

Abstract
Malignant gliomas are characterized by invasive and infiltrative behavior that generally involves the destruction of normal brain tissue. Strategies to treat infiltrating gliomas, such as chemotherapy and gene therapy, have remained largely unsuccessful. The infiltrative nature of gliomas can be attributed largely to proteases, which include serine, metallo and cysteine proteases. Our previous work and that of others strongly suggest a relationship between the expression of uPAR, MMP-9, and MMP-2; this relationship is generally indicative of the infiltrative phenotype of gliomas. In the present study, we have demonstrated that the RNAi-mediated downregulation of MMP-2 induces apoptosis in the 4910 human glioma xenograft cell line. Using western blot analysis, we observed that caspase-8 levels increased in MMP-2-downregulated cells whereas TRADD and TRAF-2 levels decreased. Further, NIK levels increased in MMP-2-downregulated cells. To determine the nuclear localization of AIF and IκB-α, we analyzed the levels of AIF, IκB-α and p-IκB-α in the cytosolic and nuclear fractions of MMP-2-downregulated cells. Western blot analysis revealed that MMP-2 downregulation resulted in the translocation of AIF to the nucleus and also inhibited the nuclear localization of p-IκB-α. To confirm the involvement of AIF, we performed FACS analysis to determine the integrity of the mitochondrial membrane using the Mito-PT method. FACS analysis showed that the downregulation of MMP-2 caused a collapse in the mitochondrial cell membrane. Immunolocalization of AIF revealed that in MMP-2-downregulated cells, AIF translocates to the nucleus, thereby enabling the induction of apoptosis. RT-PCR analysis revealed that caspase-8 was overexpressed 57-fold, whereas p73 was downregulated 28-fold. Evidence of apoptosis was determined by TUNEL assay and visualization of nuclear fragmentation by DAPI staining. In summary, it is evident from our results that MMP-2 downregulation induces caspase-8 and AIF-mediated apoptosis and, as such, shows potential for glioma therapy.
Introduction
Matrix metalloproteases (MMPs) are the largest group of ECM-degrading enzymes; so far, 22 members have been found in human tissue. MMPs are named according to the order in which they were identified (MMP 1–28), but several MMPs also have names based on functional or structural similarities. These subclasses include the collagenases (MMP-1, -8, -13, -18), which degrade collagen; the gelatinases (MMP-2, -9), which degrade denatured collagen and type IV collagen; matrilysin (MMP-7) and the stromelysins (MMP-3, -10, -11), which degrade proteoglycans; metalloelastase (MMP-12), which degrades elastin; and enamelysin (MMP-20), which degrades enamel (1). The expression and activity of MMPs increase in most human cancer types; this expression and activity is generally associated with advanced tumor stage and poor survival. For example, the expression of MMP-2 and MMP-9 strongly correlates with glioma progression.
Nuclear factor-κB (NF-κB) is responsible for the expression and regulation of pro- and anti-apoptotic genes, including MMPs (2,3). The function of NF-kB is inhibited by binding to NF-κB inhibitor (IκB), and an imbalance in the levels of NF-κB and IκB has been associated with development of many diseases, including cancer. Phosphorylation of IκB causes it to dissociate from NF-κB, thereby enabling NF-κB to enter the nucleus and activate various genes. Conversely, de-phosphorylation of the NF-κB inhibitor IκBα inactivates NF-κB, arresting cellular proliferation and functioning, and thereby causing cells to undergo apoptotic or necrotic cell death with or without the involvement of the mitochondria (2,4). IκBα is not normally known to be present in the nucleus, regardless of its phosphorylation state, and as such, its presence in the nucleus indicates a disruption in IκBα inhibitory functioning.
Mitochondria are essential for cell survival, but they also play a major role in cell death, which results from the permeabilization of their membranes, and which known to be mediated by the activation of caspase-8 (5). Once mitochondrial membrane permeabilization (Δψ collapse) occurs, cells die either by apoptosis or necrosis. Key factors regulating Δψ include calcium, cellular redox status (including levels of reactive oxygen species), and the mobilization and targeting of mitochondria Bcl-2 family members. Hence, activating the collapse of Δψ would be an important step in initiating apoptosis in glioma cells (5,6).
In this study, we demonstrate that MMP-2 downregulation in human glioma xenograft cells not only activates caspase-8 but also activates Δψ collapse, which initiates the nuclear translocation of AIF, essentially initiating the extrinsic apoptotic pathway. These events also cause the nuclear dephosphorylation of IκBα, thereby inhibiting NF-κB. This study also demonstrates the existence of a feedback mechanism in glioma xenograft cells which require MMP-2 activity for normal function. Further study of this feedback mechanism may provide insight for the development of targeted molecular therapies specific to individual malignancies.
Materials and Methods
Construction of hpRNA-expressing adenovirus
The adenovirus was constructed using an adenoviral pSuppressor kit (Imgenex) as previously described (7). The pSuppressor plasmids (pSup-3) containing the MMP-2 siRNA sequence 5′-AACGGACAAAGAGTTGGCAGTATCGATACTGCCAACTCTTTGTCCGTT-3′ were digested with PacI and co-transfected with a pAd vector backbone in 293 cells to generate an adenovirus containing MMP-2 siRNA (Ad-MMP-2). The pSV construct was used to construct an adenovirus containing the scrambled sequence (Ad-SV). Adenovirus generation, amplification, and titer were carried out as previously described (8). Viruses were plaque-purified, propagated on 293 cells, and purified by cesium chloride gradient using standard techniques. Particle titers of all adenoviruses were determined by absorbance measurements at 260 nm, and functional titers (plaque-forming units) were determined by end-point dilution titration on 293 cells using standard techniques. The amount of infective adenoviral vector per cell (plaque-forming units/cell) in the culture medium was expressed as multiplicity of infection (MOI).
Cell lines and culture conditions
4910 human glioma xenograft cells were maintained as a monolayer in DMEM/F12 medium supplemented with 10% FBS, 50 units/mL penicillin and 50 μg/mL streptomycin (Life Technologies Inc., Frederick, MD) at 37°C in a humidified 5% CO2 atmosphere.
Gelatin zymography
MMP activity in conditioned medium was determined by gelatinase zymography as described previously (9). 4910 and 5310 human glioma xenograft cells were infected with mock, Ad-SV, and the indicated doses of Ad-MMP-2 (2.5, 5 and 10 MOI) for 36 hrs. Cells were washed and incubated with serum-free medium overnight. Conditioned medium containing equal amounts of protein were electrophoresed in 7.5% SDS-polyacrylamide gels containing 1.5 mg/mL gelatin. The gels were washed and gently shaken in three consecutive washings in 2.5% Triton X-100 solution to remove SDS. The gels were then incubated at 37°C overnight in incubation buffer [50 mmol/L Tris-HCl (pH 7.5), 0.05% NaN3, 5 mmol/L CaCl2, and 1 μmol/L ZnCl2]. Next, the gels were stained with 0.1% Coomassie brilliant blue in 10% acetic acid and 10% isopropanol and subsequently destained for 1 hr. Gelatinolytic activities were identified as clear zones of lysis against a dark background.
RT-PCR analysis
4910 human glioma xenograft cells were infected with mock, Ad-SV, and the indicated doses of Ad-MMP-2 (2.5, 5, and 10 MOI) for 36 hrs. The cells were collected and total cell RNA was isolated. Reverse transcriptase PCR was set up using appropriate MMP-2 specific primers (see Table 1) using the PCR cycle [95°C-5′, (95°C-30 sec, 65°C-1′ 72°C-1′) × 30, 72°C-10′]. The PCR product was quantified and plotted relative to GAPDH expression as arbitrary units.
Table 1.
| Primers | Position | |
|---|---|---|
| MMP-2 | GTGCTGAAGGACACACTAAAGAAGA | 494 - 518 |
| CCTACAACTTTGAGAAGGATGGCAA | 1074 - 1098 | |
| GAPDH | GGAGTCAACGGATTTGGTCGTAT | 93 - 116 |
| GTCTTCACCACCATGGAGAAGGCT | 376 - 399 | |
Western blot analysis
4910 human glioma xenograft cells were infected with mock, Ad-SV, and the indicated doses of Ad-MMP-2 (2.5, 5 and 10 MOI) and incubated for 36 hrs. Cells were then collected, and total cell lysates were prepared in standard RIPA extraction buffer containing aprotinin and phenyl-methyl-sulfonyl-fluoride. Twenty micrograms of protein from these samples were separated under non-reducing conditions by 12% SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The membranes were immunoprobed for 2 hrs with antibodies against TRADD, TRAF-2, NIK and caspase-8 as per standard protocols. Next, membranes were treated with the appropriate HRP-conjugated secondary antibody and then developed according to enhanced chemiluminescence protocol (Amersham, Arlington Heights, IL). For loading control, the membranes were stripped and probed with monoclonal antibodies for GAPDH as per standard protocol.
Immunohistochemical determination of AIF nuclear localization
4910 glioma xenograft cells (1×103) were seeded in 8-well chamber slides, incubated for 24 hrs, infected with 10 MOI of Ad-SV (scrambled vector) or the indicated doses of Ad-MMP-2 (2.5, 5 or 10 MOI) for 48 hrs. The cells were then fixed and immunoprobed for AIF using a specific monoclonal antibody as per standard protocols. An HRP-conjugated secondary antibody was used to visualize AIF in 4910 cells in conjunction with a DAB reagent.
Isolation of nuclear and cytoplasmic fractions
Nuclear extracts were prepared by disrupting human xenograft cells infected with either Ad-SV or the indicated doses of Ad-MMP-2 for 36 hrs in a sucrose-HEPES buffer containing 0.5% Nonidet P40 as a detergent, protease inhibitors, and dithiothreitol. After a 5-minute incubation on ice and centrifugation at 16,000g to pellet the nuclei, the supernatant contained the cytoplasmic extract. Nuclear proteins were separated in a sodium chloride-HEPES buffer and resuspended in a glycerol-containing buffer. All procedures were carried out on ice. Protein quantification was determined using the Bradford assay. Cytoplasmic and nuclear extracts were western blotted and immunoprobed for AIF and pIκbα.
Visualization of mitochondrial permeability transition
4910 xenograft cells were cultured to 50% confluence and infected with either Ad-SV or Ad-MMP-2 (2.5, 5 and 10 MOI). Thirty-six hours after infection, the cells were trypsinized and resuspended in serum-free media containing 5,5′,6,6′-tetrachloro-1,1′3,3′tetraethylbenzamidazolocarbo cyanin iodide (JC-1) using the MitoPT kit. After a 15 to 20-minute incubation at 37°C, the cells were fixed and observed for red/green fluorescence. Cells were counted using a flow cytometer and results graphically represented.
In situ terminal-deoxy-transferase mediated dUTP nick end labeling (TUNEL) assay
A TUNEL apoptosis detection kit (Upstate Biotechnology Inc, Lake Placid, NY.) was used for DNA fragmentation fluorescence staining and carried out according to the manufacturer's protocol. Briefly, 4910 glioma xenograft cells grown on chamber slides were infected with either Ad-SV or Ad-MMP-2 (2.5, 5 and 10 MOI). Thirty-six hours after transfection, cells were fixed with 4% paraformaldehyde 0.1 M phosphate buffer (pH 7.4). Cells were then incubated with a reaction mix containing biotin-dUTP and terminal deoxynucleotidyl transferase for 60 min. Fluorescein-conjugated avidin was applied to the sample, which was then incubated in the dark for 30 min. Positively stained fluorescein-labeled cells were visualized with fluorescence microscopy and quantified.
Visualization of apoptotic cells by nuclear staining
4910 glioma xenograft cells were cultured on 6-well chamber slides and infected with either Ad-SV or Ad-MMP-2 (2.5, 5 and 10 MOI). Thirty-six hours after transfection, glass cover slips were mounted using DAPI-containing mounting media (Vector Laboratories, Burlingame, CA). The slides were allowed to rest for 20 min in the dark. Cells were observed for the nuclear fragmentation indicative of apoptosis with a fluorescent microscope and photographed using a high resolution CCD camera. Ten fields were photographed and the percentage of apoptotic cells compared to the average number of cells per field was determined and graphically represented.
Real time PCR assay
The Human Apoptosis Signaling Pathway RT2² Profiler™ PCR Array was used to profile the expression of 84 genes related to the apoptosis signaling pathway. Human glioma xenograft cells were infected with either Ad-SV or Ad-MMP-2 (10 MOI). Total RNA was isolated from treated and control cells and mRNA was further isolated using oligo dT-conjugated paramagnetic particles. cDNA was synthesized from the purified mRNA as per standard protocol. 96-well plates with appropriate PCR-primers were used and the target cDNAs amplified using SYBR green master mix. PCR reaction was set and optical acquisition was at the annealing step [95°C-5′, (95°C-30 sec, 55°C-1′ optical acquisition, 72°C-1′) × 35, 72°C-10′] using the Bio-Rad iCycler. Ct values were plotted and fold change determined by 2-Ct(experiment)-Ct(Control). Appropriate internal controls for mRNA and genomic DNA were included.
Results
RNAi-mediated MMP-2 downregulation activates caspase-8 and NIK
Overexpression of MMP-2 is characteristic of infiltrative and metastatic cancers. To retard the infiltrative character of 4910 glioma xenograft cells, we developed an siRNA-expressing adenovirus targeting MMP-2. The cells were cultured in vitro and infected with Ad-MMP-2 at the indicated MOI. From the results of gelatin zymography, we observed that infection with Ad-MMP-2 caused a dose-dependent decrease in MMP-2 activity (Fig. 1A). These findings were supported by the RT-PCR analysis where we observed that the levels of MMP-2 mRNA decreased in a dose-dependent manner, which correlates with the results of the gelatin zymography analysis (Fig. 1B). To determine the apoptotic state, we measured the activation of caspase-8 in MMP-2-downregulated cells. From the western blot analysis, caspase activation was seen to increase in a dose-dependent manner in cells downregulated for MMP-2. We also determined the levels of the TNFR-associated proteins TRADD, TRAF-2 and NIK. We observed that levels of TRADD and TRAF-2 decreased with an increase in caspase activation. NIK levels were similar to activated caspase-8 and showed an increase in a dose-dependent manner after MMP-2 downregulation (Figs. 1C-D).
Figure 1. RNAi-mediated downregulation of MMP-2 causes reduction of MMP-2 mRNA expression and protein activity, activation of caspase-8, and overexpression of NIK.
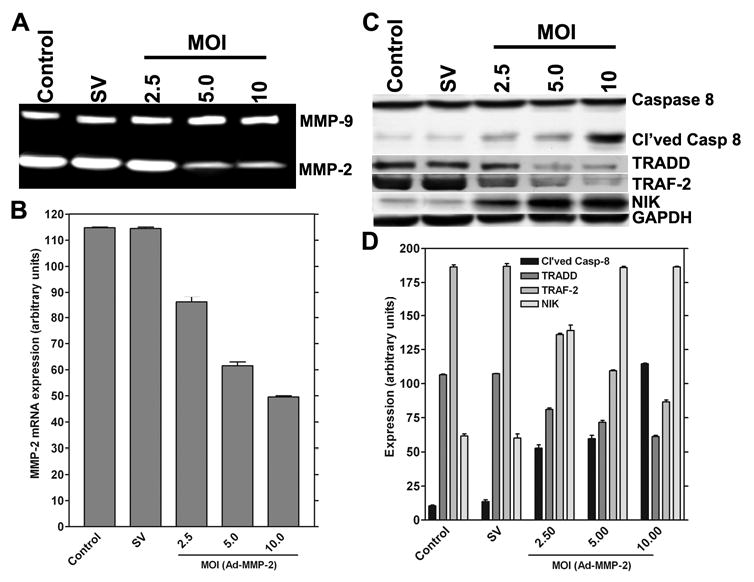
Human glioma xenograft cells 4910 were infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. MMP-2 mRNA expression was determined by RT-PCR analysis after 48 hrs of adenovirus infection; the experiment was performed in triplicate (A). For the control, we used uninfected cells or cells infected with 10 MOI of Ad-SV. The expression levels were determined by image densitometry (B). Human glioma xenograft cells 4910 were infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 48 hrs. Expression levels of cleaved caspase-8, TRADD, TRAF-2 and NIK were determined by western blot analysis (A). Expression levels of GAPDH were also determined and served as loading controls. Quantitative analysis of the expression levels of cleaved caspase-8, TRADD, TRAF-2 and NIK were determined by densitometry. All experiments were done in triplicate (B).
Downregulation of MMP-2 activates the nuclear translocation of AIF in a time- and dose-dependent manner
Initiation of apoptotic events may involve the mitochondria. A collapse in the mitochondrial membrane is accompanied by the release of AIF, which is translocated to the nucleus and activates nuclear fragmentation. To determine the nuclear translocation of AIF, we performed immunohistochemical analysis of AIF on glioma xenograft cells downregulated for MMP-2. From the immunolocalization studies of AIF in control, Ad-SV-infected and Ad-MMP-2-infected 4910 glioma xenograft cells, we observed that AIF was localized in the cell nucleus rather than in the cytoplasm in Ad-MMP-2-infected cells as compared to the controls in a dose-dependent manner (Fig. 2A). The rate of AIF translocation was determined by conducting a time course study of AIF nuclear translocation at 6, 12, 24, 48 and 72 hrs after infection with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI). We determined that, at 6 hrs after infection, cells infected with 10 MOI of Ad-MMP-2 showed 15% nuclear translocation, whereas cells infected with 2.5 and 5 MOI of Ad-MMP-2 were similar to controls (5-7%). At 12 hrs after infection, cells infected with Ad-MMP-2 at 10 MOI showed 30% nuclear translocation of AIF, whereas cells infected with 2.5 and 5 MOI showed 10% and about 15% nuclear localization, respectively. Similarly, at 24 hrs, cells infected with 10 MOI of Ad-MMP-2 showed 50% nuclear translocation, while cells infected with 2.5 and 5 MOI Ad-MMP-2 showed 12% and 22% AIF nuclear translocation, respectively. After 48 hrs, cells infected with 10 MOI showed 62% AIF nuclear translocation while cells infected with 5 and 2.5 MOI showed 28% and 17% AIF nuclear translocation, respectively. Finally, after 72 hrs, cells infected with 10 MOI showed 70%, 5 MOI showed 35% and 2.5 MOI showed 25% nuclear translocation of AIF (Fig. 2B).
Figure 2. MMP-2 downregulation causes the nuclear translocation of AIF.
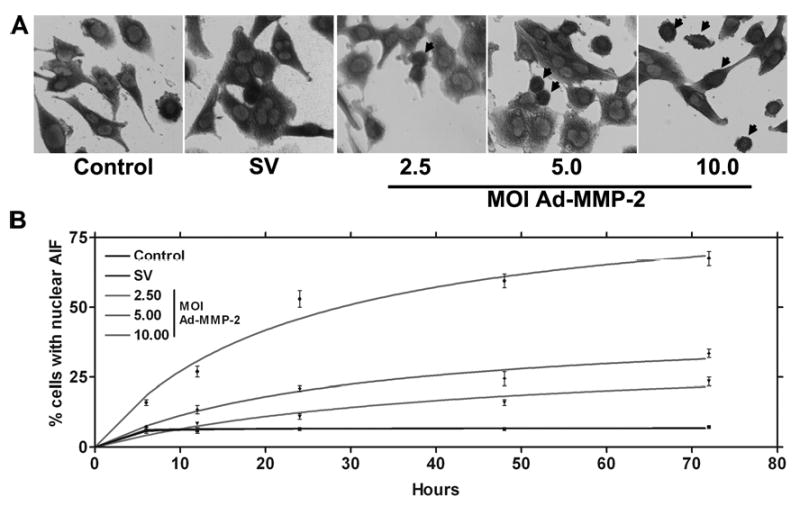
4910 glioma xenograft cells (1×103) were seeded in 8-well chamber slides, incubated for 24 hrs, and then incubated with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. Next, cells were fixed and immunoprobed for AIF using a specific monoclonal antibody as per standard protocols. HRP-conjugated secondary antibody was used to visualize AIF in 4910 cells in conjunction with a DAB reagent (A). Nuclear localization of AIF was also determined over time at 6, 12, 24, 48 and 72 hrs post-infection in triplicate (B).
Downregulation of MMP-2 causes the nuclear accumulation of AIF and inhibits the nuclear accumulation of pIκbα in a dose-dependent manner. To determine the nuclear localization of AIF and pIκbα, the expression levels of both were determined by western blot analysis of the nuclear and cytoplasmic extracts of Ad-MMP-2-infected cells. From the western blot analysis, we observed that that Ad-MMP-2 infection activated the nuclear localization of AIF and inhibits nuclear localization of pIκbα in 4910 human glioma xenograft in a dose-dependent manner (Fig. 3A). Quantitative analysis revealed that, under control conditions, no detectable levels of AIF were observed whereas after infection with Ad-MMP-2, nuclear localization of AIF increased in a dose-dependent manner. Significant levels of pIκbα were observed in the nucleus under control conditions whereas after Ad-MMP-2 infection, nuclear levels of pIκbα were reduced in a dose-dependent manner up to 2-fold, as seen in cells infected with 10 MOI of Ad-MMP-2 (Fig. 3B).
Figure 3. MMP-2 downregulation causes the nuclear translocation of AIF and the dephosphorylation of pIκbα.
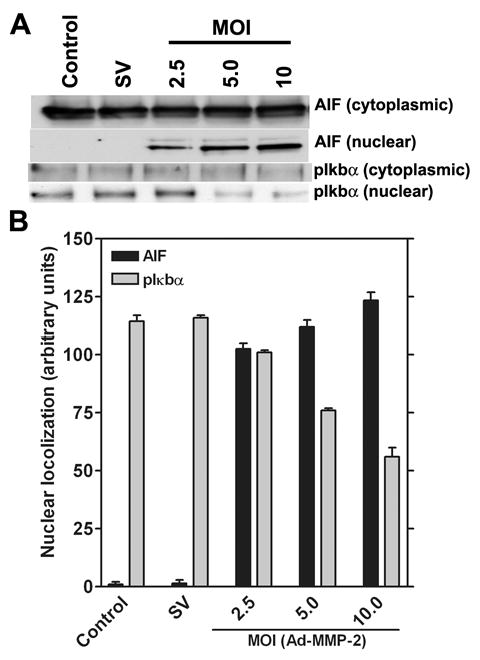
Human glioma xenograft cells 4910 were infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. After infection, cells were collected and separated into nuclear and cytoplasmic fractions and proteins extracted as per standard protocols. Expression levels of AIF and pIκbα were determined by western blot analysis (A). Levels of nuclear localization were quantified in triplicate by densitometry (B).
Downregulation of MMP-2 induces a collapse in the mitochondrial Δψ
To determine the involvement of mitochondrial membrane potential after MMP-2 downregulation, 4910 glioma xenograft cells (1×106) were seeded and infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI), followed by staining with JC-1 (Mito PT) mitochondrial Δψ indicator specific dye, and photographed using fluorescent microscopy. Quantitative analysis of mitochondrial membrane potential collapse was carried out by FACS analysis. From the results, we observed that, in Ad MMP-2 infected cells, mitochondrial Δψ collapse was demonstrated by a decrease in red fluorescence in a dose-dependent manner (Fig. 4A). FACS analysis revealed that downregulation of MMP-2 caused a dose-dependent increase in mitochondrial Δψ collapse, with the maximum effect seen after infection with 10 MOI (Fig. 4B).
Figure 4. MMP-2 downregulation induces the collapse of mitochondrial Δψ.
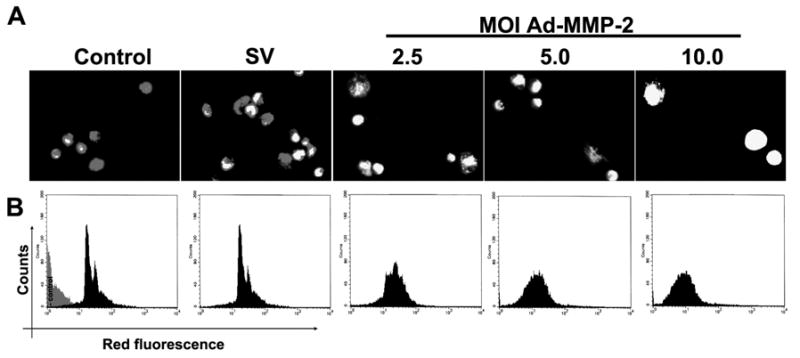
4910 glioma xenograft cells (1×103) were seeded in 8-well chamber slides, incubated for 24 hrs, and infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. Then, cells were stained with JC-1 (Mito PT) mitochondrial Δψ indicator specific dye (A). To determine initiation of apoptosis in infected 4910 human glioma xenograft cells, we carried out FACS analysis of mitochondrial membrane potential by sorting cells for decreased red fluorescence (B). A total of 10,000 cells were sorted per treatment.
Downregulation of MMP-2 induces nuclear and DNA fragmentation in a dose dependent manner
To determine the induction of nuclear fragmentation, 4910 glioma xenograft cells (1×103) were seeded in 8-well chamber slides, incubated for 24 hrs, and infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. TUNEL assay was performed to determine DNA fragmentation. In a duplicate set, cells were stained with DAPI nuclear stain to determine the extent of nuclear fragmentation. From the TUNEL assay, we determined that DNA fragmentation was initiated in a dose-dependent manner after Ad-MMP-2 infection when compared to controls. About 80% of cells infected with 10 MOI of Ad-MMP-2 were TUNEL-positive. Of cells infected with 5 MOI, 25% cells were TUNEL-positive and when infected with 2.5 MOI, 15% cells were TUNEL-positive (Fig. 5A). DAPI staining revealed nuclear condensation in a dose-dependent manner in cells infected with Ad-MMP-2 (Fig. 5B). Quantitative analysis of nuclear condensation revealed that cells infected with 10 MOI of Ad-MMP-2 showed up to 70% cells with nuclear condensation, and cells infected with 5 and 2.5 MOI showed up to 50% and 35% nuclear condensation, respectively (Fig. 5C).
Figure 5. MMP-2 downregulation induces nuclear condensation.
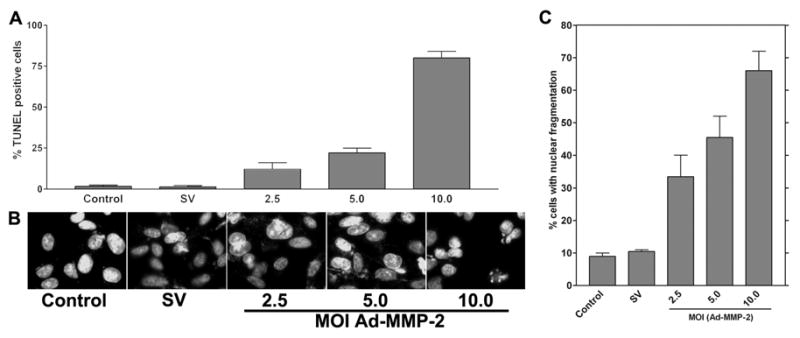
4910 glioma xenograft cells (1×103) were seeded in 8-well chamber slides, incubated for 24 hrs, and infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. Next, cells were fixed and subjected to TUNEL assay (A) or DAPI staining to visualize nuclear morphology (B). DAPI-stained cells showing nuclear condensation, which is indicative of apoptosis, were quantified and graphically represented (C).
Downregulation of MMP-2 causes upregulation of caspase-8 mRNA and downregulation of TP73 mRNA
To survey the molecular events in relation to apoptosis, we used the Human Apoptosis Pathway RT2² Profiler™ PCR Array to profile the expression of genes related to the apoptotic pathway after MMP-2 downregulation in human glioma xenograft cells. The results show that the expression of caspase-8 increased 57-fold while levels of TP73 decreased 28-fold when compared to controls (Fig. 6A). Scatter plot of the test versus control samples are shown to demonstrate the validity of the experiment (Fig. 6B).
Figure 6. MMP-2 downregulation induces the overexpression of caspase-8 mRNA and the downregulation of TP73 mRNA.
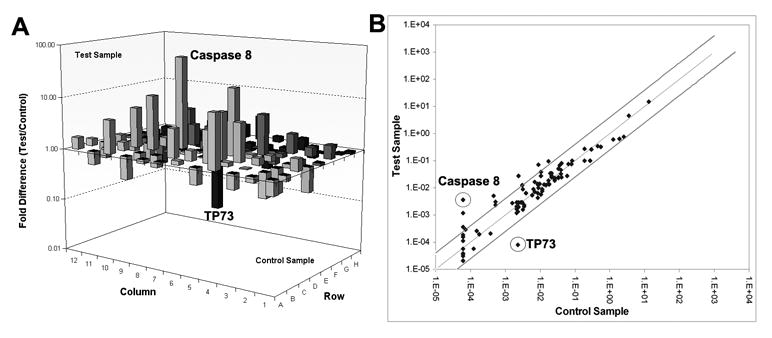
We used the RT2² Profiler™ PCR Array to profile the expression of genes related to the apoptotic signaling pathway. Human glioma xenograft cells 4910 were infected with Ad-SV (10 MOI) or Ad-MMP-2 (2.5, 5 or 10 MOI) for 36 hrs. Total RNA was isolated from treated and control cells, and mRNA were further isolated using oligo dT conjugated paramagnetic particles. cDNA was synthesized from the purified mRNA as per standard protocol. Real time PCR was performed and Ct values were plotted and fold change determined by 2-ΔCt (experiment-control) (A). Scatter plot analysis was carried out to validate experimental results (B).
Discussion
Matrix metalloproteinases (MMPs) play an important role in the degradation of extracellular matrix (ECM) under various physiological and pathological conditions. Accumulated evidence suggest that MMPs contribute to tumor cell invasion of the surrounding normal tissue and metastasis through cell surface ECM degradation. Strong correlations have been reported between elevated MMP levels and tumor cell invasiveness in human gliomas. Among them, attention has been focused on gelatinases (MMP-2 and MMP-9) and membrane type MMPs (MT-MMPs) (10). Previously, researchers have demonstrated that targeting MMP-2 via siRNA caused reduction in invasion and growth of laryngeal squamous cell carcinoma tumors (11). In the present study, we have used adenovirus-based siRNA to target MMP-2 expression. Our results demonstrate that specific targeting of MMP-2 via siRNA caused a decrease in enzymatic activity accompanied by a decrease in mRNA expression levels, which indicates degradation of mRNA molecules and the RNAi effect of our construct.
Apoptosis is a programmed form of cell death with well-defined morphological traits that are often associated with activation of caspases. Apoptotic pathways are activated by death receptors of the tumor necrosis factor (TNF) family, such as Fas, TNFR1, or the TRAIL receptors DR4 and DR5. These are the most well characterized apoptosis pathways, and many of our ideas about apoptosis regulation come from studying them. Apoptotic cell death from such receptors occurs because of the recruitment of the adaptor protein FADD, which in turn recruits the pro-form of caspase-8. Aggregation of pro-caspase-8 leads to its activation and the subsequent activation of effector caspases such as caspase-3. These apoptotic signals can be amplified through the mitochondria and inhibited through the action of competing molecules such as the inhibitor c-FLIP, which binds to the receptor complex in place of caspase-8 (12). Researchers have even attempted to overexpress caspase-8 molecules in glioma cells for therapeutic purposes (13). In the present study, we downregulated MMP-2, which is known to be overexpressed in malignant tumors, and determined the activation of caspase-8 both at the mRNA and protein levels using western blot analysis and real time RT-PCR.
Researchers have shown that overexpression of caspase-8 induced apoptosis in U251 and U-373MG glioma cells, but it did not induce apoptosis in human endothelial cells, fibroblasts, and nerve growth factor-treated PC12 cells (13). As such, overexpression of a protein may possess deleterious effects when considered in vivo, though the addition of a pro-apoptotic molecule in an in vivo system does not rule out off target effects. Recently, researchers have demonstrated a direct role for caspase-8-mediated proteolysis in promoting gene transcription (14). These studies indicate that caspase-8 overexpression could have unforeseen effects in clinical settings.
RNAi-mediated targeting of MMP-2 gene expression targets the mRNA expression levels and thereby causes a decrease in MMP-2 activity. Since gliomas overexpress large quantities of MMP-2, targeting MMP-2 expression causes a decrease in invasion and migration of tumors both in vitro and in vivo (15). Here, we have demonstrated that MMP-2 downregulation also causes the activation of caspase-8. We have also observed that the overexpression of caspase-8 is accompanied by the upregulation of NIK and the downregulation of TRADD and TRAF-2. Researchers have demonstrated that pretreatment of B16F10 melanoma cells with anti-MMP-2 antibody along with anti-MMP-9 antibody drastically inhibited the OPN-induced cell migration and chemo invasion, whereas cells pretreated with anti-MMP-2 antibody had no effect on OPN-induced pro-MMP-9 activation, suggesting that OPN induces pro-MMP-2 and pro-MMP-9 activation through two distinct pathways (16). These results indicate that NIK and MMP-2 may possess feedback inhibition where MMP-2 is the dominant molecule.
TNF and Fas-L have been shown to induce the caspase cascade by binding and activating their membrane receptors, TNF receptor-1 (TNFR1) and Fas, respectively. TNFR1 associates with the TNFR-associated death domain protein (TRADD) through the death domain-death domain interaction in the cytoplasm. Similarly, Fas associates with the Fas-associated death domain protein (FADD) through the death domain-death domain interaction. FADD has another important domain, the death effector domain (DED) (17). It is known that the interaction of caspase-8, an initiator caspase, with FADD results in the activation of caspase-8, thereby leading to the activation of downstream caspases, including caspase-3. In the present study, the activation of caspase-8 is accompanied by decreases in the levels of the adaptor molecules TRADD and TRAF-2. The activation of caspase-8 may not require the association with TRADD or TRAF-2. Its activation may actually be enhanced by its dissociation from the adaptor molecules. The reason for the dissociation of pro-caspase-8 from TRADD and TRAF-2 is still not clear but may be due to the conformational changes of TNFR, which would cause the dissociation of TRADD and TRAF-2 and thereby activate caspase-8.
In the present study, MMP-2 downregulation has apparently caused the activation of caspase-8. Among MMPs, the expression of the gelatinases MMP-2 and MMP-9 strongly correlates with glioma progression (18). Since MMP-2 is also a transient cell surface protease bound to receptors like CD44 via the PEX domain, it may provide a protective role to Fas receptors. Researchers have shown that the conserved binding ability of the HPX domains suggests that CD44H may act as a core molecule assembling multiple MT-MMPs and possibly MMP-2 and MMP-9 on the cell surface (19). Researchers have also shown that the PEX/HPX domain of MT1-MMP was indispensable in promoting cell migration and CD44H shedding (20). All these studies support the fact that the presence of MMP-2 on the cell surface somehow prevents the activation of caspase-8 via the TRADD/TRAF-2 association.
We have also observed that MMP-2 downregulation is accompanied by the nuclear translocation of AIF from the mitochondria. AIF (apoptosis-inducing factor) is another mitochondrial protein that is released into the cytoplasm during apoptosis (21). AIF, which is identical to programmed cell death 8, is a 57 kDa flavoprotein that is homologous to oxidoreductases in bacteria. It contains two mitochondrial localization sequences as well as two nuclear localization signal sequences. AIF resides in the intermembrane space of the mitochondria, and when apoptotic signals arrive at the mitochondria, it can translocate to the cytoplasm and nucleus. In the nucleus, AIF is thought to increase chromatin condensation and large-scale DNA fragmentation by unknown mechanisms. AIF also seems to be involved in an increase in the release of cytochrome c, a dissipation of mitochondrial transmembrane potential and an exposure of phosphatidylserine to the cell surface. These effects of AIF are not inhibited by inhibitors of caspases, suggesting that AIF induces a caspase-independent apoptosis (17).
From our immunohistochemical studies and western blot analysis of nuclear and cytoplasmic proteins, it is evident that AIF shows nuclear translocation with MMP-2 downregulation in a dose-dependent manner. The release of AIF from the mitochondria is initiated by pro-apoptotic signals to the mitochondria. In our previous study, we have shown that activation of caspase-8 is accompanied by the release of cytochrome c and cleavage of PARP, which is indicative of Fas-mediated apoptosis (22). The mechanism for initiating apoptotic events via the mitochondria caused by downregulating cell surface components is still not clear.
From the results of the present study, it is clear that MMP-2 downregulation caused mitochondrial Δψ collapse accompanied by the nuclear translocation of AIF. We also observed that pIκbα levels in the nucleus decreased with increasing MOI of Ad-MMP-2, indicating that downregulation of MMP-2 caused the decrease in pIκbα expression in the nucleus. It is known that pIκbα regulates NF-κB nuclear transport. NF-κB is a key regulator of stress-induced transcriptional activation and has been implicated in mediating primary or acquired apoptosis resistance in various cancers (23). By retarding the nuclear transport of NF-κB, survival molecules may be underexpressed and may trigger mitochondrial or caspase mediated apoptosis as seen in our experiments. Our Mito-PT experiments confirm a collapse in mitochondrial Δψ. The results from western blot, immunolocalization and nuclear condensation studies show that MMP-2 downregulation triggers activation of caspase-8 and mitochondrial-mediated apoptosis. Activation of pre-existing caspase-8 could be due to the influence of cell surface TNFR and associated molecules TRADD and TRAF-2, which need not account for the upregulation of caspase-8 mRNA levels. We observed that levels of caspase-8 mRNA were overexpressed 57-fold, thereby indicating an active regulation of caspase-8 gene and the initiation of apoptotic machinery.
In addition, the levels of TP73 were downregulated more than 28-fold. TP73 was initially described as a homologue of the tumor suppressor p53. TP73 drew the attention of tumor biologists because it is rarely mutated in human cancers and can induce cell cycle arrest and apoptosis by activating genes also regulated by p53. However, TP73 harbors an additional promoter that produces a dominant negative p73 protein (ΔNp73), which has the opposite effect of the TAp73 protein. Thus, the regulation of p53-responsive genes in the absence of p53 relies on a critical balance between different p73 gene-derived proteins. The molecular mechanism through which p73 induces apoptosis involves (i) expression and changes in subcellular localization of scotin, producing an endoplasmic reticulum (ER) stress; and (ii) transactivation of PUMA and Bax, thus determining cell fate. On the contrary, ΔNp73 inhibits apoptosis, thus contributing to the oncogenic potential of neuroblastoma cells (24). The identification of the TP73 isoform in our cell line would shed light on the mechanisms of TP73 regulation and its role in cell cycle regulation. Taken together, our results suggest the importance of modulating protease regulation as a potential apoptotic inducer with therapeutic possibilities.
Acknowledgments
We thank Shellee Abraham for assistance in manuscript preparation and Diana Meister and Sushma Jasti for manuscript review.
This research was supported by National Cancer Institute Grant CA75557, CA116708, CA138409 (to J.S.R.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Reference List
- 1.Lakka SS, Gondi CS, Rao JS. Proteases and glioma angiogenesis. Brain Pathol. 2005;15:327–341. doi: 10.1111/j.1750-3639.2005.tb00118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Y, Dutta J, Gupta N, Fan G, Gelinas C. Regulation of programmed cell death by NF-kappaB and its role in tumorigenesis and therapy. Adv Exp Med Biol. 2008;615:223–250. doi: 10.1007/978-1-4020-6554-5_11. [DOI] [PubMed] [Google Scholar]
- 3.St-Pierre Y, Couillard J, Van TC. Regulation of MMP-9 gene expression for the development of novel molecular targets against cancer and inflammatory diseases. Expert Opin Ther Targets. 2004;8:473–489. doi: 10.1517/14728222.8.5.473. [DOI] [PubMed] [Google Scholar]
- 4.Cortes SM, Rodriguez FV, Sanchez PI, Perona R. The role of the NFkappaB signalling pathway in cancer. Clin Transl Oncol. 2008;10:143–147. doi: 10.1007/s12094-008-0171-3. [DOI] [PubMed] [Google Scholar]
- 5.Chalah A, Khosravi-Far R. The mitochondrial death pathway. Adv Exp Med Biol. 2008;615:25–45. doi: 10.1007/978-1-4020-6554-5_3. [DOI] [PubMed] [Google Scholar]
- 6.Herzig S, Martinou JC. Mitochondrial dynamics: to be in good shape to survive. Curr Mol Med. 2008;8:131–137. doi: 10.2174/156652408783769625. [DOI] [PubMed] [Google Scholar]
- 7.Chetty C, Bhoopathi P, Joseph P, Chittivelu S, Rao JS, Lakka SS. Adenovirus-mediated siRNA against MMP-2 suppresses tumor growth and lung metastasis in mice. Mol Cancer Ther. 2006;5:2289–2299. doi: 10.1158/1535-7163.MCT-06-0169. [DOI] [PubMed] [Google Scholar]
- 8.Mohan PM, Chintala SK, Mohanam S, Gladson CL, Kim ES, Gokaslan ZL, Lakka SS, Roth JA, Fang B, Sawaya R, Kyritsis AP, Rao JS. Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Res. 1999;59:3369–3373. [PubMed] [Google Scholar]
- 9.Mohanam S, Chandrasekar N, Yanamandra N, Khawar S, Mirza F, Dinh DH, Olivero WC, Rao JS. Modulation of invasive properties of human glioblastoma cells stably expressing amino-terminal fragment of urokinase-type plasminogen activator. Oncogene. 2002;21:7824–7830. doi: 10.1038/sj.onc.1205893. [DOI] [PubMed] [Google Scholar]
- 10.Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci. 2003;8:e261–e269. doi: 10.2741/1016. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Liu M, Yang B, Li B, Lu J. Role of siRNA silencing of MMP-2 gene on invasion and growth of laryngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2008 doi: 10.1007/s00405-008-0684-y. [DOI] [PubMed] [Google Scholar]
- 12.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Shinoura N, Koike H, Furitu T, Hashimoto M, Asai A, Kirino T, Hamada H. Adenovirus-mediated transfer of caspase-8 augments cell death in gliomas: implication for gene therapy. Hum Gene Ther. 2000;11:1123–1137. doi: 10.1089/10430340050015185. [DOI] [PubMed] [Google Scholar]
- 14.Scott FL, Fuchs GJ, Boyd SE, Denault JB, Hawkins CJ, Dequiedt F, Salvesen GS. Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J Biol Chem. 2008 doi: 10.1074/jbc.M800331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kargiotis O, Chetty C, Gondi CS, Tsung AJ, Dinh DH, Gujrati M, Lakka SS, Kyritsis AP, Rao JS. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene. 2008;27:4830–4840. doi: 10.1038/onc.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rangaswami H, Bulbule A, Kundu GC. Nuclear factor-inducing kinase plays a crucial role in osteopontin-induced MAPK/IkappaBalpha kinase-dependent nuclear factor kappaB-mediated promatrix metalloproteinase-9 activation. J Biol Chem. 2004;279:38921–38935. doi: 10.1074/jbc.M404674200. [DOI] [PubMed] [Google Scholar]
- 17.Cho SG, Choi EJ. Apoptotic signaling pathways: caspases and stress-activated protein kinases. J Biochem Mol Biol. 2002;35:24–27. doi: 10.5483/bmbrep.2002.35.1.024. [DOI] [PubMed] [Google Scholar]
- 18.Levicar N, Nuttall RK, Lah TT. Proteases in brain tumour progression. Acta Neurochir (Wien) 2003;145:825–838. doi: 10.1007/s00701-003-0097-z. [DOI] [PubMed] [Google Scholar]
- 19.Suenaga N, Mori H, Itoh Y, Seiki M. CD44 binding through the hemopexin-like domain is critical for its shedding by membrane-type 1 matrix metalloproteinase. Oncogene. 2005;24:859–868. doi: 10.1038/sj.onc.1208258. [DOI] [PubMed] [Google Scholar]
- 20.Mori H, Tomari T, Koshikawa N, Kajita M, Itoh Y, Sato H, Tojo H, Yana I, Seiki M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002;21:3949–3959. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 22.Gondi CS, Kandhukuri N, Dinh DH, Gujrati M, Rao JS. Down-regulation of uPAR and uPA activates caspase-mediated apoptosis and inhibits the PI3K/AKT pathway. Int J Oncol. 2007;31:19–27. [PMC free article] [PubMed] [Google Scholar]
- 23.La Ferla-Bruhl K, Westhoff MA, Karl S, Kasperczyk H, Zwacka RM, Debatin KM, Fulda S. NF-kappaB-independent sensitization of glioblastoma cells for TRAIL-induced apoptosis by proteasome inhibition. Oncogene. 2007;26:571–582. doi: 10.1038/sj.onc.1209841. [DOI] [PubMed] [Google Scholar]
- 24.Rossi M, Sayan AE, Terrinoni A, Melino G, Knight RA. Mechanism of induction of apoptosis by p73 and its relevance to neuroblastoma biology. Ann NY Acad Sci. 2004;1028:143–149. doi: 10.1196/annals.1322.015. [DOI] [PubMed] [Google Scholar]