

Abstract
Mature neurons have diminished intrinsic regenerative capacity. Axotomy of the peripheral branch of adult dorsal root ganglia (a “conditioning” lesion) triggers a transcription-dependent axon growth program. Here, we show that this growth program requires the function of the transcription factor Smad1. After peripheral axotomy, neuronal Smad1 is upregulated, and phosphorylated Smad1 accumulates in the nucleus. Both events precede the onset of axonal extension. Reducing Smad1 by RNA interference in vitro impairs axonal growth, and the continued presence of Smad1 is required to maintain the growth program. Furthermore, intraganglionic injection of BMP2 or 4, which activates Smad1, markedly enhances axonal growth capacity, mimicking the effect of a conditioning lesion. Thus, activation of Smad1 by axotomy is a key component of the transcriptional switch that promotes an enhanced growth state of adult sensory neurons.
Introduction
Injured adult neurons in the mammalian peripheral nervous system (PNS) can regenerate axons, but this requires reversal of an age-dependent loss of intrinsic axonal growth capability (Makwana and Raivich, 2005). Neurons in the CNS do not regenerate, in part because of an unfavorable environment for growth (Schwab and Bartholdi, 1996; Filbin, 2003; McGee and Strittmatter, 2003), but also because of an irreversible reduction in the intrinsic regenerative capacity of mature neurons (Goldberg et al., 2002). The molecular mechanisms regulating the intrinsic growth state remain incompletely understood (Zhou and Snider, 2006). Such an understanding may assist in attempts to further increase regeneration of PNS axons and to encourage regeneration of CNS axons.
A useful system for studying the rekindling of axonal growth capacity after injury is provided by sensory neurons in dorsal root ganglia. Dorsal root ganglia (DRG) neurons have two branches originating from a unipolar axon: a peripheral branch that innervates sensory targets, and a central branch that extends in the spinal cord and relays information to the CNS. Axotomy of the peripheral branch, but not the central branch, triggers axonal growth: these neurons extend axons more readily in dissociated culture (Smith and Skene, 1997), and they are able to regenerate their central branches after a later spinal cord lesion (Richardson and Issa, 1984; Neumann and Woolf, 1999). Importantly, axotomy-induced regeneration requires activation of a transcription-dependent program that reactivates the neuron's intrinsic axonal growth capacity (Smith and Skene, 1997). Gene microarray analyses have identified a number of genes whose expression is changed after injury, including several transcription factors (Costigan et al., 2002; Méchaly et al., 2006). For example, the immediate early gene ATF3 is rapidly induced after axotomy, and increasing ATF3 enhances the rate of peripheral regeneration (Seijffers et al., 2007). However, the remainder of the transcriptional cascade for regeneration remains poorly understood. In this study, we turned our attention to earlier time points than those previous studied, and we came to focus on Smad1 as a key component of the axon growth program.
Smads are the intracellular mediators of TGFβ/bone morphogenetic protein (BMP) signaling pathways. TGFβ/BMP ligands activate membrane receptor serine/threonine kinases, which in turn signal through C-terminal phosphorylation of Smads at an SXS serine motif. The phosphorylation triggers nuclear translocation of pSmads to activate downstream target genes. Particular Smads are dedicated to different ligands, with Smad1, 5, and 8 mediating signals from members of the BMP subfamily (Massagué et al., 2005).
Here, we identify axotomy-induced Smad1 activation and nuclear translocation as key components of the transcriptional switch for adult sensory neurons to enter into an active growth state.
Materials and Methods
Peripheral lesion, thoracic hemisection, and DRG microinjection.
All surgeries were performed on adult C57BL/6 mice (Neumann and Woolf, 1999). Two microliters of vehicle, BMP2, or BMP4 (10 ng) were injected into L4 or L5 DRGs.
Dissociated DRG cultures.
DRG neurons were cultured (Neumann et al., 2002) in Neurobasal-A and B-27 (Invitrogen) at a low density of 500–800 cells per 1 cm2 on slides or 96-well plates (Becton Dickinson) precoated with poly-d-lysine (100 μg/ml, Sigma) and laminin (10 μg/ml, Invitrogen). To remove myelin debris and cell clumps, a partial-purification step was performed by centrifugation through a 15% BSA cushion. For replating, DRG neurons from 3 d in vitro (DIV) were trypsinized (0.025% Trypsin-EDTA, Invitrogen) for 5 min at 37°C, gently triturated in fresh medium with 5% FCS, then cultured.
In situ hybridization, immunostaining, and Western blotting.
Radioactive in situ hybridization (ISH) was performed with mRNAlocator kit (Ambion). Antibodies for fluorescent IHC were: Smad1 (1:200; Millipore Bioscience Research Reagents), pSmad1/5/8 (Cell Signaling, 1:1000–1:2000), Tuj1 (Covance,1:1000), ATF3 (Santa Cruz Biotechnology, 1:200) and Alexa-conjugated 2° Ab (Invitrogen, 1:400); for ICC: Smad1 (1:200, RabMAb Epitomics) and pSmad1/5/8 (1:2000, Cell Signaling). Western blots (WBs) were immunoblotted with RabMAb against Smad1 (Epitomics, 1:200), pSmad1/5/8 (Cell Signaling, 1:200), or β-actin (Sigma, 1:5000).
Plasmids and siRNA.
Smad1 siRNA #1 (Dharmacon): (1293–1311): UC,ACA,GAU,CCG,UCC,AAC,AA. Smad1 full-length cDNA and a 964 bp 3′ UTR used for ISH (1835–2798 bp) were amplified from an adult mouse DRG cDNA library. For RNAi-resistant Smad1, 7 silent point mutations were introduced: TC,ACG,GAC,CCC,AGT,AAT,AA. SSVS→AAVA mutant Resistant-Smad1 was made (QuickChange XL-Site-Directed Mutagenesis, Stratagene).
siRNA transfection and plasmid electroporation of DRG neurons.
siRNAs (100 nm) were transfected (Dharmacon) and plasmids were electroporated into adult DRG neurons (Amaxa). The longest axons were quantified with an automated Image Express machine (Molecular Devices) or MetaMorph. At least three sets of experiments were performed for each condition. Data are presented as mean ± SEM; statistical analyses were performed by ANOVA.
Results
Expression profiling reveals early axotomy-induced changes in transcription factors
To search for transcriptional regulators involved at early steps of the axonal growth response, we profiled mRNA from DRGs of mice with either sciatic nerve transection (SNT; peripheral axotomy) or dorsal spinal column transection (central axotomy, as control). At 12 h post-SNT, 114 transcripts were upregulated and 19 downregulated. Of these, 13 were known transcription factors. At 24 h, 290 were upregulated and 139 downregulated, presumably reflecting the emergence of a second wave of downstream effector genes. Of these, 11 were known transcription factors (supplemental Table S1, available at www.jneurosci.org as supplemental material). Combining the two time points, a total of 19 distinct transcription factors were identified.
Induction and nuclear translocation of Smad1 precede the onset of axonal extension by DRG neurons in vitro
This microarray analysis identified Smad1 as being induced 1.8-fold 12 h after peripheral axotomy (compared with a central axotomy), a change confirmed by ISH (Fig. 1 A). Smad1 transcripts in lesioned DRG were also upregulated compared with those in contralateral, uninjured DRGs as assed by quantitative reverse transcriptase (qRT)-PCR and ISH (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Immunohistochemistry (IHC) and Western blots showed that Smad1 protein was up by approximately two-fold in ipsilateral compared with contralateral DRGs (Fig. 1 B–C; supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). The increase of Smad1 protein was evident at 1 d postlesion, peaked at 2 or 3 d, and by 7 d settled to a lower plateau but still remained above baseline (Fig. 1 C). The induction of Smad1 was neuronal (Fig. 1 A,B; supplemental Fig. S1B,C, available at www.jneurosci.org as supplemental material).
Figure 1.
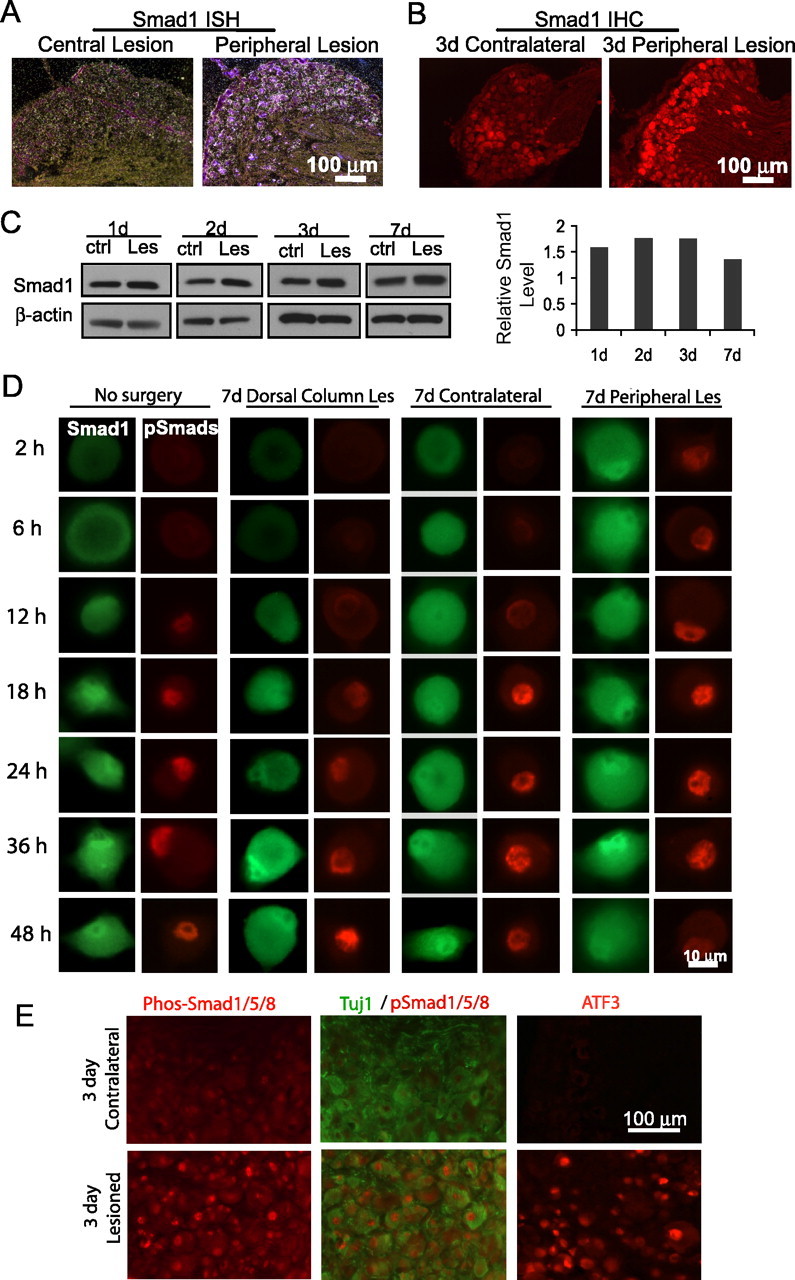
Induction and activation of Smad1 precede the onset of axonal extension by adult DRG neurons in culture. A, In situ hybridization demonstrates an upregulation of Smad1 mRNA in DRG after peripheral axotomy (SNT) compared with a central dorsal column lesion. B, Fluorescence immunohistochemistry shows induced Smad1 in ipsilateral versus contralateral DRG 3 d after axotomy. C, Western blots demonstrate upregulation of Smad1 in injured compared with contralateral DRGs from mice 1, 2, 3, and 7 d after SNT (β-actin as control, quantification on the right). D, Fluorescent immunocytochemistry of Smad1 (green) or pSmad1/5/8 (red) in dissociated DRG neurons. Naive (no surgery) or contralateral neurons showed induction and activation of Smad1 after 12–18 h. Neurons conditioned by peripheral, but not by central, axotomy displayed these features from the onset. E, An increase of nuclear pSmad1/5/8 was evident in DRGs at 3 d post-SNT by IHC (ATF3 as positive control). Tuj1 labels entire neurons. ctrl, Control; Les, lesion.
To analyze the function of Smad1, we turned to dissociated cultures. Before DRG neurons extend long axons, there is a “priming” period of 24 h requiring active transcription (Smith and Skene, 1997) (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). Focusing on this period, fluorescent immunostaining showed that total Smad1 began to increase in DRG neurons after 18 h in culture. Strikingly, Smad1 started to accumulate in the nucleus after 12 h (Fig. 1 D, first column). This was observed in virtually all neurons examined (>95%; data not shown). Western blots confirmed the increase in total Smad1 after 1 DIV (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Nuclear translocation of Smad1 requires its activation by phosphorylation at its C terminus. Using an antibody to the phosphorylated epitope, a time-dependent accumulation of nuclear pSmad1/5/8 was observed (Fig. 1 D, second column; supplemental Fig. S4A, available at www.jneurosci.org as supplemental material) [seen in virtually all (92%) neurons].
We hypothesized that these changes in Smad1 seen in vitro are triggered by the axotomy involved in dissociation. If so, then DRG neurons that have been axotomized in vivo one week before explantation should already have Smad1 induced and pSmad1 translocated into the nucleus when examined immediately after plating. Such changes were indeed observed in neurons conditioned in vivo by peripheral, but not central, axotomy (Fig. 1 D, 3rd–4th columns, 7th–8th columns). For both antibodies, neurons from the contralateral DRG showed similar staining patterns to those from uninjured animals (Fig. 1 D, 5th–6th columns), indicating that the change is triggered by axotomy, and not by systemic response to injury. An increase of nuclear pSmad1/5/8 was also seen when in vivo conditioned DRGs were examined directly in situ by immunohistochemistry at 3 d postlesion (Fig. 1 E).
Smad1 knockdown reduces axonal growth in adult sensory neurons
Smad1 knock-out mice die at embryonic day 9.5 because of allantois defects (Lechleider et al., 2001). We therefore turned to RNAi-mediated Smad1 knockdown, resulting in a 90% reduction of the transcript, and an 80% decrease of the protein (Fig. 2 A). Immunostaining similarly revealed dramatically reduced Smad1 (Fig. 2 B) and pSmad1/5/8 (Fig. 2 E; supplemental Fig. S2A, available at www.jneurosci.org as supplemental material).
Figure 2.
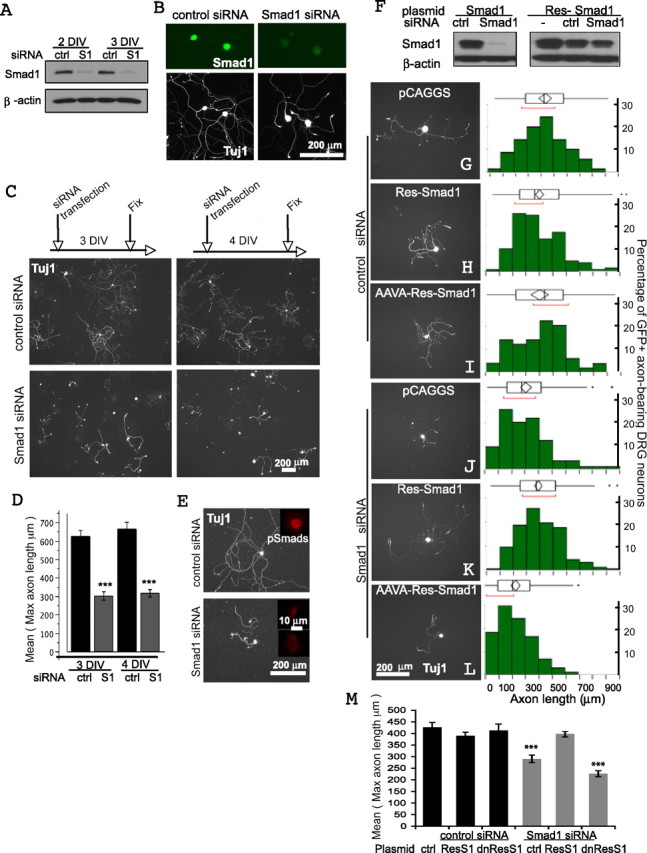
Smad1 is essential for axonal extension by adult DRG neurons. A, B, siRNA-mediated Smad1 knockdown in DRGs assessed by WB and immunocytochemistry. Tuj1 labeled the entire neurons, and its immunoreactivity was unaffected. C, D, Neurite outgrowth assay. Approximately 100 neurons were randomly picked for each condition, and the mean of the maximal axonal length was quantified. A total of 183 and 172 neurons were measured for 3 and 4 DIV, respectively (***p < 0.001, difference = 322.98 μm, SE difference = 37.94). E, Decreased pSmad1/5/8 immunoreactivity after Smad1 siRNA transfection for 3 DIV. Tuj1 highlights shorter neurites. F, Western blots of lysates of Cos-1 cells cotransfected with siRNA plus either the wild-type or an RNAi-resistant Smad1 (Res-Smad1) plasmid (β-actin as loading control). K–M, The Smad1 knockdown phenotype can be fully rescued using Res-Smad1 (K), but not using a mutant form of the Res-Smad1 with C-terminal SSVS changed to AAVA (L); quantification in M (***p < 0.001). The mutant form also appeared to function as a dominant negative to enhance the effect of the Smad1 siRNA (L). All GFP positive (coelectroporated) neurons (> 100) were counted for each condition. The quantile and outlier box plots summarize the distribution of the longest axonal length of the entire population of GFP positive neurons. The ends of the box are the 25th and 75th quantiles. The line across the middle is the median. The diamond is the mean with 95% confidence interval. Additional statistics are described in supplemental material, available at www.jneurosci.org. ctrl, Control; S1, Smad1; Res, resistant; dnRes1, dominant-negative resistant, i.e., AAVA-resistant Smad1.
Smad1 knockdown caused a marked decrease (50% reduction) in neurite length after 3 or 4 DIV (Fig. 2 C–E). Expression of one of the growth-associated proteins (GAPs), GAP-43, is widely correlated with axonal regeneration (Skene, 1989), and its transcript and protein were decreased after Smad1 knockdown (supplemental Fig. S2B,C, available at www.jneurosci.org as supplemental material).
The Smad1 knockdown phenotype did not seem to stem from decreased viability since the number of surviving neurons was comparable in both cultures (data not shown). The morphology of neurons also appeared normal, as judged by Tuj1 and DAPI staining (Figs. 2, 3) (data not shown). The phenotype instead reflected a reduction in the intrinsic axonal growth capacity of these neurons; for example, addition of NGF, known to stimulate growth of wild-type sensory axons, did not rescue the phenotype (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material). Four different Smad1 siRNAs were tested: three resulted in similar phenotypes, and they all caused a large reduction in Smad1, whereas the fourth had no functional effect but also did not produce effective knockdown (data not shown).
Figure 3.
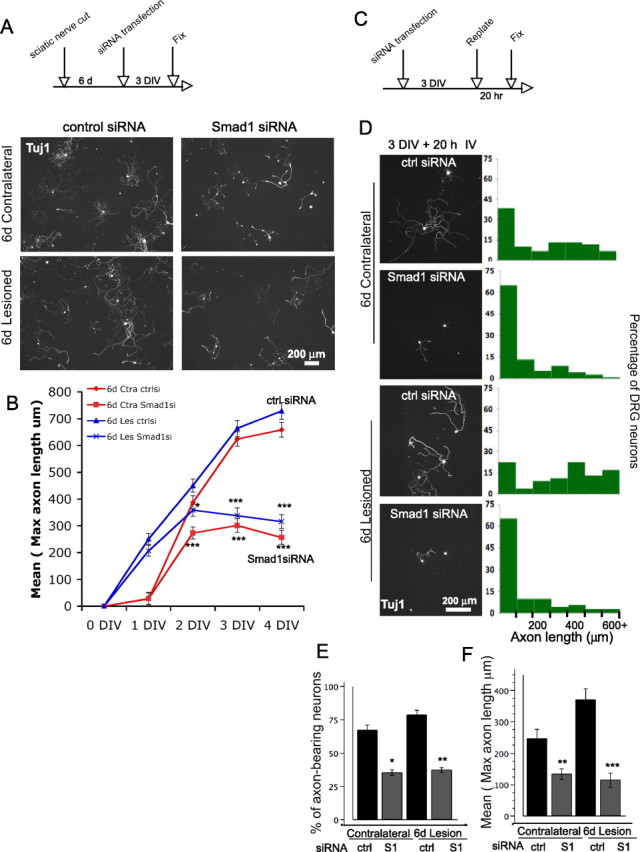
Smad1 is required to maintain the axon growth program. A, Both the contralateral and the conditioned neurons have reduced axonal length at 3 DIV after Smad1 knockdown. B, Time course analyses demonstrate that conditioned DRG neurons re-extended axons immediately after plating. In contrast, nonconditioned neurons have a 24 h delay before reextending axons. From 2 DIV, both the contralateral (red) and the conditioned DRG neurons (blue) have reduced axonal length with Smad1 knockdown. ∼100 neurons were measured for each data point. A total of 1451 neurons were counted. (*p < 0.01; **p < 0.001; and ***p < 0.0001, Student's t tests to compare Smad1 vs control siRNA and One-way ANOVA followed by Dunnett's post tests to compare all four groups). C, Schematic of the replating experiments. The distribution of the entire population is shown in D. y-axis is the % of neurons and x-axis is the maximal axonal length. E, F, A total of 276 neurons were measured. Smad1 knockdown resulted in a significant decrease of the % of the axon-bearing neurons (E), and of the mean of the longest axonal length (F) (*p < 0.01; **p < 0.001; and ***p < 0.0001). Ctra, Contralateral; Les, lesion; ctrlsi, control siRNA; S1, Smad1.
These results suggested that the phenotype was caused by specific knockdown of Smad1. This was further supported by rescue experiments using a plasmid encoding an RNAi-resistant Smad1 with seven silent point mutations introduced into the siRNA targeting region (Fig. 2 F). Electroporation of this plasmid into the neurons fully rescued the Smad1 knockdown phenotype (Fig. 2 J,K). In contrast, a mutant form of the RNAi-resistant Smad1 in which the C-terminal serines were mutated to alanines (SSVS to AAVA), which is predicted to be nonfunctional, did not rescue (Fig. 2 L,M). Over-expression of Smad1 or AAVA-Smad1 did not cause any significant changes in axonal length in the presence of control siRNA (Fig. 2 G–I) but, interestingly, the AAVA-Smad1 enhanced the effect of the Smad1 siRNA in reducing axonal length (Fig. 2 L,M), presumably by interfering with residual Smad1 through a dominant-negative effect.
In the time course study (Fig. 3 B, red curves), by 1 DIV, DRG neurons have just begun to form short neurites, and there was no visible effect of Smad1 siRNA. By 2 DIV, Smad1 knockdown resulted in significantly shorter neurites, which then did not appear to extend any further over the next 2 d.
Conditioned DRG neurons continue to require Smad1 for axonal growth
Smad1 upregulation and phosphorylation were maintained a week after SNT (Fig. 1 C,D), raising the possibility that continued presence of sufficient Smad1 may be required to maintain the growth program. When the conditioned and the contralateral neurons were subjected to Smad1 knockdown, both showed a significant decrease in axonal length (Fig. 3 A). As expected, at 1 DIV, the conditioned neurons treated with control siRNA showed a tremendous growth advantage over their naive counterparts. Smad1 siRNA had no effect at 1DIV; but at 2 DIV, the axonal length of the conditioned neurons leveled off, although they were still slightly longer than their naive counterparts; at 3 and 4 DIV, they seemed to have even retracted their already extended axons, bringing their lengths in line with those of the nonconditioned counterparts (Fig. 3 B, blue curves).
An alternative way of testing the continued requirement for Smad1 for axonal extension is to use a replating protocol. DRG neurons were cultured for 3 d (equivalent to a 3 d in vitro conditioning), then replated and grown for an additional 20 h. Unlike the freshly dissociated neurons, replated neurons had extended long neurites after only 20 h. Replated neurons with Smad1 knockdown extended much shorter neurites, and a much lower percentage of neurons had neurites. A similar effect was observed for neurons preconditioned by SNT in vivo (Fig. 3 C–F).
Smad1 is a transcription factor, so our model predicts that the axon growth program requires ongoing transcription. DRG neurons preconditioned in vivo are known to be able to extend axons in a transcription-independent manner for the initial 16 h in vitro (Smith and Skene, 1997) (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). We found that transcription is required beyond that, since in the continued presence of the transcription inhibitor DRB, the conditioned neurons eventually stopped extending axons at 2 DIV, the axons started to degenerate and retract by 3 DIV, and Smad1 was depleted (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Intraganglionic injection of BMP4 or BMP2 can mimic the effect of a conditioning lesion
We next tested whether Smad1 is sufficient to serve as a molecular switch. Gain-of-function studies by addition of BMPs to the cultures in the first 18 h did not prove informative, because of loss of BMP receptors during enzymatic dissociation (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). However, since the receptors are present on the neurons in vivo, we reasoned that we could likely activate Smad1 in vivo by exposing the neurons to BMPs. We therefore delivered BMPs by injection into the DRGs, and allowed neurons to be “primed” for 3 d in vivo. The neurons were then grown in dissociated culture for only 16 h. In the control cultures derived from vehicle-injected DRGs, very little axonal growth was observed. In contrast, cultures of neurons exposed to BMP2 or 4 in vivo showed a significant increase in the percentage of neurite-bearing neurons and in neurite length (Fig. 4 A–C). Intraganglionic BMP4 injection activated Smad1 in sensory neurons: 52% of neurons showed early nuclear accumulation of pSmad1/5/8, compared with 19% from vehicle-injected and 8% from DRGs with no injection (Fig. 4 D–E).
Figure 4.
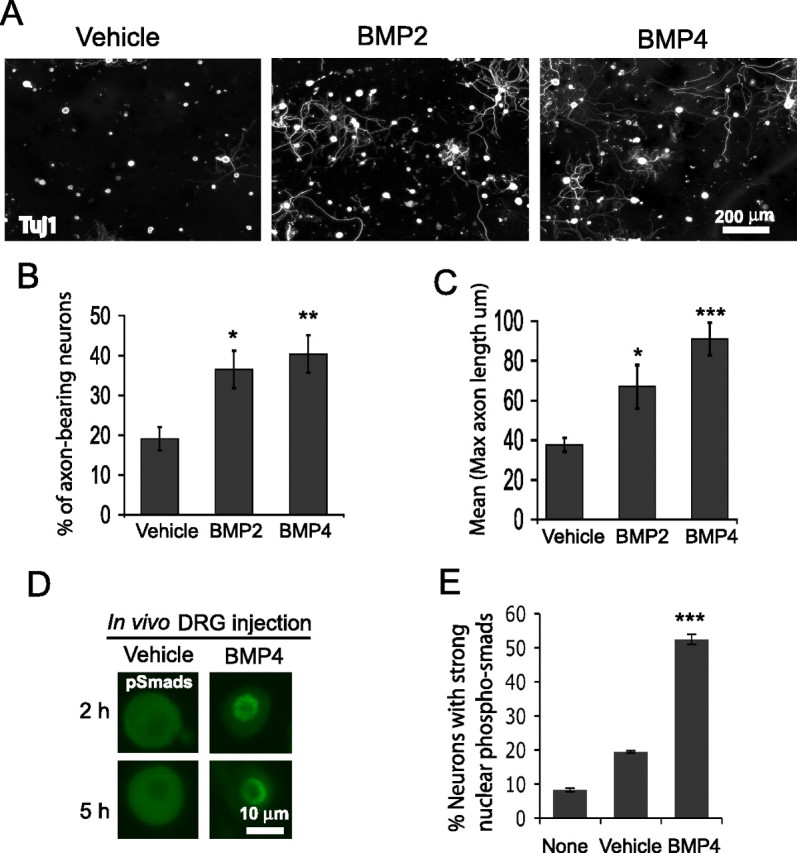
In vivo intraganglionic injection of BMP2 or 4 activates Smad1 and promotes axonal growth. A–C, DRG neurons injected with vehicle (n = 7 mice), BMP2 (10 ng, n = 4), or BMP4 (10 ng, n = 8), primed for 3 d, then cultured for 16 h. The percentage of axon-bearing neurons (defined as neurite length >50 μm) and the mean of the longest axonal length were both increased by BMP2/4 injection. (*p < 0.05; **p < 0.001; and ***p < 0.0001). D,E, Immunocytochemistry for pSmad1/5/8 (in dissociated sensory neurons from vehicle vs BMP4-injected DRGs at early time points with quantification. Error bars represent SEM (***p < 0.0001). n = 4, 4, 6 for the number of repeats for each group (none-, vehicle-, and BMP4-injected).
Discussion
Our results establish a key role for Smad1 activation in rekindling axon growth after injury: peripheral axotomy triggers the phosphorylation and nuclear translocation of Smad1, which appears to be necessary and sufficient to push the neurons into a growth state.
Early changes in transcription factors in vivo and in vitro
We have extended previous studies by showing that peripheral axotomy induces a rapid (<12 h) and robust change in expression of a number of transcription factors, including ATF3 and Smad1, in DRG neurons in vivo; similar changes are seen in dissociated neurons in vitro, triggered by the axotomy during dissociation. The parallels between the two suggest that the reprogramming process may be largely neuron-autonomous and directly linked to injury per se, since neurons were semipurified and plated at low density, thus making the influence from residual non-neuronal cells lower than might be expected in vivo. Furthermore, the dissociation step severs the axon so close to the soma that retrograde transport of messages from the injury site seems inessential. How peripheral, but not central axonal injury triggers Smad1 induction, activation and nuclear translocation awaits future study. In preliminary experiments, we found that blocking extracellular BMPs with the antagonist noggin (which binds multiple BMPs) or with antibodies to BMP2/4 did not alter the rate of Smad1 activation nor the time course of axon extension (data not shown). This suggests that Smad1 is activated by axotomy independently of extracellular BMPs; a precedent for direct activation of Smads by a cytoplasmic kinase independent of extracellular TGFβs has been reported (Zhu et al., 2007); it is also possible that an intracrine mechanism (intracellular BMPs activating their receptors while both are in the endoplasmic reticulum) operates here, as documented for other peptide growth factors like VEGF (vascular endothelial growth factor) (Lee et al., 2007). Whether one of these mechanisms operates here, or whether some other mechanism is at play, remains to be determined.
It is, of course, conceivable that activation of Smad1 may also be triggered at the peripheral lesion site in vivo, with pSmad1 then retrogradely transported to the cell body and into nucleus. Smad1 has been implicated in mediating BMP signaling from axon terminals, through retrograde transport, to regulate neuronal differentiation and synaptic maturation (Hodge et al., 2007), and in topographic mapping of neural circuits (Ting et al., 2007). Moreover, retrograde transport of factors like JNK (c-Jun N-terminal protein kinase) (Goldstein, 2001), MAP (mitogen-activated protein) kinase (Perlson et al., 2005), and pSTAT3 (Lee et al., 2004) may further enhance the neuronal responses to peripheral axotomy.
Our microarray analyses did not identify an upregulation of Smad1 24 h postlesion, but we believe that this may reflect the detection threshold of the microarray analysis, since sustained induction of Smad1 up to a week after axotomy was readily detected by qRT-PCT, ISH, and Western blotting.
Smad1 functions as a key regulator of the axon growth program
With Smad1 knockdown, adult DRG neurons show reduced axonal growth. Because maximal depletion of Smad1 by RNAi takes ∼2 d (Fig. 2 A) (data not shown), we believe some Smad1-dependent activation of the growth program occurs initially and likely accounts for the early relatively normal growth. After 2 DIV, however, the siRNA-treated neurons do not extend axons further although the knockdown is not complete (an ∼80% reduction), indicating a central requirement for Smad1. Thus the timeframe within which the axon growth program is halted coincides approximately with the time course of Smad1 knockdown.
Consistent with the in vivo relevance of this mechanism, priming adult sensory neurons by exposure to exogenous BMP2 or 4 in vivo in the absence of axotomy both activates Smad1 in the neurons and triggers the switch to an axonal growth state, providing support for sufficiency. It is possible that the effect of injected BMPs is partly attributable to an indirect effect, with the BMPs acting on non-neuronal cells which in turn signal the neuron, although the fact that injected BMPs activate Smad1 in neurons suggests that the effect is at least partly, if not entirely direct on the neurons. It should also be noted that the effect of intraganglionic injection of BMP2 or 4 is not as strong (as assessed by a lower percentage of neurite-bearing neurons) as seen with a conditioning lesion. Similarly, a lower percentage (52%) of neurons exhibit a strong nuclear pSmads signal after in vivo BMP “priming” than after sciatic nerve transection (>90%). This partial activation may result from only a subpopulation of DRG neurons being exposed to adequate concentrations of the exogenous BMPs. Alternatively, the Smad1 pathway might be only one of several components that are activated by peripheral axotomy (Cao et al., 2006).
It is possible that Smad1 activation also controls expression of other injury-induced genes, beyond those that regulate axonal growth. For example, galanin is highly upregulated after axotomy, and a recent study highlighted the presence of Smad-binding sites in a region of the galanin promoter required for this upregulation, as well as in the promoters of other injury-induced genes (Bacon et al., 2007), consistent with a central role for Smad1 in the axotomy-induced response.
Although we have focused on the role of Smad1 in mediating the response to axotomy, our results do not exclude functions for BMPs at the tips of axons, independent of nuclear signaling. Indeed, noncanonical BMP signaling pathways are implicated in mediating local effects of BMPs–regulating actin dynamics during dendritogenesis (Lee-Hoeflich et al., 2004), synaptic stability (Eaton and Davis, 2005), and acute induction of growth cone collapse (Butler and Dodd, 2003). Other TGFβ regulated Smads, e.g., Smad2, are also expressed in adult DRG neurons; their function remains to be reported.
Our data implicate Smad1 as a key component of the transcriptional mechanism underlying the switch of adult sensory neurons into a growth state. Whether the same transcriptional cascade that rekindles peripheral axonal growth also enable the central branch to navigate through the hostile environment of CNS after a conditioning peripheral lesion remains to be determined. It is certainly conceivable that distinct transcriptional programs regulate intrinsic axon growth capacity and axonal sensitivity to extrinsic CNS inhibitors. Regardless, our finding of a key role of Smad1 in the transcriptional switch to a growth state provides a further handle on this crucial step in the regeneration of adult axons.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke (Grant 1F32NS056587 to H.Z. and K08-NS048058 to C.H.) and by Genentech. We are indebted to Zhigang He (Harvard University) for sharing the replating technique, M. Tessier-Lavigne's laboratory members for insightful discussions, Edwin Oh for technical support for qRT-PCR and electroporation, and Hannes Vogel (Stanford University) for advice on histochemistry.
References
- Bacon A, Kerr NC, Holmes FE, Gaston K, Wynick D. Characterization of an enhancer region of the galanin gene that directs expression to the dorsal root ganglion and confers responsiveness to axotomy. J Neurosci. 2007;27:6573–6580. doi: 10.1523/JNEUROSCI.1596-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SJ, Dodd J. A role for BMP heterodimers in roof plate-mediated repulsion of commissural axons. Neuron. 2003;38:389–401. doi: 10.1016/s0896-6273(03)00254-x. [DOI] [PubMed] [Google Scholar]
- Cao Z, Gao Y, Bryson JB, Hou J, Chaudhry N, Siddiq M, Martinez J, Spencer T, Carmel J, Hart RB, Filbin MT. The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J Neurosci. 2006;26:5565–5573. doi: 10.1523/JNEUROSCI.0815-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Befort K, Karchewski L, Griffin RS, D'Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 2002;3:16. doi: 10.1186/1471-2202-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BA, Davis GW. LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- Goldstein LS. Transduction. When worlds collide–trafficking in JNK. Science. 2001;291:2102–2103. doi: 10.1126/science.1059766. [DOI] [PubMed] [Google Scholar]
- Hodge LK, Klassen MP, Han BX, Yiu G, Hurrell J, Howell A, Rousseau G, Lemaigre F, Tessier-Lavigne M, Wang F. Retrograde BMP signaling regulates trigeminal sensory neuron identities and the formation of precise face maps. Neuron. 2007;55:572–586. doi: 10.1016/j.neuron.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Lechleider RJ, Ryan JL, Garrett L, Eng C, Deng C, Wynshaw-Boris A, Roberts AB. Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev Biol. 2001;240:157–167. doi: 10.1006/dbio.2001.0469. [DOI] [PubMed] [Google Scholar]
- Lee N, Neitzel KL, Devlin BK, MacLennan AJ. STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J Comp Neurol. 2004;474:535–545. doi: 10.1002/cne.20140. [DOI] [PubMed] [Google Scholar]
- Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med. 2007;4:e186. doi: 10.1371/journal.pmed.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 2004;23:4792–4801. doi: 10.1038/sj.emboj.7600418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana M, Raivich G. Molecular mechanisms in successful peripheral regeneration. Febs J. 2005;272:2628–2638. doi: 10.1111/j.1742-4658.2005.04699.x. [DOI] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26:193–198. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- Méchaly I, Bourane S, Piquemal D, Al-Jumaily M, Ventéo S, Puech S, Scamps F, Valmier J, Carroll P. Gene profiling during development and after a peripheral nerve traumatism reveals genes specifically induced by injury in dorsal root ganglia. Mol Cell Neurosci. 2006;32:217–229. doi: 10.1016/j.mcn.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene JH. Axonal growth-associated proteins. Annu Rev Neurosci. 1989;12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CY, Herman T, Yonekura S, Gao S, Wang J, Serpe M, O'Connor MB, Zipursky SL, Lee CH. Tiling of r7 axons in the Drosophila visual system is mediated both by transduction of an activin signal to the nucleus and by mutual repulsion. Neuron. 2007;56:793–806. doi: 10.1016/j.neuron.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Wang W, Clarke DC, Liu X. Activation of Mps1 promotes transforming growth factor-beta-independent Smad signaling. J Biol Chem. 2007;282:18327–18338. doi: 10.1074/jbc.M700636200. [DOI] [PubMed] [Google Scholar]