

Abstract

Thiocarboxylates, prepared conveniently by cleavage of 9-fluorenylmethyl or trimethoxybenzyl thioesters, react at room temperature with isocyanates and isothiocyanates to give amide bonds in good to excellent yield. A carboxylate salt is also shown to react with an electron-deficient isocyanate to give the corresponding amide in excellent yield at room temperature.
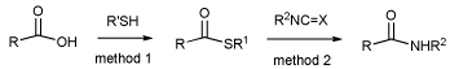
Given the widespread availability of amines and of carboxylic acids, amide bond forming reactions are necessarily some of the most widely applied transformations in parallel synthesis and combinatorial chemistry. The needs for atom ecomony, ever milder conditions, and greater selectivity combine to drive the search for new and improved methods for the formation of amide bonds.1 The condensation of isocyanides with carboxylic acids has recently received much interest in this regard,2 and this despite the relatively limited range of commercially available isocyanides and the somewhat forcing reaction conditions. Our attention was caught by a little known reaction3 involving condensations of the much more widely available isocyanates and isothiocyanates with monothiocarboxylates which result in the formation of amides with loss of a simple, volatile byproduct, carbon oxysulfide or carbon disulfide, respectively. Although this reaction was described in 1973 for use with very simple substrates it has seen very limited application subsequently,4 perhaps due to the relative paucity of commercially available thioacids.5 We reasoned that the advent of convenient mild methods for thioacid synthesis6 would combine with this reaction to afford a convenient and mild method for amide formation, and report here that this is indeed the case. In addition, we report promising preliminary results of a comparable reaction between simple carboxylic acids and electron-deficient isocyanates, with loss of carbon dioxide, also leading to the formation of amide bonds.
The 9-fluorenylmethyl (Fm) thioesters are a readily prepared class of self-stable thioesters whose treatment with piperidine provides a convenient in situ means of entry into thiocarboxylates.6b Their complement, the equally readily available 2,4,6-trimethoxybenzyl (Tmob) thioesters, release thioacids on exposure to trifluoroacetic acid.6a Accordingly, the Fm and Tmob thioesters were selected as the precursors of choice for thioacids in this study and several were prepared as outlined in Table 1. The thiocarboxylates were released with piperidine or trifluoroacetic acid and triethylsilane, according to the thioester employed, and then exposed to either isocyanates or an isothiothiocyanate at room temperature leading to the results set out in Table 1.
Table 1.
Preparation of Thioesters and Their Reaction with Isocyanates and Isothiocyanatesa
 | |||||||
|---|---|---|---|---|---|---|---|
| preparation of RCOSR1 | R2NC=X | preparation of RCONHR2 | |||||
| method 1 | thioester | yield | method 2 | amide | yield | ||
| 1 | NaH, (COCl)2 FmSH, pyr | 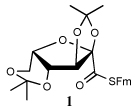 |
79% | 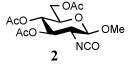 |
i) RCOSR1, piperidine in DMF, 15 min, rt | 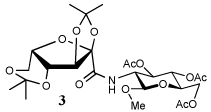 |
73% |
| ii) R2NCO, CH2Cl2, 6 h, rt | |||||||
| 2 | 1 | 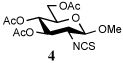 |
i) RCOSR1, piperidine in DMF, 15 min, rt | 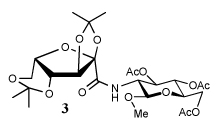 |
70% | ||
| ii) R2NCS, CH2Cl2, 6 h, rt | |||||||
| 3 | 1 | 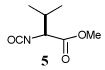 |
i) RCOSR1, piperidine in DMF, 15 min, rt | 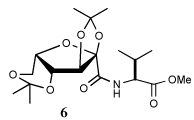 |
76% | ||
| ii) R2NCO, CH2Cl2, 6 h,rt | |||||||
| 4 | FmSH, EDCI, DMAP | 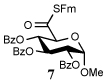 |
83% | 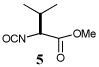 |
i) RCOSR1, piperidine in DMF, 15 min, rt | 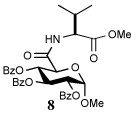 |
52% |
| R2NCO, CH2Cl2, 6 h, rt | |||||||
| 5 | TmobSH, EDCI, DMAP | 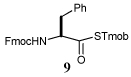 |
85% | PhNCO, 10 | i) RCOSR1, TFA, Et3SiH, CH2Cl2, 4 h, rt | 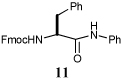 |
66% |
| ii) R2NCO, i-Pr2NEt, CH2Cl2, 6 h, rt | |||||||
| 6 | 9 | C6H13NCO, 12 | i) RCOSR1, TFA, Et3SiH, CH2Cl2, 4 h, rt | 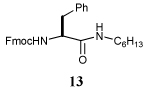 |
73% | ||
| ii) R2NCO, i-Pr2NEt, CH2Cl2, 6 h, rt | |||||||
| 7 | 9 | 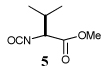 |
i) RCOSR1, TFA, Et3SiH, CH2Cl2, 4 h, rt | 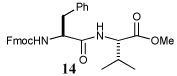 |
68% | ||
| ii) R2NCO, i-Pr2NEt, CH2Cl2, 6 h, rt | |||||||
| 8 | 9 | 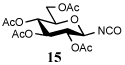 |
i) RCOSR1, TFA, Et3SiH, CH2Cl2, 4 h, rt | 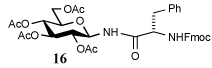 |
36% | ||
| ii) R2NCO, i-Pr2NEt, CH2Cl2, 6 h, rt | |||||||
| 9 | 9 | 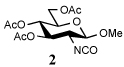 |
i) RCOSR1, TFA, Et3SiH, CH2Cl2, 4 h, rt | 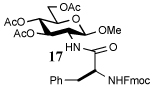 |
30% | ||
| ii) R2NCO, i-Pr2NEt, CH2Cl2, 6 h, rt | |||||||
The isocyanates used in this study were either commercial or prepared by reaction of the corresponding amine with phosgene in the presence of aqueous sodium bicarbonate in excellent yield (2, 100%). The isothiocyanate 4 was similarly prepared in quantitative yield by reaction of the amine with thiophosgene under phase transfer conditions.
Entry 1 of Table 1 establishes the viability of the method and nicely illustrates the potential for application to a hindered tertiary thioacid. The release of the thioacid from the 9-fluorenylmethyl thioester by simple treatment with piperidine in DMF at room temperature is an important feature of this reaction and one that ensures compatibility with the two acetonide groups of the substrate. A comparison of entries 1 and 2 of Table 1 indicates that isocyanates and isothiocyanates behave more or less analogously in this chemistry; this observation led us to favor the more readily available isocyanates in the subsequent studies. Examples 3 and 4 of Table 1 illustrate the coupling of 9-fluorenylmethyl thioester-derived thiocarboxylates with an α-isocyanato ester and hint at the potential for the application of this chemistry in the formation of neoglycopeptides.7 Entries 5–9 of Table 1 use the 2,4,6-trimethoxybenzylthioester technology, with release of the thioacid by treatment with trifluoroacetic acid and triethylsilane, thereby enabling the generation of the thioacid in the presence of the 9-fluorenylmethoxy carbamate protecting group favored in solid phase peptide synthesis. Entries 5 and 6 (Table 1) indicate that both aromatic and aliphatic isocyanates function correctly in this chemistry. Entry 7 of Table 1 is directed at the formation of a peptide bond, while entries 8 and 9 are again directed at the neoglycopeptide area. Only modest yields were obtained in entries 8 and 9 and in the latter case the symmetrical urea 18 was isolated as the major byproduct in 45% yield.

To further extend the scope of this reaction we have also applied it to thioacids generated in situ through the nucleophilic ring opening of succinic monothioanhydride (Table 2). Here too, generally good yields were obtained with the exception of the use of the sugar based isocyanate 2 and isothiocyanate 4 (Table 2, entries 4 and 5). Entries 4 and 5 of Table 2 serve as a second comparison between the isocyanates and the isothiocyanates and support the earlier conclusion that the two have comparable reactivity in this chemistry.
Table 2.
Three component coupling reactions of amines with succinic thioanhydride and isocyanates or isothiocyanates
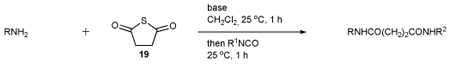 | |||||
|---|---|---|---|---|---|
| RNH2 | R1NC=X | base | product | yield | |
| 1 | Ph(CH2)2NH2 | PhNC=O | i-Pr2NEt | Ph(CH2)2NHCO(CH2)2CONHPh, 20 | 90% |
| 2 | Ph(CH2)2NH2 | C6H13NC=O | i-Pr2NEt | Ph(CH2)2NHCO(CH2)2CONHC6H13, 21 | 96% |
| 3 | PhNH2 | C6H13NC=O | sym-collidine | PhNHCO(CH2)2CONHC6H13, 22 | 66% |
| 4 | PhNH2 | 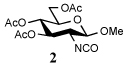 |
i-Pr2NEt | 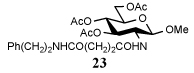 |
30% |
| 5 | PhNH2 | 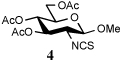 |
i-Pr2NEt | 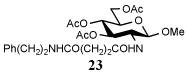 |
27% |
The general mechanism of the reaction of thiocarboxylates with isocyanates and isothiocyanates likely involves attack by the thiocarboxylate at the sp carbon to give an intermediate adduct 24 that undergoes rearrangement with loss of COS or CS2 to give the final product 25 (Scheme 1, path a).
Scheme 1.

Anticipated reaction mechanism
In view of this mechanism the somewhat poorer yields obtained with the carbohydrate based isocyanates 2 and 15 (Table 1, entries 8 and 9, Table 2, entry 4) and the related isothiocyanate 4 (Table 2, entry 5) must be viewed in the light of the intermediate adduct, particularly in view of the good yield obtained with one of these isocyanates and a different thioacid (Table 1, entry 1). We speculate the reduced yields in these cases are due to the decomposition of the intermediate adduct 24 to the product with loss of COX being retarded by the multiplicity of electron withdrawing β-C-O (acetoxy groups and/or acetals) which serve to reduce the nucleophilicity of the nitrogen atom in the adduct. A slower decomposition enables competing processes to intervene such as the attack of a further equivalent of thiocarboxylate at the thiocarboxyl center in the adduct leading to the generation of a thioanhydride and liberation of the amine (Scheme 1, path b). The symmetric urea byproducts (e.g., 18) are then formed by attack of the liberated amine on the initial isocyanate. The good yield obtained with the more hindered thioacid 1 (Table 1, entry 1) is the result of the steric hindrance about the thiocarboxylate which retards attack of external nucleophiles on the intermediate adduct, thereby providing the time for the desired rearrangement to take place.8
We have also attempted the reaction of simple carboxylate esters with isocyanates, with a view to eliminating the dependency on thiocarboxylates. In accord with the earlier findings of Kricheldorf and Leppert, however, we find this to be an unproductive avenue for most isocyanates (and isothiocyanates) surveyed owing to the much reduced nucleophilicity of the carboxylates. However, in the case of suitably electron-deficient isocyanates such reactions proceed in a satisfactory manner over a period of several hours at room temperature as illustrated in Scheme 2.
Scheme 2.

Reaction of a carboxylate with an electron-deficient isocyanate
The reaction that we describe overall is closely related to the reaction of thiocarboxylic acids with azides5l-w but is potentially of broader scope for combinatorial and parallel synthesis applications owing to the much wider commercial availability of isocyanates. While we have not investigated the possibility we anticipate that, as in the case of the azide reaction, selenocarboxylates will also perform as suitable nucleophiles in this chemistry.
Supplementary Material
Acknowledgment
We thank the NIH (GM62160) for partial support of this work.
Footnotes
Supporting Information Available. Full experimental details and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent advances see: Bode JW. Curr. Opin. Drug Discovery Dev. 2006;9:765–775.Montalbetti CAGN, Falque V. Tetrahedron. 2005;61:10827–10852.Han S-Y, Kim Y-A. Tetrahedron. 2004;60:2447–2467.
- 2.a) Li X, Yuan Y, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:13225–13227. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:13222–13224. doi: 10.1021/ja8047078. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li X, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:5446–5448. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jones GO, Li X, Hayden AE, Houk KN, Danishefsky SJ. Org. Lett. 2008;10:4093–4096. doi: 10.1021/ol8016287. [DOI] [PubMed] [Google Scholar]; e) Restrop P, Rebek JR. J. Am. Chem. Soc. 2008;130:11850–11851. doi: 10.1021/ja803854r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kricheldorf HR, Leppert E. Makromol. Chem. 1973;167:47–68. [Google Scholar]
- 4.Beyond the original paper,3 which itself has only been cited 14 times to date, we are only aware of four isolated examples of the reaction of isothiocyanates with simple thioacids (three with thioacetic acid and one with indolethioacetic acid) and of none between isocyanates themselves and thioacids. The four examples isothiocyanates and thioacids all make use of heating to 60 or 80 °C to achieve the reactions described, and none of the four papers makes reference to the original work of Kricheldorf and Leppert.3 Gonda J, Martinkova M, Walko M, Zavacka E, Budesinsky M, Cisarova I. Tetrahedron Lett. 2001;42:4401–4404.Gonda J, Zavacka E, Budesinsky M, Cisarova I, Podlaha J. Tetrahedron Lett. 2000;41:525–529.Gonda J, Bednarikova M. Tetrahedron Lett. 1997;38:5569–5572.Schoepfer J, Marquis C, Pasquier C, Neier R. J. Chem. Soc., Chem. Commun. 1994:1001–1002.
- 5.For other amide bond forming reactions employing thioacids see: Blake J. Int. Pept. Prot. Res. 1981;17:273–274. doi: 10.1111/j.1399-3011.1981.tb01992.x.Yamashiro D, Blake JF. Int. J. Pept. Prot. Chem. 1981;18:383–392. doi: 10.1111/j.1399-3011.1981.tb02996.x.Blake J, Yamashiro D, Ramasharma K, Li CH. Int. J. Peptide Protein Res. 1986;28:468–476. doi: 10.1111/j.1399-3011.1986.tb03281.x.Yamashiro D, Li CH. Int. J. Peptide Protein Res. 1988;31:322–334. doi: 10.1111/j.1399-3011.1988.tb00040.x.Mitin YV, Zapevalova NP. Int. J. Pept. Prot. Chem. 1990;35:352–356. doi: 10.1111/j.1399-3011.1990.tb00060.x.Høeg-Jensen T, Olsen CE, Holm A. J. Org. Chem. 1994;59:1257–1263.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629.Tam JP, Lu Y-A, Liu C-F, Shao J. Proc. Natl. Acad. Sci. USA. 1995;92:12485–12489. doi: 10.1073/pnas.92.26.12485.Messeri T, Sternbach DD, Tomkinson NCO. Tetrahedron Lett. 1998;39:1673–1676.Messeri T, Sternbach DD, Tomkinson NCO. Tetrahedron Lett. 1998;39:1669–1672.Crich D, Sharma I. Angew. Chem. Int. Ed. 2009;48:2355–2358. doi: 10.1002/anie.200805782.Hakimelahi GH, Just G. Tetrahedron Lett. 1980;21:2119–2122.Rosen T, Lico IM, Chu DTW. J.Org. Chem. 1988;53:1580–1582.Rakotomanomana N, Lacombe J-M, Pavia A. Carbohydr. Res. 1990;197:318–323.McKervey MA, O'Sullivan MB, Myers PL, Green RH. J. Chem. Soc., Chem. Commun. 1993:94–96.Dudkin VY, Crich D. Tetrahedron Lett. 2003;44:1787–1789.Kolakowski RV, Shangguan N, Sauers RR, Williams LJ. J. Am. Chem. Soc. 2006;128:5695–5702. doi: 10.1021/ja057533y.Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J. Am. Chem. Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919.Fazio F, Wong CH. Tetrahedron Lett. 2003;44:9083–9085.Barlett KN, Kolakowski RV, Katukojvala S, Williams LJ. Org. Lett. 2006;8:823–826. doi: 10.1021/ol052671d.Merkx R, van Haren MJ, Rijkers DTS, Liskamp RMJ. J. Org. Chem. 2007;72:4574–4577. doi: 10.1021/jo0704513.Zhu XM, Pachamuthu K, Schmidt RR. Org. Lett. 2004;6:1083–1085. doi: 10.1021/ol036186z.Merkx R, Brouwer AR, Rijkers DTS, Liskamp RMJ. Org. Lett. 2005;7:1125–1128. doi: 10.1021/ol0501119.
- 6.a) Vetter S. Synth. Commun. 1998;28:3219–3223. [Google Scholar]; b) Crich D, Sana K, Guo S. Org. Lett. 2007;9:4423–4426. doi: 10.1021/ol701583t. [DOI] [PubMed] [Google Scholar]; c) Crich D, Bowers AA. Org. Lett. 2007;9:5323–5325. doi: 10.1021/ol702570x. [DOI] [PubMed] [Google Scholar]; d) Crich D, Sasaki K, Rahaman MdY, Bowers J. Org. Chem. 2009;74:3886–3893. doi: 10.1021/jo900532e. [DOI] [PubMed] [Google Scholar]
- 7.a) Nicotra F, Cipolla L, Peri F, La Ferta B, Redaelli C. Adv. Carbohydr. Chem. Biochem. 2007;61:353–398. doi: 10.1016/S0065-2318(07)61007-5. [DOI] [PubMed] [Google Scholar]; b) Roy R. In: Carbohydrate Chemistry. Boons G-J, editor. London: Blackie Academic and Professional; 1998. pp. 243–321. [Google Scholar]; c) Lee YC, Lee RT, editors. Neoglycoconjugates:Preparation and Applications. San Diego: Academic Press; 1994. [Google Scholar]; d) Haase C, Seitz O. Top. Curr. Chem. 2007;267:1–36. [Google Scholar]
- 8.In an attempt to accelerate the decomposition of the intermediate and improve the yield of 2 and 9 a number of Lewis acids (nBu2BOTf, BF3·OEt2, Sc(OTf)3, Sn(OTf)2, Yb(OTf)3, TiCl4, CuBr2, Fe(acac)3, Cu(acac)2, Ti(OiPr)4, Zr(OiPr)4, and Sn(2-ethylhexanoate)2 were screened both with and without the addition of Hunig’s base, unfortunately to no avail.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.