

Abstract
T cell receptors (TCR) recognize antigenic peptides displayed by MHC molecules. Whereas T-cell recognition of foreign peptides is essential for immune defense against microbial pathogens, recognition of self-peptides can cause autoimmune disease. Structural studies of anti-foreign TCR showed remarkable similarities in the topology of TCR binding to peptide–MHC, which maximize interactions with the ligand. However, recent structures involving autoimmune and tumor-specific TCR have revealed that they engage self-peptide–MHC with different topologies, which are suboptimal for TCR binding. These differences might reflect the distinct selection pressures exerted on anti-microbial versus autoreactive T cells. The structures also provide new insights into TCR cross-reactivity, which can contribute to autoimmunity by increasing the likelihood of self-peptide–MHC recognition.
T-cell recognition of non-self, self and mutant self
The immune system has evolved in response to the need for distinguishing non-self pathogens from self tissues. Discrimination is mediated by T-cell receptors (TCR), which recognize peptide fragments (epitopes) bound to MHC molecules on the surface of antigen-presenting cells (APC) (Box 1). These peptides are generated by proteolytic degradation of self or foreign proteins within cells expressing MHC class I or II molecules in a process known as antigen presentation [1]. Whereas T-cell recognition of foreign peptides is vital for defense against invading microorganisms, recognition of self-peptides can lead to autoimmune disease. A third category of T-cell epitopes involves self-peptides resulting from mutations accumulated during aging or disease [2,3]. However, in terms of T-cell recognition, the boundaries separating foreign, self, and altered-self epitopes are not necessarily absolute. For example, immunity to cancer can arise from mutations in self-proteins, which render them visible to T cells [2,3], a process that might also induce autoimmunity [4]. Similarly, autoimmunity can be triggered by viral or bacterial peptides that mimic self-peptides [5,6].
Box 1.
Structure of TCR and MHC molecules
The TCR is composed of two chains, α and β, each of which comprises an N-terminal variable (V) region and a C-terminal constant (C) region (Figure I). The Vα and Vβ regions are structurally related to the V domains of the light and heavy chains of antibody molecules. Loops corresponding to the CDR of antibodies are located at the membrane-distal ends of Vα and Vβ, where they collectively form the binding site for peptide–MHC. The first and second CDR (CDR1 and CDR2) are encoded within TCR V gene segments, whereas the third CDR (CDR3) is formed by DNA recombination involving juxtaposition of Vα and Jα segments for α-chain genes, and of Vβ, D and Jβ segments for β-chain genes. During recombination, the coding ends of the V, D and J segments are subjected to base addition and/or deletion, introducing considerable junctional diversity. As a result, the Vα and Vβ CDR3 loops account for most of the variability of TCR binding sites, whereas variability contributed by CDR1 and CDR2 is restricted to that already existing in the germline. MHC molecules consist of two polypeptide chains (α and β2m for MHC class I; α and β for MHC class II). The α and β chains of MHC class II each contain two extracellular domains (α1, α2, β1, β2); the two that are distal from the membrane (α1 and β1) compose the peptide-binding site. This site is a long cleft with walls formed by two α-helices, and with a floor consisting of a β-sheet derived from the pairing of the α1 and α2 domains. The TCR interacts with the compound surface created by MHC α-helices and the MHC-bound peptide.
Although much is known about TCR recognition of foreign antigens [7], the structural and biophysical principles governing TCR recognition of self and mutant-self have only recently begun to be elucidated. Here we review structural studies of self-recognition by autoimmune TCR [8–11], and of altered self-recognition by tumor-specific TCR [12]. We focus on MHC class II-restricted TCR because susceptibility to autoimmune diseases is more strongly linked to MHC class II than class I. The strength of the disease associations of particular MHC class II alleles makes them the primary genetic risk factors for autoimmune disorders [13].
Sources of autoimmunity
The ability to distinguish between self and non-self is known as immunological tolerance, and involves the removal of autoreactive T cells by negative selection during T-cell ontogeny in the thymus, thereby avoiding immune responses to self. However, in organ-specific auto-immune diseases, such as type I (juvenile) diabetes and multiple sclerosis (MS), the existence of T cells reactive with self-antigens in peripheral blood demonstrates that negative selection is imperfect. In type I diabetes, destruction of insulin-secreting pancreatic β cells is mediated by MHC class II-restricted, CD4+ T cells specific for various β-cell proteins, including insulin and glutamic acid decarboxylase [14,15]. Similarly, MHC class II-restricted T cells reactive with central nervous system antigens, such as myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein, are key to the induction and perpetuation of MS [16]. Susceptibility to autoimmune diseases is often associated with specific MHC class II alleles; for example, human leukocyte antigen (HLA)-DR4 in type I diabetes and HLA-DR2 in MS [14–16]. These associations might arise from differences in the ability of different MHC alleles to present self-peptides to autoreactive T cells [13]. Alternatively, some MHC molecules might drive the positive selection of developing T cells specific to particular autoantigens.
Various mechanisms have been put forward to explain why some autoreactive T cells escape thymic deletion [17]. In certain cases, escape from negative selection might result from simple lack of self-antigen expression in the thymus. However, most self-antigens, including those implicated in autoimmune disorders (insulin, MBP) are produced in the thymus [18]. A different, yet related, mechanism for avoiding thymic deletion involves expression, in the thymus, of splice variants of self-proteins that do not contain the relevant T-cell epitope [19]. However, these mechanisms do not account for most autoreactive T cells in peripheral blood. Instead, thymic selection is based primarily on recognition of self-peptide–MHC complexes, whereby weak interactions with self-peptide–MHC permit T-cell survival (positive selection), whereas strong interactions induce apoptosis (negative selection) [20,21]. Failure of negative selection could result from reduced TCR affinity for self-peptide–MHC ligands, such that the complex with TCR is too short-lived to enable negative selection [17]. Alternatively, unusually weak binding of the self-peptide to MHC could effectively destabilize the complex with TCR [22]. Both these affinity-based mechanisms are supported by data from animal models of autoimmune disease [17,23]. They are also supported by binding measurements using surface plasmon resonance (SPR), which have demonstrated correlations between the longevity (half-life) of TCR–peptide–MHC complexes and selection outcome [21,24,25]. Typically, TCR recognizing foreign epitopes bind peptide or MHC with dissociation constants (KDs) in the 1–100 μM range [26], whereas autoreactive TCR that have escaped negative selection exhibit weaker affinities.
It has also been proposed that autoimmunity might result from mutations in self-proteins that render them immunogenic [3,4]. Although animals have sophisticated machinery for protecting their genetic integrity, this machinery (like negative selection) is imperfect, and errors accumulate during disease processes, most notably cancer. For example, a recent study using combinatorial cDNA libraries showed that immunizing mice with mutated pigment-related proteins associated with melanoma elicited rejection of melanoma tumors and autoimmune hypopigmentation by generating T-cell responses to self [4]. Such mutant self-proteins are situated at the interface between truly self, and foreign, antigens. In other cases, immune tolerance might be broken by post-translational modification of self-antigens, creating neo-antigens [27].
Binding topology of TCR specific for foreign epitopes
Until recently, structural studies of TCR–peptide–MHC complexes had been restricted to TCR specific for microbial and other foreign epitopes, or displaying alloreactivity [7]. These studies demonstrated remarkable similarities in the overall topology of TCR binding to peptide–MHC, irrespective of MHC class I or class II restriction. Typically, the TCR is positioned diagonally across the composite surface created by the peptide and the MHC α-helices that flank the peptide-binding groove, with the Vα domain situated over the N-terminal half of the peptide, and the Vβ domain over the C-terminal half [7]. The diagonal orientation is exemplified by the structure of human TCR HA1.7 bound to an influenza virus hemagglutinin (HA) peptide and the MHC class II molecule HLA-DR1 (Figure 1a,b) [28]. However, significant variations from this topology have been observed [29]. Notably, MHC class I-restricted TCR that recognize viral epitopes with no upward-facing side chains [30], or that bulge from the peptide-binding groove [31], have been found to bind orthogonally.
Figure 1.
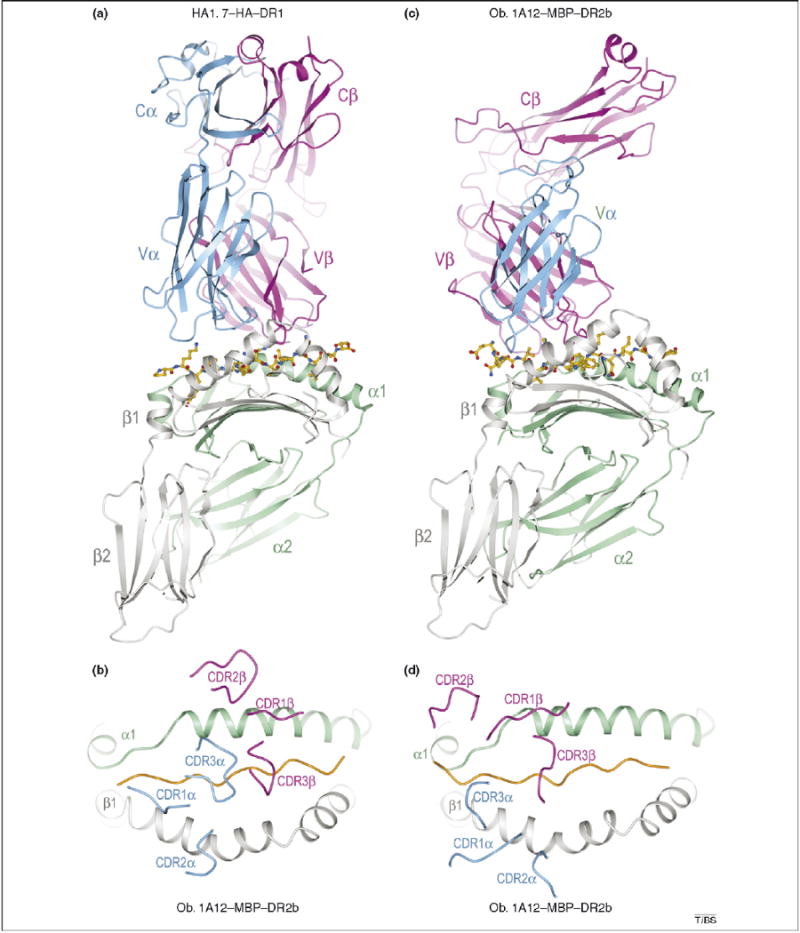
Structures of TCR–peptide–MHC class II complexes. The structures demonstrate that different binding topologies can exist between anti-foreign and autoimmune complexes (compare a and b with c and d). (a) Ribbon diagram showing a side view of the anti-microbial HA1.7–HA–DR1 complex (Protein Database Accession No. 1FYT). TCR α-chain, blue; β-chain, magenta; MHC α-chain, green; β-chain, gray. The peptide is drawn in ball-and-stick representation with carbon atoms in yellow, nitrogen atoms in blue, and oxygen atoms in red. (b) Top view of the HA1.7–HA–DR1 complex showing positions of the Vα and Vβ CDR loops on the peptide–MHC surface. The peptide is represented as a solid orange line. (c) Side view of the autoimmune Ob.1A12–MBP–DR2b complex (Protein Database Accession No. 1YMM). The Cα domain is disordered in the crystal and is not shown. (d) Top view of the Ob.1A12–MBP–DR2b complex.
For anti-foreign TCR, the most structurally diverse complementarity-determining regions (CDR) of the receptor, CDR3α and CDR3β, are usually located over the central residue of the MHC-bound peptide (residue P5), but might also focus on the C-terminal end of the peptide [7]. For example, a TCR recognizing a cytomegalovirus peptide bound to the MHC class Ib molecule HLA-E was found to be centered on residue P8 [32]. In the case of MHC class II-restricted TCR specific for foreign epitopes, the CDR3α and CDR3β loops form a pocket that accommodates the side chain of residue P5 [28,33]. This docking orientation maximizes interactions between the CDR3 loops and the antigenic peptide. The less diverse CDR1 and CDR2 loops mediate the prevalence of contacts to the MHC α-helices, but might also participate in peptide recognition (Figure 1b).
Structural studies of peptide–MHC class II recognition by autoreactive TCR
The overall similarities between the initial structures of TCR bound to MHC class I and II led to the expectation that all TCR bind peptide–MHC complexes in a similar way, and that peptide–MHC recognition by autoreactive TCR would be indistinguishable from that by anti-foreign TCR. However, several structures of autoimmune TCR– peptide–MHC complexes have now been reported: (i) the complex between human TCR Ob.1A12 and MBP 85–99 presented by HLA-DR2b [9]; (ii) the complex between human TCR 3A6 and MBP 89–101 presented by HLA-DR2a [10]; and (iii) the complex between mouse TCR 172.10 and MBP 1–11 presented by I-Au [8]. TCR Ob.1A12 and 3A6 were isolated from MS patients, and humanized mice transgenic for these TCR and for DR2b or DR2a develop symptoms typical of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. TCR 172.10 was derived from a T-cell clone that causes EAE following transfer into mice. Significantly, all three autoimmune TCR engage peptide–MHC class II with unconventional binding topologies compared with TCR specific for foreign antigens. In addition, the structure of a tumor-specific TCR bound to a mutant self-peptide presented by HLA-DR1 showed features intermediate between anti-foreign and autoimmune TCR, which might reflect the hybrid nature of altered self [12]. These studies are described in the following sections. An important limitation to the data presented is that the current structural database for MHC class II-restricted TCR (which is considerably smaller than that for class I-restricted TCR) [7], might not define the full range of docking topologies for class II-restricted TCR.
Non-canonical binding mode of a myelin-reactive TCR from an MS patient
The first structure of a human autoimmune TCR in complex with self-peptide–MHC showed a markedly different docking topology from those of all previously reported TCR–peptide–MHC complexes involving non-self ligands [9]. As expected for a TCR that has escaped negative selection, Ob.1A12 binds MBP–DR2a with low affinity (KD > 100 μM) [34]. In the complex (Figure 1c), the TCR is not centered over peptide–MHC and only contacts the N-terminal portion of the MBP self-peptide (Figure 1d). In addition, in contrast to HA1.7 and other anti-foreign TCR, Ob.1A12 exhibits a counterclockwise rotation relative to peptide–MHC (Figure 2a,d) resulting in a highly asymmetrical interaction with MHC. Thus, the orientation angle of Ob.1A12 to peptide–MHC (defined as the angle between a line connecting the Cα atoms of residues P1 and P9 of the peptide and a line connecting the centers of mass of the Vα and Vβ domains) [33] is 110°, compared with 70° for HA1.7 [28]. Notably, this orientation angle lies far outside the range for all reported MHC class I- or class II-restricted TCR (45°–80°) [7], including autoimmune TCR 3A6 and 172.10 (see following section).
Figure 2.
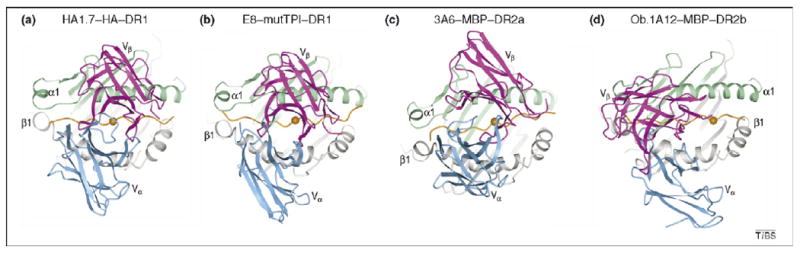
Comparison of human TCR–peptide–MHC class II complexes. The structures show the range of docking topologies observed for MHC class II-restricted TCR. (a) Top view of the anti-microbial HA1.7–HA–DR1 complex. Colors of TCR, MHC and peptide are the same as in Figure 1. The central P5 residue of the peptide is shown as an orange sphere. (b) The anti-tumor E8–mutTPI–DR1 complex (Protein Database Accession No. 2IAM). (c) The autoimmune 3A6–MBP–DR2a complex (Protein Database Accession No. 1ZGL). (d) The autoimmune Ob.1A12–MBP–DR2b complex.
In contrast to anti-microbial TCR, which contact the MHC α-helices mostly through the germline-encoded CDR1 and CDR2 loops of Vα and Vβ (Figure 1b), the majority of contacts to MHC made by Ob.1A12 are mediated by the more structurally diverse CDR3 loops. In particular, CDR3β spans the peptide-binding groove and contacts both α-helices of HLA–DR2b (Figure 1d). Because most MHC-contacting residues in CDR3β of Ob.1A12 are encoded by N-region nucleotides that were randomly inserted at the Vβ-D and D-Jβ junctions during rearrangement of the Ob.1A12 β-chain, it seems that sequence variability in the CDR3 loops makes unconventional binding topologies possible.
Other features distinguish the Ob.1A12–MBP–DR2a complex from complexes involving anti-foreign TCR. Because of the overall shift in the Ob.1A12 footprint on MBP–DR2b (Figure 2d), the TCR is tilted towards the DR2b β-chain, and it makes many more interactions with this chain than the α-chain. Only CDR1α and CDR2α are located in similar positions on the MHC β-chain in the Ob.1A12 and HA1.7 structures (Figure 1b,d). In addition, the two CDR3 loops of Ob.1A12 form a broad pocket that accommodates the P2 side chain of MBP, as well as a side chain from the MHC molecule (His81β) (Figure 3d,h). By contrast, this pocket accommodates only a single peptide residue (P5) in class II-restricted anti-foreign TCR (Figure 3a,e) [28,33]. As discussed in the following section, the focus of Ob.1A12 on the N-terminal, rather than central, portion of the self-peptide might be frequent among autoreactive TCR.
Figure 3.
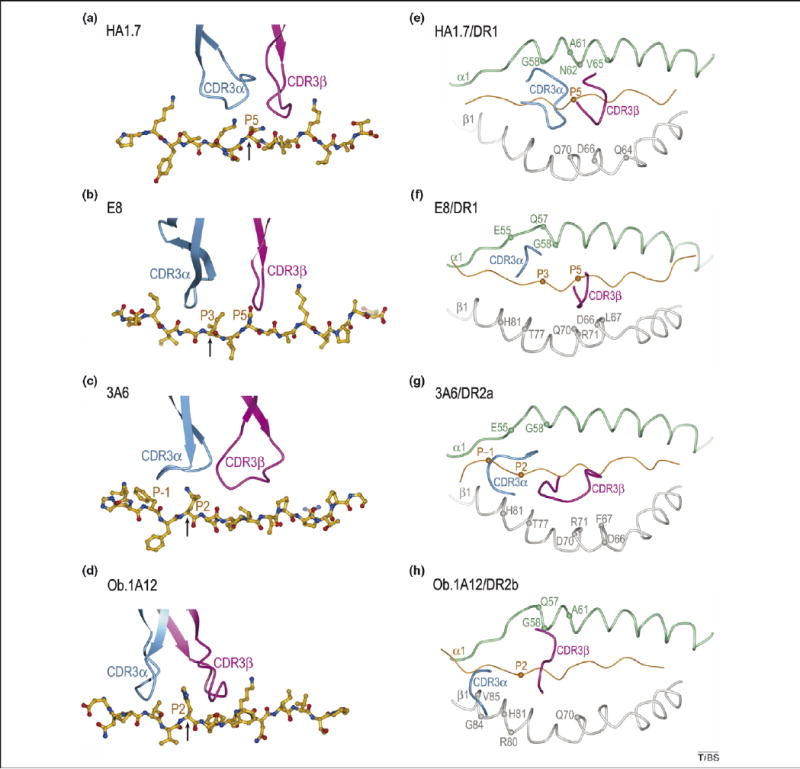
Position of TCR CDR3 loops over foreign, self or mutant self-peptide antigens in human TCR–peptide–MHC class II complexes. The structures show that the anti-foreign TCR focuses on the central portion of the MHC-bound peptide, whereas the autoreactive TCR focus on its N-terminal half [compare (a) with (b), (c) and (d)]. Color codes for TCR, MHC and peptide are the same as in Figure 1. (a) In the HA1.7–HA–DR1 complex, CDR3α and CDR3β are positioned above the central P5 residue (arrow) of the influenza HA peptide. (b) In the E8–mutTPI–DR1 complex, the CDR3 loops are centered over the P3 residue (arrow) of the mutant TPI self peptide, while maintaining contacts with P5. In the 3A6–MBP–DR2a (c) and Ob.1A12–MBP–DR2b (d) complexes, the two CDR3s converge over the P2 residue (arrow) of MBP, farther still toward the N-terminus of the peptide. The HA, mutTPI, and MBP peptides are aligned according to the P5 residue. (e)–(h) Positions of the CDR3 loops of TCRs HA1.7, E8, 3A6, and Ob.1A12 on the peptide/MHC surface. Peptide residues located within the pocket formed by CDR3α and CDR3β in the four complexes are indicated by spheres; MHC residues contacted by the CDR3 loops are labeled.
The non-canonical binding topology present in the Ob.1A12–MBP–DR2b structure is supported by T-cell stimulation experiments with peptide analogs demonstrating that substitutions in the C-terminal half of the MBP peptide do not affect TCR recognition (unless binding to MHC is decreased), and that MBP residues P2 and P3 are crucial TCR contacts [9]. Moreover, because other MBP-reactive T-cell clones from the same MS patient exhibit fine specificities similar to Ob.1A12, the docking mode of this TCR might not be an anomaly, but instead might highlight the presence of a wider range of possible TCR orientations on peptide–MHC than was earlier appreciated [11].
Autoimmune recognition of MBP by a TCR from a different MS patient
The 3A6–MBP–DR2a complex provides further evidence of unusual interactions between autoimmune TCR and self-peptide–MHC. Similar to Ob.1A12, 3A6 binds its self-ligand with low affinity (KD > 200 μM) [10]. In the 3A6–MBP–DR2a structure (Figure 2c), the orientation angle of TCR to peptide–MHC is 65°, compared with 70° and 110° for the HA1.7–HA–DR1 and Ob.1A12–MBP–DR2b complexes, respectively [10]. However, as with Ob.1A12 (Figure 2d), the CDR footprint of 3A6 on MBP–DR2a (Figure 2c) is shifted towards the N-terminus of the bound peptide, and towards the MHC β1 α-helix, relative to the CDR footprint of HA1.7 on its class II ligand (Figure 2a).
In the 3A6–MBP–DR2a complex, CDR3α interacts with the N-terminal portion of MBP, in which the central and C-terminal portions contact all three CDR loops of Vβ. Compared with HA1.7 (Figure 3a,e), substantial differences are observed in the placement of both CDR3 loops along the MBP peptide, such that residue P1 is enveloped by the CDR3α loop (Figure 3c,g). The pocket formed by CDR3α and CDR3β, which accommodates a single peptide side chain in other TCR, including Ob.1A12 (Figure 3d,h), accommodates residues P1 and P2 in 3A6 (Figure 3c,g).
Although 3A6 and Ob.1A12 engage peptide–MHC at different orientation angles (Figure 2c,d), it is striking that both TCR primarily recognize the N-terminal, rather than the central, portion of the peptide. Could this focus on the N-terminal half of the self-peptide be broadly characteristic of autoimmune TCR? It has been proposed that the N-terminal site is intrinsically suboptimal for TCR binding, favoring escape from negative selection [10]. Conversely, anti-microbial TCR achieve higher affinities by focusing on the central, or sometimes C-terminal, portion of peptides [7,32], which presumably has more favorable sites for TCR engagement. It is also conceivable that TCR that mainly recognize the N-terminal portion of peptides are more inherently cross-reactive than those targeting the central portion, because the overall conformation of peptides bound to MHC class II molecules is more highly conserved for residues P1 to P4 than for P5 to P9 [10,35]. Such cross-reactivity could enhance the pathogenic potential of T cells expressing these TCR because it would increase the likelihood of self-peptide–MHC recognition, resulting in autoimmunity.
The mouse 172.10–MBP–I-Au complex
In the 172.10–MBP–I-Au complex [8], the CDR3 loops overlay the central region of the peptide-binding groove in the conventional manner. However, the MBP–I-Au ligand is unusual in that the N-terminal third of the binding groove is empty [36]. As a consequence, 172.10 recognizes only six peptide residues (P3 to P8), compared with nine (P1 to P8) in the case of HA1.7. Furthermore, only two CDR of 172.10 –CDR3α and CDR3β – contact MBP, whereas HA1.7 uses four CDR to engage HA. Thus, all three autoimmune TCR engage peptide–MHC with suboptimal topologies compared with TCR specific for foreign antigens.
Selection of anti-microbial versus autoimmune T cells
The different ways in which anti-foreign and autoimmune TCR recognize peptide–MHC might reflect the distinct selection pressures exerted on anti-microbial versus autoreactive T cells [9–12]. In this view, the central diagonal orientation commonly observed for TCR recognizing microbial epitopes represents an optimal binding mode for maximizing interactions between TCR and the MHC-bound peptide, resulting in high affinity for peptide–MHC (KD ∼ 1–100 μM) [26]. As such, this docking mode confers a selective advantage during the intense competition among anti-microbial T cells following an infection [37]. By contrast, autoreactive T cells face different selection pressures, whereby cells expressing TCR with too high an affinity for self-peptide–MHC are deleted or inactivated by central tolerance mechanisms [17,23]. The suboptimal binding mode of autoimmune TCR [8–11] enables certain autoreactive T cells to escape thymic deletion, without necessarily precluding their activation in the periphery at appropriate antigen concentrations or inflammatory conditions [23,38,39]. As noted previously, both Ob.1A12 and 3A6 bind self-peptide–MHC with lower affinities than TCR recognizing foreign peptide–MHC do. Although TCR 172.2 binds MBP–I-Au relatively tightly (KD ∼ 5 μM), the short half-life of the MBP–I-Au complex probably explains the escape of 172.2 T cells from negative selection [40].
TCR degeneracy and autoimmunity
Structural studies of autoimmune TCR have also provided insights into the phenomenon of TCR degeneracy (or cross-reactivity), whereby a single TCR can recognize diverse peptide–MHC ligands [41,42]. Given the large numbers of clonotypically unique TCR that can be generated by gene rearrarrangements and N-region substitutions (Box 1), it was initially believed that the immune system might be capable of generating a unique TCR for almost every antigenic peptide. However, subsequent estimates of the size of the peptide repertoire recognized by TCR showed that there are far more potentially immunogenic peptides in the environment of an animal than the number of T cells the animal has [43]. Accordingly, immunologists have evoked TCR degeneracy as a mechanism for expanding the effective size of the TCR repertoire [41,42]. TCR degeneracy also underlies the molecular-mimicry hypothesis for autoimmune disease, whereby autoimmunity can be triggered by viral or bacterial peptides that mimic self-peptides [5,6]. However, the structural basis for TCR degeneracy has remained unclear.
Degenerate peptide recognition by 3A6 and other TCR has been investigated using synthetic combinatorial peptide libraries [6,44]. In the case of 3A6, numerous peptides have been isolated from combinatorial libraries that stimulate 3A6 T cells up to 10 000-fold more efficiently than MBP [44]. Based on the 3A6–MBP–DR2a structure, these super-agonist peptides contain multiple substitutions at all TCR-contacting positions, while retaining MHC-binding anchor residues. A notable feature of the 3A6–MBP–DR2a interface is the complete absence of hydrogen bonds or salt bridges involving either main-chain or side-chain atoms of the TCR or peptide between the CDR loops of 3A6 and MBP, in contrast to all other TCR–peptide–MHC complexes [7,10] (Figure 4). Interactions between TCR and peptide are mainly restricted to van der Waals contacts, with poor fit. Hence, numerous opportunities exist for improving shape and chemical complementarity with TCR through variations in the peptide. For example, because the volume of the interfacial cavity occupied by MBP residue P1 phenylalanine seems sufficient to accommodate the bulkier tryptophan side chain found in superagonists, the stimulatory effect of this substitution probably results from additional hydrophobic or van der Waals interactions with 3A6, augmenting affinity. Similar to 3A6–MBP–DR2a, the interface of the 172.10–MBP–I-Au complex is characterized by a paucity of hydrogen bonds between TCR and peptide [8], again suggesting degeneracy.
Figure 4.
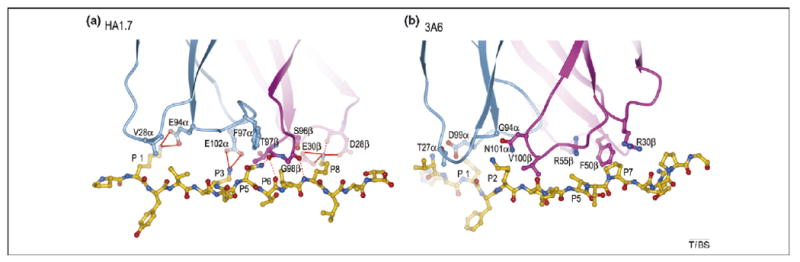
Basis for degenerate peptide recognition by an autoimmune TCR. The structures show the paucity of specific interactions between TCR and peptide in the autoimmune complex compared with the anti-foreign complex. (a) Interaction of anti-microbial TCR HA1.7 with influenza virus HA peptide. TCR α-chain, blue; β-chain, magenta; peptide, yellow. Contact residues are drawn and labeled. Hydrogen bonds are indicated by red dotted lines and salt bridges by red solid lines. (b) Interaction of autoimmune TCR 3A6 with MBP peptide. Contact residues are labeled. No hydrogen bonds or salt bridges are observed between TCR 3A6 and the MBP self-peptide.
Two recent crystallographic studies of MHC class I-restricted TCR provided additional insights into TCR degeneracy [45,46]. Thus, TCR BM3.3 recognizes three distinct peptides bound to the mouse class I molecule H-2Kb. Comparison of the corresponding BM3.3–peptide–H-2Kb complexes revealed that each docking solution achieved by BM3.3 involves a highly specific hydrogen-bonding network, with non-specific interactions and structural adjustments in the CDR loops making less important contributions to degeneracy [45]. Similarly, TCR 2C recognized two globally similar, yet distinct, peptide–MHC class I ligands via unique interatomic contacts, involving both shared and polymorphic residues on H-2Ld and H-2Kb, as well as the unrelated peptide antigens QL9 and dEV8 [46]. In agreement with results from studies of autoimmune class II-restricted TCR [8–11], 2C seems to engage its self-ligand, QL9–H-2Ld, through suboptimal interactions compared with its foreign ligand, dEV8–H-2Kb.
Recognition of altered self by a human melanoma-specific TCR
Tumor-specific antigens resulting from mutations in autologous gene products are biologically important examples of mutant self-proteins, belonging to a category between truly self and foreign antigens. These mutations might become visible to the immune system if they are incorporated into the recognized peptide epitope, or if they lead to aberrant processing of a normally cryptic wild-type epitope. An example of the former mechanism is a unique HLA-DR1-restricted human melanoma antigen derived from the glycolytic enzyme triosephosphate isomerase (TPI), in which a naturally occurring point mutation replaced a threonine residue with isoleucine within the recognized epitope [47]. Presentation of wild-type and mutant TPI (mutTPI) peptides to melanoma-specific CD4+ tumor-infiltrating lymphocytes by HLA-DR1 results in dramatically different T-cell responses, such that recognition is enhanced 100 000-fold for the mutant relative to the wild-type peptide.
The structure of a human tumor-specific TCR (E8) bound to mutTPI and HLA-DR1 has provided information on T-cell recognition of a naturally mutated self-antigen compared with recognition of native self or foreign antigens [12]. The E8–mutTPI–DR1 complex revealed several features intermediate between those of anti-foreign and autoimmune TCR–peptide–MHC class II complexes that might reflect the hybrid nature of altered self. These include a shift of E8 towards the N-terminus of the bound peptide compared with anti-foreign TCR (Figure 3b), although not as far as for autoimmune TCR, while maintaining the diagonal docking orientation of anti-foreign TCR and autoimmune TCR 3A6 (Figure 2b). As a consequence of this displacement, the CDR3 loops of E8 are positioned directly over the substituted P3 residue of mutTPI (Figure 3b,f), whereas in MBP-specific TCR 3A6 and Ob.1A12 the CDR3 loops converge on residue P2 (Figure 3c,d). Consistent with the idea that the N-terminal portion of peptides is intrinsically unfavorable for TCR binding [10], E8 resembles autoimmune TCR in binding TPI–DR1 (wild-type or mutant peptide) with low affinity, although affinity is increased by the Thr-to-Ile mutation at TCR-contacting position P3 of TPI [12].
Also in common with autoimmune TCR 3A6 and Ob.1A12, the CDR3 loops of E8 form a broad pocket that accommodates two ligand residues (P3 and P5) (Figure 3b,f), whereas the corresponding, but narrower, pocket of anti-foreign TCR generally contains only a single residue (P5). As for anti-foreign complexes, the E8–mutTPI–DR1 structure indicates that residue P5 is crucial for TCR recognition, whereas the P5 position is relatively tolerant of substitutions in the 3A6–MBP–DR2a and Ob.1A12–MBP–DR2b complexes, in which P5 lies outside the CDR3 pocket (Figure 3c,d). Finally, E8 is tilted towards the DR1 β-chain, and makes many more contacts with this than with the α-chain (80% of total contacts), a feature that also distinguishes the autoimmune 3A6–MBP–DR2a and Ob.1A12–MBP–DR2b complexes from the anti-microbial HA1.7–HA–DR1 complex. This tilt precludes formation of a conserved salt bridge between CDR2β Asp56 and invariant class II residue Lys39α [28,33], which might contribute to the low affinity of E8 for its self-ligand and the resulting escape of E8 T cells from thymic deletion.
Concluding remarks and future perspectives
The TCR–peptide–MHC class II complexes described here revealed unusual features of peptide–MHC binding by auto-immune and tumor-specific TCR, including a focus on the N-terminal half of the self-peptide and suboptimal TCR interactions with peptide. However, whether these features distinguish class II-restricted TCR recognizing self or altered self from those recognizing non-self must await determination of additional TCR–peptide–MHC class II structures. There are only seven of these to date, compared with >20 TCR–peptide–MHC class I structures [7]. These studies will be necessary to establish the full spectrum of binding modalities for MHC class II-restricted TCR.
We, and others [17,23], have emphasized reduced TCR affinity for self-peptide–MHC ligands as the main reason autoreactive T cells sometimes escape negative selection. However, it is also possible that, in certain cases, altered binding topologies might affect the interaction of TCR or MHC with accessory molecules, such as CD4, that are required for normal T-cell function. The predicted geometry of CD4 engagement by the autoimmune Ob.1A12–MBP–DR2b complex is substantially different from that by the anti-microbial HA1.7–HA–DR1 complex [9]. It is also noteworthy that low affinity for self-peptide–MHC does not necessarily preclude T-cell activation by low concentrations of the self-ligand. Various models have been proposed to explain this phenomenon, including peptide-driven formation of TCR dimers for enhancing T-cell sensitivity [12,48–50]. However, the actual existence of such signaling intermediates on the T-cell surface remains to be established.
Figure I.
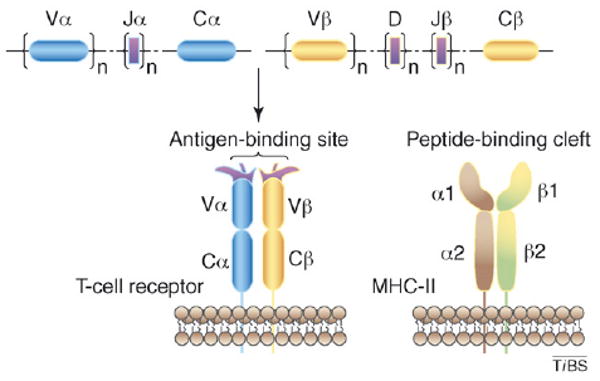
Structures of TCR and MHC class II molecules. The genes coding for TCR are split into several subgenes (V, D, J, C); V, D, and J exist in numerous copies. Somatic recombination of these subgenes during T-cell development generates vast numbers of rearranged TCR genes that encode the α and β chains of the receptor.
Acknowledgments
R.A.M. is supported by grants from the National Multiple Sclerosis Society and the National Institutes of Health. L.D. is a Cancer Research Institute Postdoctoral Fellow.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Davis MM, Chien YH. T-cell antigen receptors. In: Paul WE, editor. Fundamental Immunology. 5th. Lippincott Williams & Wilkins; 2003. pp. 227–258. [Google Scholar]
- 2.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 3.Houghton AN, Guevara-Patino JA. Immune recognition of self in immunity against cancer. J Clin Invest. 2004;114:468–471. doi: 10.1172/JCI22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelhorn ME, et al. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med. 2006;12:198–206. doi: 10.1038/nm1363. [DOI] [PubMed] [Google Scholar]
- 5.Wucherpfennig KW, Strominger JL. Molecular mimicry in T-cell mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hemmer B, et al. Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J Exp Med. 1997;185:1651–1659. doi: 10.1084/jem.185.9.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudolph MG, et al. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 8.Maynard J, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Hahn M, et al. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, et al. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson MJ, et al. Unusual features of self-peptide/MHC binding by autoimmune T cell receptors. Immunity. 2005;23:351–360. doi: 10.1016/j.immuni.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng L, et al. Structural basis for recognition of mutant self by a tumor-specific. MHC class II-restricted T cell receptor. Nat Immunol. 2007;8:398–408. doi: 10.1038/ni1447. [DOI] [PubMed] [Google Scholar]
- 13.Jones EY, et al. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6:271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 14.Nepom GT, et al. Identification and modulation of a naturally processed T cell epitope from the diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65) Proc Natl Acad Sci U S A. 2001;98:1763–1768. doi: 10.1073/pnas.98.4.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kent SC, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435:224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 16.Sospedra M, et al. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 17.Ohashi PS. Negative selection and autoimmunity. Curr Opin Immunol. 2003;15:668–676. doi: 10.1016/j.coi.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Derbinski J, et al. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 19.Klein L, et al. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6:56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- 20.Kappler JW, et al. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 21.Alam SM, et al. T-cell receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 22.Anderton SM, et al. Negative selection during peripheral immune response to antigen. J Exp Med. 2001;193:1–11. doi: 10.1084/jem.193.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodnow CC, et al. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 24.Williams CB, et al. A kinetic threshold between negative and positive selection based on the longevity of the T cell receptor–ligand complex. J Exp Med. 1999;189:1531–1544. doi: 10.1084/jem.189.10.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daniels MA, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signaling. Nature. 2006;444:724–729. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
- 26.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 27.Doyle HA, Mamula MJ. Posttranslational modification of self-antigens. Annu New York Acad Sci. 2005;1050:1–9. doi: 10.1196/annals.1313.001. [DOI] [PubMed] [Google Scholar]
- 28.Hennecke J, et al. Structure of a covalently stabilized complex of a human αβ T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 2000;19:5611–5624. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clements CS, et al. Specificity on a knife-edge: the αβ T cell receptor. Curr Opin Struct Biol. 2006;16:787–795. doi: 10.1016/j.sbi.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Stewart-Jones GBE, et al. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol. 2003;4:657–663. doi: 10.1038/ni942. [DOI] [PubMed] [Google Scholar]
- 31.Tynan FE, et al. T cell receptor recognition of a ‘super-bulged’ major histocompatibility complex class I-bound peptide. Nat Immunol. 2005;6:1114–1122. doi: 10.1038/ni1257. [DOI] [PubMed] [Google Scholar]
- 32.Hoare HL, et al. Structural basis for a major histocompatibility complex class Ib-restricted T cell response. Nat Immunol. 2006;7:256–264. doi: 10.1038/ni1312. [DOI] [PubMed] [Google Scholar]
- 33.Reinherz EL, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 34.Appel H, et al. Kinetics of T-cell receptor binding by bivalent HLA-DR peptide complexes that activate antigen-specific human T-cells. J Biol Chem. 2000;275:312–321. doi: 10.1074/jbc.275.1.312. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh P, et al. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 36.He XL, et al. Structural snapshots of antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity. 2002;17:83–94. doi: 10.1016/s1074-7613(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 37.Kedl RM, et al. Epitope dominance, competition and affinity maturation. Curr Opin Immunol. 2003;15:120–127. doi: 10.1016/s0952-7915(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 38.Gronski MA, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. 2004;10:1234–1239. doi: 10.1038/nm1114. [DOI] [PubMed] [Google Scholar]
- 39.Zehn D, Bevan MJ. Cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia KC, et al. Kinetics and thermodynamics of T cell receptor-autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2001;98:6818–6823. doi: 10.1073/pnas.111161198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holler PD, Kranz DM. T cell receptors: affinities, crossreactivities, and a conformer model. Mol Immunol. 2004;40:1027–1031. doi: 10.1016/j.molimm.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 42.Wucherpfennig KW. T cell crossreactivity as a general property of T cell recognition. Mol Immunol. 2004;40:1009–1017. doi: 10.1016/j.molimm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Mason D. A very high level of cross-reactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 44.Hemmer B, et al. Contribution of individual amino acids within MHC molecule or antigenic peptide to TCR ligand potency. J Immunol. 2000;164:861–871. doi: 10.4049/jimmunol.164.2.861. [DOI] [PubMed] [Google Scholar]
- 45.Mazza C, et al. How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? EMBO J. 2007;26:1972–1983. doi: 10.1038/sj.emboj.7601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colf LA, et al. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 47.Pieper R, et al. Biochemical identification of a mutated human melanoma antigen recognized by CD4+ T cells. J Exp Med. 1999;189:757–766. doi: 10.1084/jem.189.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krogsgaard M, et al. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 49.Cebecauer M, et al. CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex-peptide dimers. J Biol Chem. 2005;280:23820–23828. doi: 10.1074/jbc.M500654200. [DOI] [PubMed] [Google Scholar]
- 50.Minguet S, et al. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:43–54. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]