissociation of the first and binding of the second PDR molecules. This proposed mechanism is likely not relevant for reduction of the Rieske centers under steady-state turnover conditions when the Rieske center, after delivering an electron to the mononuclear center at 40 s−1, would become available to receive the second electron from the same partially oxidized PDR without the need for its dissociation from the PDO. The slower phase of electron transfer from a Fdx center in PDR to the Rieske center in PDO was essentially unaffected by the substitutions in the 105–125 loop. This result could be due to the dissociation and/or binding of the second PDR being the rate limiting step for the reduction of the second Rieske center.
Positioned in van der Waals contact with both the Rieske center and the residues linked to the [2Fe-2S] center of the Fdx center (including those in the 105–125 loop), Trp94 appeared to be in a position to facilitate electron transfer between the Fdx center in PDR and the Rieske center in PDO. However, substitutions of this well conserved residue failed to achieve major disruption of this electron transfer. The changes observed in these variants are probably the result of mutation-induced structural modifications of the Rieske center environment and argue against direct involvement of Trp94 in electron transfer pathway. Nevertheless, all three variants (Trp94Tyr, Phe, and Ala) exhibited significantly increased rates of the reduction of Rieske center by sodium dithionite, with the effect especially prominent in the case of the substitution with alanine (6,000-fold increase in rates of reduction of Rieske centers). Such changes due to the solvent accessibility of the Rieske center could be the result of mutation-induced disruption of the interaction between Trp94 and the adjacent residues on the neighboring oxygenase subunit (Ile354 and Phe356 in CarDO, and Leu384 in NDO, which in PDO may be equivalent to Ile379 and/or Thr380). Thus, it appears that Trp94 plays a structural role in PDO and contributes to the development of a hydrophobic environment of the Rieske center. Its replacement with tyrosine or phenylalanine, and to an even greater extent by alanine, loosens the structure, possibly by allowing the neighboring subunit to move father away, making the Rieske center more accessible to solvent.
Because Trp94 only indirectly affects electron transfer between the Fdx in PDR and the Rieske center in PDO, other residues must fulfill this role. The sequence alignment of PDO with various other Rieske proteins and the analysis of crystal structures of related enzymes reveals that Arg73 and Asp377 are well-conserved and could also be facilitators of electron transfer between PDR and PDO. In particular, the CarDO-Fdx crystal structure (2DE5) indicates that in CarDO Arg72 (corresponds to Arg73 in PDO and Arg84 in NDO) is located within van der Waals distance of its oxygenase Rieske center and of Glu353 on the neighboring oxygenase subunit (Asp377 in PDO, Asp 382 in NDO), which, in turn, is within 5 Å of the [2Fe-2S] of the Fdx center. Another possible, but not highly conserved electron transfer pathway, could involve Asp359 (possibly corresponding to Asp386 in PDO and Glu391 in NDO), located about 7 Å from the Rieske center on the adjacent oxygenase subunit. The Asp359 neighbor, Glu360, (possibly similar to Asp392 in NDO) appears to be linked to Ile50 of the Fdx, which is van der Waals contact with the [2Fe-2S] center of the Fdx. This complex arrangement, with electron transfer from Fdx to the Rieske center in the oxygenase that is facilitated by residues located on two different oxygenase subunits can potentially be stabilized in CarDO by Glu357 (possibly correlated to Glu381in PDO). Future studies will examine the role these residues play in electron transfers in PDO system and in the catalysis of phthalate dioxygenation.
Acknowledgments
We thank Dr. T. Huston, University of Michigan Geological Services for his help in obtaining the iron content data for PDO using ICP, Alex Pinto for his assistance in expression and purification of the HIS-tagged PDR and Dr. L. Rider for general assistance and advice.
Supported by NIH grant (GM20877 to D.P.B.)
Abbreviations
- PDO
phthalate dioxygenase
- WT
wild type enzyme with no substitutions in the sequence
- PDR
phthalate dioxygenase reductase
- Fdx
[2Fe-2S] ferredoxin center in PDR
- DHD
cis-4,5-dihydrodiol of phthalate
- FAS
ferrous ammonium sulfate
- NDO
naphthalene dioxygenase
- CarDO
carbazole 1,9a-dioxygenase
Footnotes
One unit of activity is defined as μM of NADH oxidized in the reaction of PDR with cytochrome c per minute.
As mentioned previously, the steady-state activity assay does not determine the maximum rate of turnover, but the rate at specific, reproducible conditions. See, Materials and Methods.
References
- 1.Tarasev M, Kaddis CS, Yin S, Loo JA, Burgner J, Ballou DP. Similar enzymes, different structures: phthalate dioxygenase is an alpha3alpha3 stacked hexamer, not an alpha3beta3 trimer like “normal” Rieske oxygenases. Arch Biochem Biophys. 2007;466:31–9. doi: 10.1016/j.abb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gassner GT. Mechanistic Studies of the Phthalate Dioxygenase System, University of Michigan. Ph.D. Thesis. 1995 [Google Scholar]
- 3.Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure. 1998;6:571–586. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 4.Nojiri H, Ashikawa Y, Noguchi H, Nam JW, Urata M, Fujimoto Z, Uchimura H, Terada T, Nakamura S, Shimizu K, Yoshida T, Habe H, Omori T. Structure of the terminal oxygenase component of angular dioxygenase, carbazole 1,9a-dioxygenase. Journal of Molecular Biology. 2005;351:355–70. doi: 10.1016/j.jmb.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 5.Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T. Crystal structure of the terminal oxygenase component of cumene dioxygenase from Pseudomonas fluorescens IP01. Journal of Bacteriology. 2005;187:2483–90. doi: 10.1128/JB.187.7.2483-2490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. Journal of Molecular Biology. 2004;342:1041–52. doi: 10.1016/j.jmb.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 7.Jiang H, Parales RE, Lynch NA, Gibson DT. Site-directed mutagenesis of conserved amino acids in the alpha subunit of toluene dioxygenase: potential mononuclear non-heme iron coordination sites. J Bacteriol. 1996;178:3133–3139. doi: 10.1128/jb.178.11.3133-3139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beharry ZM, Eby DM, Coulter ED, Viswanathan R, Neidle EL, Phillips RS, Kurtz DM., Jr Histidine ligand protonation and redox potential in the rieske dioxygenases: role of a conserved aspartate in anthranilate 1,2-dioxygenase. Biochemistry. 2003;42:13625–36. doi: 10.1021/bi035385n. [DOI] [PubMed] [Google Scholar]
- 9.Parales RE, Parales JV, Gibson DT. Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity. J Bacteriol. 1999;181:1831–7. doi: 10.1128/jb.181.6.1831-1837.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaganaman S, Pinto A, Tarasev M, Ballou DP. High levels of expression of the iron-sulfur proteins phthalate dioxygenase and phthalate dioxygenase reductase in Escherichia coli. Protein Expr Purif. 2007;52:273–9. doi: 10.1016/j.pep.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarasev M, Pinto A, Kim D, Elliott SJ, Ballou DP. The “bridging” aspartate 178 in phthalate dioxygenase facilitates interactions between the Rieske center and the iron(II)--mononuclear center. Biochemistry. 2006;45:10208–16. doi: 10.1021/bi060219b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto A, Tarasev M, Ballou DP. Substitutions of the “bridging” aspartate 178 result in profound changes in the reactivity of the Rieske center of phthalate dioxygenase. Biochemistry. 2006;45:9032–41. doi: 10.1021/bi060216z. [DOI] [PubMed] [Google Scholar]
- 13.Haigler BE, Gibson DT. Purification and properties of NADH-ferredoxinNAP reductase, a component of naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J Bacteriol. 1990;172:457–464. doi: 10.1128/jb.172.1.457-464.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukuda M, Yasukochi Y, Kikuchi Y, Nagata Y, Kimbara K, Horiuchi H, Takagi M, Yano K. Identification of the bphA and bphB genes of Pseudomonas sp. strains KKS102 involved in degradation of biphenyl and polychlorinated biphenyls. Biochem Biophys Res Commun. 1994;202:850–6. doi: 10.1006/bbrc.1994.2008. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian V, Liu TN, Yeh WK, Narro M, Gibson DT. Purification and properties of NADH-ferredoxinTOL reductase. A component of toluene dioxygenase from Pseudomonas putida. J Biol Chem. 1981;256:2723–2730. [PubMed] [Google Scholar]
- 16.Ashikawa Y, Fujimoto Z, Noguchi H, Habe H, Omori T, Yamane H, Nojiri H. Crystallization and preliminary X-ray diffraction analysis of the electron-transfer complex between the terminal oxygenase component and ferredoxin in the Rieske non-haem iron oxygenase system carbazole 1,9a-dioxygenase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:577–80. doi: 10.1107/S1744309105014557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarasev M, Rhames F, Ballou DP. Rates of the phthalate dioxygenase reaction with oxygen are dramatically increased by interactions with phthalate and phthalate oxygenase reductase. Biochemistry. 2004;43:12799–808. doi: 10.1021/bi0490587. [DOI] [PubMed] [Google Scholar]
- 18.Batie CJ, Ballou DP. Phthalate dioxygenase. Methods Enzymol. 1990;188:61–70. doi: 10.1016/0076-6879(90)88013-z. [DOI] [PubMed] [Google Scholar]
- 19.Batie CJ, LaHaie E, Ballou DP. Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia. J Biol Chem. 1987;262:1510–8. [PubMed] [Google Scholar]
- 20.Massey V, Hemmerich P. A photochemical procedure for reduction of oxidation-reduction proteins employing deazariboflavin as catalyst. J Biol Chem. 1977;252:5612–5614. [PubMed] [Google Scholar]
- 21.Massey V, Hemmerich P. Photoreduction of flavoproteins and other biological compounds catalyzed by deazaflavins. Biochemistry. 1978;17:9–16. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- 22.Bevington PR. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill Inc; New York: 1996. pp. 235–42. [Google Scholar]
- 23.Tarasev M, Ballou DP. Chemistry of the catalytic conversion of phthalate into its cis-dihydrodiol during the reaction of oxygen with the reduced form of phthalate dioxygenase. Biochemistry. 2005;44:6197–207. doi: 10.1021/bi047724y. [DOI] [PubMed] [Google Scholar]
- 24.Kuila D, Fee JA. Evidence for a redox-linked ionizable group associated with the [2Fe- 2S] cluster of Thermus Rieske protein. J Biol Chem. 1986;261:2768–2771. [PubMed] [Google Scholar]
- 25.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Crystal structure of naphthalene dioxygenase: side-on binding of dioxygen to iron. Science. 2003;299:1039–42. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 26.Hegg E, Que LJ. The 2-His-1-carboxylate Facial-triad - an Emerging Structural Motif in Mononuclear Non-heme Iron(II) Enzymes. Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 27.Bertini I, Luchinat C, Mincione G, Parigi G, Gassner GT, Ballou DP. NMRD studies on phthalate dioxygenase: evidence for displacement of water on binding substrate. J Biol Inorg Chem. 1996:468–475. [Google Scholar]