

Abstract
During the course of examining the feasibility of using an adenoviral vector to deliver a potential anti-angiogenic agent to endothelial cells, we discovered that adenoviruses, themselves, have pro-angiogenic activities. Thus, an adenoviral vector containing a green fluorescent protein transgene (Ad-GFP) stimulated the growth, migration, tube formation, and phosphorylation of focal adhesion kinase (FAK) of human lung microvascular endothelial cells. However, adenovirus-mediated endothelial cell mitogenesis, tube formation, and FAK phosphorylation were completely reduced and migration was partially reversed by the addition of a Fak-Related Non-Kinase (FRNK) transgene to the vector. Because FRNK inhibits focal adhesion kinase (FAK) activity, this suggests that the adenoviral effects on endothelial cells are in part mediated through FAK. These data, as well as data obtained in other laboratories suggest that adenoviruses should be used with caution in cancer gene therapy due to potential pro-angiogenic effects. However, some of these untoward effects may be modulated by concurrent use of a FAK inhibitor.
Keywords: Angiogenesis, adenovirus, FAK, focal adhesion kinase, FRNK, tube formation, migration, endothelial cell
INTRODUCTION
Adenoviruses are an efficient means of transferring foreign genes into cells having the coxsackie-adenovirus receptor. Consequently, adenovirus-mediated gene transfer is appealing for gene therapy (Kanerva and Hemminki, 2005). Although systemic treatment with large amounts of adenoviruses can result in a massive inflammatory response, which was linked to one death (Kanerva and Hemminki, 2005; Raper et al., 2003), adenoviruses have an excellent safety record in treatment of advanced cancers (Kanerva and Hemminki, 2005; Roth, 2006).
Adenoviruses are appealing gene therapy agents for several reasons. Wild-type viruses produce mild, well-characterized disease. Because adenoviral DNA does not integrate into the host genome, there is little chance of accidentally activating an oncogene or inactivating a tumor suppressor gene. Additionally, it is relatively easy to produce the high quality stocks needed for clinical use (Kanerva and Hemminki, 2005). Finally, adenoviruses, themselves, can be somewhat cytotoxic (Nielsen and Maneval, 1998), which is advantageous for local delivery of an anti-cancer agent.
Because angiogenesis is required for nascent tumors to grow beyond 1–2 mm, there is considerable interest in developing anti-angiogenic agents (Harper and Moses, 2006). Angiogenesis is a complex process involving endothelial cell adhesion, proliferation, survival, migration, tube formation, and protease secretion. These events are influenced by cell surface receptors for adhesion molecules and growth factors and the local environment, which includes the extracellular matrix, growth factors/cytokines, and inflammatory infiltrates (Harper and Moses, 2006). Molecules interfering with any of these processes are potential anti-angiogenic agents. Because signal transduction molecules may act at the cross roads of multiple pathways, they are appealing therapeutic targets.
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase, which is phosphorylated and activated in response to a variety of stimuli including integrin-mediated adhesion and many growth factors. Consequently, FAK serves as a point of convergence between extracellular matrix-mediated and growth factor-mediated signaling pathways. Studies originating in a variety of cell types show that FAK, in part, regulates the cell cycle, survival, migration, invasion, and metastasis of a variety of cell types including those derived from the endothelium (Gabarra-Nieko, et al, 2003). Consequently, we proposed that FAK may be a good target for anti-angiogenic agents (Kornberg et al., 2004).
Fak Related Non Kinase (FRNK) is an autonomously expressed splice variant of FAK corresponding to a large portion of the FAK non-catalytic carboxy-terminal domain (Schaller et al., 1993). Under some circumstances, FRNK acts as an endogenous inhibitor of FAK (Taylor, et al, 2001) by competing with FAK for binding to a common intracellular receptor (Richardson and Parsons, 1996). Consequently, FRNK has been used experimentally to inhibit FAK activity in a variety of cell types (Gabarra-Nieko, et al, 2003).
During the course of using adenoviral-mediated gene transfer of FRNK to study the feasibility of using FAK targeting to inhibit angiogenesis, we discovered that adenoviruses, themselves, stimulated human lung microvascular endothelial cell growth (HMVEC-L), migration, and tube formation. However, the insertion of a FRNK transgene into the adenovirus partially reversed the pro-angiogenic effects of adenoviruses on these cells. We conclude that adenoviruses should be used with caution in cancer gene therapy due to potential pro-angiogenic effects within tumors. However, it may be possible to partially attenuate this untoward effect with inhibitors of FAK.
MATERIALS AND METHODS
Cells
Cryropreserved human microvascular endothelial cells (lung; HVMEC-L) were purchased from Cambrex (Walkersville, MD). The cells were cultured in EGM 2-MV (Cambrex) as described by the supplier. The cells were rendered quiescent by replacing the EGM 2-MV growth medium with EGM 2 basal medium lacking growth supplements, but containing antibiotics.
Adenovirus
Adenovirus-green fluorescent protein (Ad-GFP) and Adenovirus Green fluorescent protein-FRNK fusion protein (Ad-GFP-FRNK), which were produced as previously described (Taylor et al 2000), were a gift from Joan Taylor (University of North Carolina-Chapel Hill). A control adenovirus lacking an insert (Ad-Empty) was generated by us as previously described (Kornberg, 2005). These are E1−E4+ type 5 adenoviruses.
Mitogenesis assay
Cells (2 × 104 per well) were plated in 24-well plates and incubated overnight. The growth medium was removed and replaced with EGM 2, which contained nothing, Ad-GFP (5 pfu/cell), or Ad-FRNK (5 pfu/cell). The plates were incubated at 37C for 18 hours and infection was confirmed by viewing the cells under a fluorescent microscope. The cells were exposed to VEGF (0, 100 ng/ml) for 3 days and then manually counted. The treatments were performed in triplicate.
Migration Assay
When the cells reached 70% confluence, the growth media was replaced with EGM 2 containing nothing, Ad-GFP (5 pfu/cell), or Ad-FRNK (5 pfu/cell) and the cells were incubated for 18 hours. After the virus infection was verified by viewing the cells under a fluorescent microscope, the cells were dissociated using trypsin-EDTA (# T4299, Sigma-Aldrich, St Louis, MO). The cells were washed, resuspended in DMEM to a concentration of 1000 cells/ul, and 30uL of cells were then added to each well of a blind well chemotaxis chamber. The wells were overlaid with a porous polyvinyl- and pyrrolidine-free polycarbonate membrane (12 uM pores) that had been coated with 10 % bovine collagen. The chemotaxis chamber was inverted and incubated at 37C for 4 hours in a humidified incubator. The chambers were then placed upright and 50 uL of a cocktail containing VEGF (25 ng/ml), bFGF (25 ng/ml) and various concentrations of IGF (1, 10, 100 ng/ml) were added to the upper wells. DMEM was used as a negative control to evaluate random cell migration, whereas 10% FBS served as a positive control. The chambers were incubated for an addition 12 hours. The membranes were collected and cells on the attachment (lower) side were scrapped leaving behind cells that migrated through the pores of the membrane. The membranes were fixed, stained with Leukostat Solution (Fisher, Springfield, NJ), and attached to glass slides. The cells were counted under a light microscope and the number of migrating cells per well was calculated by averaging the number of cells counted in three separate high power fields. The counts for three replicate wells were averaged.
Tube Formation Assay
When the cells reached 70% confluence, the growth media was replaced with EGM 2, which contained nothing, Ad-GFP (5 pfu/cell), or Ad-FRNK (5 pfu/cell) and the cells were incubated for 18 hours. After the virus infection was verified by viewing the cells under a fluorescent microscope, the cells were dissociated using trypsin-EDTA. The suspended cells were washed, resuspended in EGM 2 containing 0.25% fetal bovine serum, and plated in triplicate into 24-well plates whose wells had been previously coated with Matrigel (BD Biosciences, Bedford, MA). The plates were incubated at 37C for 18 hours and the wells were viewed and photographed under visible light and using a GFP filter.
Western blotting
Cells were incubated at 37C for 24 hours with 5 pfu/cell Ad-GFP, Ad-GFP-FRNK, or Ad-Empty. Where applicable, infection was confirmed by viewing the cells under a fluorescent microscope. The cell monolayers were rinsed in phosphate-buffered saline (PBS) and lysed in ice-cold lysis buffer (50mM HEPES, pH 7.4; 150 mM NaCl; 1% NP40; 0.5% sodium deoxycholate; 1mM sodium orthovanadate; 5mM EDTA; 5 mM NaF) containing a 1:20 dilution of mammalian proteinase inhibitor cocktail (Sigma P 8340). Protein was assayed using the BCA reagent (Pierce, Rockford, IL) and equal amounts of protein per lane were electrophoresed on a 10% polyacrylamide gel under denaturing and reducing conditions. The resolved proteins were electrophoretically transferred to PVDF membranes which were subsequently cut into high and low molecular weight sections
PhosphoFAK was detected on the high molecular weight section of the membrane using an antibody directed against against FAK pY397 (BioSource International/Invitrogen, Carlsbad, CA; Cat # 44-624). Immunoreactive proteins were detected using the ECL system (Amersham, Piscataway, NJ) as per the manufacturer’s instructions. The membrane was then stripped by heating (50C/10 min) in 6mM glycine; 1 % SDS, pH 2.0. After extensive washing, FAK was detected with anti-FAK clone 4.47 (cat # 05-537; Upstate Biotechnology/Millipore, Charlottesville, VA). Actin was detected on the low molecular weight section of the blot using anti-β-actin clone AC-15 (Sigma, St Louis, MO).
Statistical Analysis
The data were analyzed using JMP (SAS Institute) Statistical Discovery Software Version 4. The difference between means was determined using a one-way analysis of variance (ANOVA) followed by the Student’s t test for each pair. Differences were considered significant where p ≤ 0.05.
RESULTS
HMVEC-L were infected with increasing amounts (0–50 pfu/cell) of Ad-GFP or Ad-GFP-FRNK and GFP expression was detected within 18 hours of infection. The number of infected cells and the intensity of fluorescence was proportional to the amount of virus added to the media (data not shown). Cells, which were infected with the higher concentrations of Ad-GFP-FRNK rounded and detached. As such, subsequent experiments used a concentration of virus (5 pfu/cell) yielding maximal infection (~70–80%) and minimal toxicity (data not shown).
When cells maintained in basal medium were infected with Ad-GFP, there was an increase in cell number relative to uninfected cells (Fig 1). Although the absolute amount of growth stimulation was variable (range 1.4–6.9-fold) depending on the type of assay (cell counting vs MTT), this observation was reproducible. In general, cells, which were infected with Ad-GFP-FRNK, behaved similarly to uninfected cells (Fig 1).
Figure 1. Effect of Adenoviruses on HMVEC-L growth.
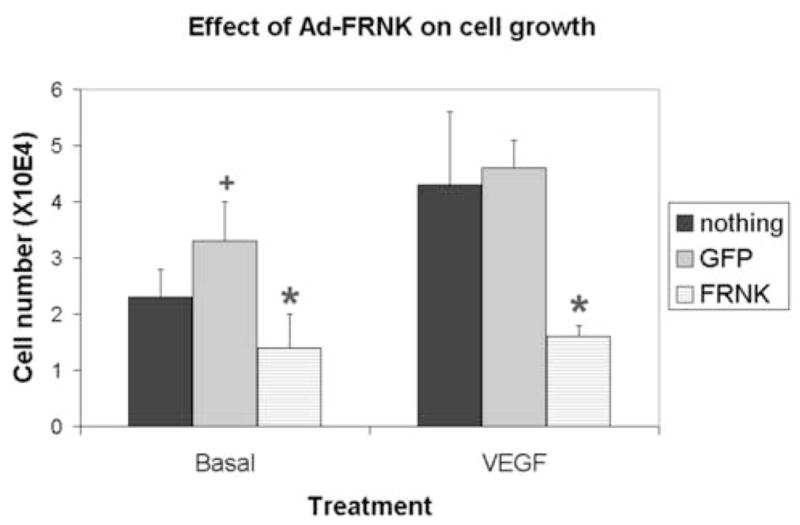
Cells (passage 2–3) were trypsinized, replated into 12-well plates (2 × 104 cells per well), and incubated overnight. The cells were washed and the medium was replaced with serum-free medium, which contained nothing, Ad-GFP (5 pfu/cell), or Ad-GFP-FRNK (5 pfu/cell). The cells were then incubated overnight. After virus infection was confirmed using a fluorescent microscope VEGF (100 ng/ml) was added to some wells. After 72 hours, the cells were trypsinzed and manually counted using a hemotocytometer. Shown is mean ± standard error for triplicate wells of a representative experiment.
*, significantly different than Ad-GFP; +, significantly different than no virus
As expected, VEGF (100 ng/ml) was mitogenic towards HMVEC-L cells (Fig 1). Depending on the assay, VEGF stimulated cell growth 1.8–4-fold. Infection of cells with Ad-GFP had no affect on the cellular response to VEGF, whereas Ad-GFP-FRNK inhibited VEGF-induced mitogenesis (Fig 1).
In order to assess the effect of Ad-GFP-FRNK on cell migration, a chemotaxis assay was performed. The assay measures the ability of cells to migrate towards increasing concentrations of IGF-1 in the presence of a constant amount of VEGF and bFGF. Infection of HMVEC-L with Ad-GFP substantially increased both basal and IGF-1 stimulated migration relative to non-infected cells (Fig 2). Although Ad-GFP-FRNK, also, stimulated cell migration, it was to a lesser extent than Ad-GFP. Thus, the presence of the FRNK transgene partially reverses adenoviral stimulated migration.
Figure 2. Effect of adenoviruses on HMVEC-L migration.
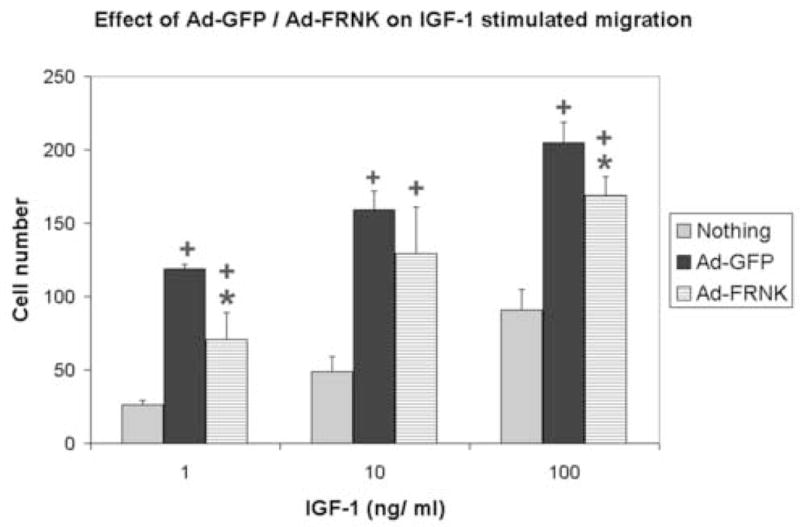
Cells were incubated overnight with nothing, Ad-GFP (5 pfu/cell), or Ad-GFP-FRNK (5 pfu/cell). After infection was confirmed using a fluorescent microscope, the cells were trypsized and a migration assay was performed as described in Methods. Each point represents the mean ± standard error of 3 replicate wells. DMEM was used as a negative control to evaluate random cell migration, whereas 10% FBS served as a positive control.
*, significantly different than Ad-GFP; +, significantly different than no virus
The ability of HMVEC-L to form tubes on Matrigel was also determined. When uninfected cells were plated on Matrigel, tubes were apparent about 12 hours following cell plating. Cells, which had been infected with GFP, formed large, complex aggregates of tubes with numerous branch points. The individual tubes formed by Ad-GFP infected cells appeared thicker than control cells and the tubular network seemed larger with more branch points than uninfected cells (Fig 3). Tube formation relative to both control cells and Ad-GFP infected cells was greatly inhibited by infection with Ad-GFP-FRNK. Although some tubes were evident, these were relatively short and contained few branches (Fig 3).
Figure 3. Effect of adenoviruses on tube formation.
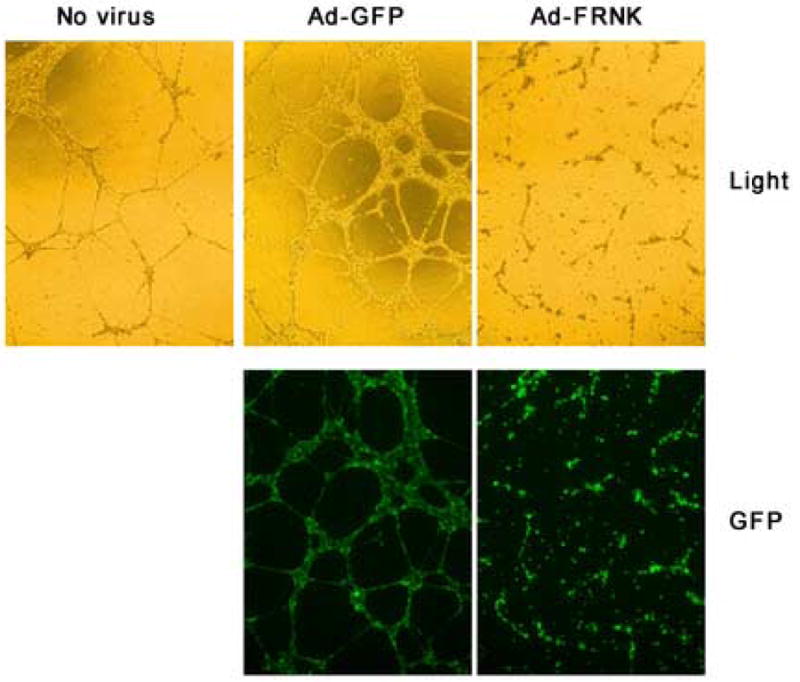
Cells were incubated overnight with nothing, Ad-GFP (5 pfu/cell), or Ad-GFP-FRNK (5 pfu/cell). After infection was confirmed using a fluorescent microscope, the cells were trypsized and replated into 24-well plates, which had been pre-coated with 400 uL of Matrigel. Each treatment was performed in triplicate. The cells were photographed using visible light and a GFP filter.
FAK becomes autophosphorylated on tyrosine 397 (Y397) in response to integrin-mediated cell adhesion or treatment with certain growth factors (Gabarra-Nieko, et al, 2003). Likewise, Y397 becomes dephosphorylated in response to cell detachment (unpublished data). As expected, adhered HMVEC-L contain readily detectable FAK pY397 (Fig 4, lanes 1–2) and this signal is increased upon infection of cells with Ad-Empty, which lacks a transgene (Fig 4, lanes 3–4) or Ad-GFP (Fig 4, lanes 5–6). Infection of cells with Ad-GFP-FRNK (Fig 4, lanes 7–8) caused a large decrease in FAK pY397 phosphorylation relative to both virus-infected cells and control cells. These results show that infection of HMVEC-L with E1−E4+ adenoviruses led to increased FAK phosphorylation on Y397 and this coincides with the observed pro-angiogenic phenotype.
Figure 4.
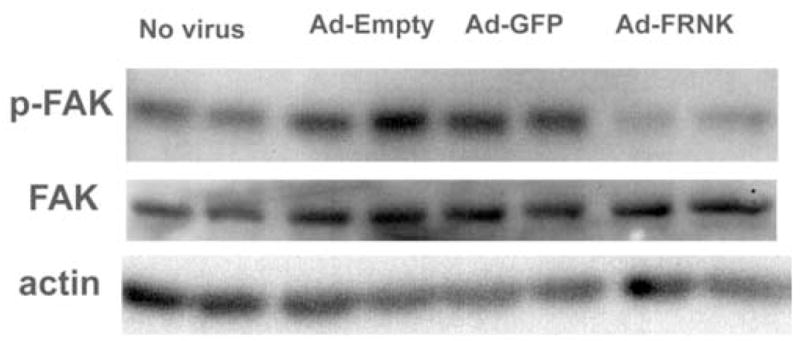
Effect of adenoviruses on FAK phosphorylation. Duplicate flasks of cells (~ 9.0 × 105cells/well) wells of cells were incubated overnight with nothing, Ad-Empty (5 pfu/cell), Ad-GFP (5 pfu/cell), or Ad-GFP-FRNK (5 pfu/cell). After infection was confirmed using a fluorescent microscope, the cells were lysed and the extracts were electrophoresed. FAK, pY397 FAK, and actin were detected as described in methods.
DISCUSSION
Angiogenesis is a complicated process involving endothelial cell proliferation, migration, and tube formation. These events involve the coordinated interactions between soluble mediators, endothelial cell surface receptors, and the extracellular matrix. During the course of examining the role of focal adhesion kinase (FAK) in angiogenesis, it became apparent that the control gene transfer vector, Ad-GFP, stimulated endothelial cell proliferation, migration, tube formation, and FAK pY397 phosphorylation. This is likely an effect of the adenoviruses itself, rather than GFP because infection with an adenovirus lacking a transgene (Ad-Empty) stimulated tyrosine phosphorylation of FAK (Fig 4).
There is now a body of evidence showing that adenoviruses affect many aspects of endothelial cell physiology. For example, adenoviral gene transfer vectors promote the survival of primary human endothelial cells (Ramalingam et al., 1999a). Adenovirus- infected endothelial cells can be maintained in a quiescent state in the absence of exogenous growth factors for at least 30 days, whereas uninfected cells die after 15 days (Ramalingam et al., 1999a). This quiescent state, which required the adenoviral E4 gene, was associated with an increase in the ratio of Bcl2 to Bax Ramalingam et al., 1999a). Adenoviruses were pro-survival regardless of whether the adenovirus contained GFP, β-galactosidase, or no transgene upstream of a CMV promotor (Ramalingam et al., 1999a), thus, supporting the notion that the effects observed in this work are not due to the GFP transgene.
Although the primary effect of E1−E4+adenovirus infection of human endothelial cells maintained under serum-free conditions was that of survival, there was initially a small increase in cell number (Ramalingam et al., 1999a). This is consistent with the results shown in Figure 1, which shows the results of 72-hour growth assay. We have now shown that the presence of the FRNK transgene inhibits the mitogenic effect of the adenovirus.
The mechanism of the adenoviral pro-survival response is not known. However, infection of endothelial cells with E1−E4+ adenoviruses induces the expression of several genes. These genes are involved in intracellular signaling (LBC guanine nucleotide exchange protein, Gα-S, MEK 5) (Ramalingam et al., 1999b), Ca2+ regulation/cytoskeleton (calpactin p11, calpactin p36, vinculin, SCA1) (Ramalingam et al., 1999a), growth regulation (IGFBP4, TGF-β2) (Ramalingam et al., 1999b), housekeeping (G6PDH) (Ramalingam et al., 1999b), and adhesion (ICAM-1, VCAM, CD34 (Rafii, et al. 2001). Likewise, adenoviral infection of human saphenous vein endothelial cells and foreskin micro-vascular endothelial cells caused up-regulation of adhesion molecules (CD54, CD62, CD106) and cytokines (Th1, Il-12, IFN-γ), as well as enhanced secretion of cytokines (TNF-α, IL-8, IL-1β, IL-6) (Tan et al., 2005).
Adenoviral infection of endothelial cells also activates several signal transduction pathways including IκB, PKR, and PI3K/Akt pathways (Tan et al., 2005). This effect occurred regardless of whether the adenovirus contained GFP Ad-β-Gal, or no transgene (Tan et al., 2005), further supporting the notion that the observation reported herein were caused by the adenovirus and not the GFP transgene. Intriguingly, adenovirus type 2 infection of colon adenocarcinoma cells (SW480) caused increased tyrosine phosphorylation FAK, CAS, and p85/PI3K (Li et al., 2000; Li et al., 1998). This effect was also caused by interaction of purified adenoviral penton base protein, but not fiber protein with αv integrin (Li et al., 2000; Li et al., 1998). Phosphorylation of FAK was stimulated by infection of corneal fibroblasts with adenovirus type 19 (Natarajan et al., 2002). However, phosphorylation of FAK was not detected in HeLa cells that were infected with adenovirus type 2 or type 5 (Rauma et al., 1999). These conflicting results may be due to differences in cell type or the amount of virus used for infection.
Although adenoviral mediated activation of FAK depends on penton base protein, many of the responses of endothelial cells to infection depend on the presence of the adenoviral E4 region (Ramalingam et al, 1999a; Ramalingam et al, 1999a; Rafii et al., 2001). This region contains seven open reading frames (Shenk, 1996). The E4ORF4 protein activates protein phosphatase 2A (Kleinberger and Stenk, 1993) and may regulate DNA synthesis and AP-1 activity (Muller et al., 1992), E4ORF6 inhibits p53-mediated transcription (Dobner et al., 1996), and E4ORF6/7 promotes E2 promoter activity (Hardy et al., 1989). As such, these genes may cooperate to induce adenovirus mediated survival and gene expression in endothelial cells.
In this work, we demonstrate that Ad-GFP stimulates growth, migration, tube formation, and FAK pY397 of HMVEC-L cells. Interestingly, adenoviral-mediated expression of FRNK partially suppressed adenoviral-mediated migration (Fig 2) and completely inhibited adenoviral-mediated cell growth (Fig 1), tube formation (Fig 3), and FAK Y397 phosphorylation (Fig 4). The precise mechanism whereby FRNK inhibits the pro- angiogenic actions of adenoviruses is not known, but our data suggests that FRNK in part acts by inhibiting the adenovirus-mediated FAK activation.
FAK is involved in the regulation of the cell cycle, survival, migration, invasion, and metastasis (Gabarra-Nieko et al., 2003). As such, FAK-mediated signal transduction is extremely complex with FAK modulating cell survival via p53-dependent and p53-independent means (Gabarra-Nieko et al., 2003). FAK mediates p-53 independent cell survival through the PI3K/Akt, MAPK/Erk and Jun pathways (Gabarra-Nieko et al., 2003), whereas FAK modulates p53-dependent cell survival by binding to and reducing the transcriptional activity of p53 (Golubovskaya, et al., 2005). As such, ectopic expression of FRNK in endothelial cells may oppose adenoviral-mediated PI3K/AKT activation while increasing the transcriptional activation of p53, which would have a pro-apoptotic effect.
Adenoviruses are being tested in clinical trials of new anti cancer agents (Kanerva and Hemminki, 2005). However, a body of work now suggests that adenoviruses are pro-angiogenic. As such, we suggest that adenoviruses should be used with caution in cancer gene therapy due to potential pro-angiogenic effects within tumors. However, it may be possible to partially attenuate this untoward effect on the tumor vasculature by using inhibitors of FAK.
Acknowledgments
This work was supported by the Flight Attendant Medical Research Institute (LJK), Grader Foundation (LJK), National Eye Institute (5R01EY007739-16; MBG), and the University of Florida Opportunity Fund (LJK and MBG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Dobner T, et al. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. 1996;272:1470–1473. doi: 10.1126/science.272.5267.1470. [DOI] [PubMed] [Google Scholar]
- Gabarra-Nieko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer and Metastasis Reviews. 2003;22:359–374. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–25021. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- Hardy S, Engel DA, Shenk T. An adenovirus early region 4 gene product is required for induction of the infection-specific form of cellular E2F activity. Genes Dev. 1989;3:1062–1077. doi: 10.1101/gad.3.7.1062. [DOI] [PubMed] [Google Scholar]
- Harper J, Moses MA. Molecular regulation of tumor angiogenesis: mechanisms and therapeutic implications. EXS. 2006;96:223–68. doi: 10.1007/3-7643-7378-4_10. [DOI] [PubMed] [Google Scholar]
- Kanerva A, Hemminki A. Adenoviruses for treatment of cancer. Annals of Medicine. 2005;37:33–43. doi: 10.1080/07853890410018934. [DOI] [PubMed] [Google Scholar]
- Kleinberger T, Shenk T. Adenovirus E4orf4 protein bi8nds to protein phosphatase 2A, and the complex down regulates E1A-enhanced jun transcription. J Virol. 1993;67:7556–7560. doi: 10.1128/jvi.67.12.7556-7560.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg LJ, et al. Focal adhesion kinase overexpression induces enhanced pathological retinal angiogenesis. Inv Opthal Vis Sci. 2004;45:4463–4469. doi: 10.1167/iovs.03-1201. [DOI] [PubMed] [Google Scholar]
- Kornberg LJ. Adenovirus-mediated transfer of FRNK augments drug-induced cytotoxicity in cultured SCCHN cells. Anticancer Res. 2005;25:4349–4356. [PubMed] [Google Scholar]
- Li E, et al. Adenovirus endocytosis via alpha(v) integrins requires phosphatidylinositol-3-OH kinase. J Virol. 1998;72:2055–2061. doi: 10.1128/jvi.72.3.2055-2061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, et al. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275:14729–14735. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- Muller U, Kleinberger T, Shenk T. Adenovirus E4orf4 protein reduces phosphorylation of c-Fos and E1A proteins while simultaneously reducing the level of Ap-1. J Virol. 1992;66:5867–5878. doi: 10.1128/jvi.66.10.5867-5878.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, et al. Activation of focal adhesion kinase in adenovirus-infected human corneal fibroblasts. Invest Ophthalmol Vis Sci. 2002;43:2685–2690. [PubMed] [Google Scholar]
- Nielsen LL, Maneval D. p53 tumor suppressor gene therapy for cancer. Cancer Gene Therapy. 1998;5:52–63. [PubMed] [Google Scholar]
- Rafii S, et al. Infection of endothelium with E1(−)E4(+), adenovirus gene transfer vectors enhances leukocyte adhesion and migration by modulation of ICAM-1, VCAM-1, CD34, and chemokine expression. Circ Res. 2001;88:9093–910. doi: 10.1161/hh0901.089884. [DOI] [PubMed] [Google Scholar]
- Ramalingam R, et al. E1−E4+ adenoviral gene transfer vectors function as a “pro-life” signal to promote survival of primary human endothelial cells. Blood. 1999a;93:2936–2944. [PubMed] [Google Scholar]
- Ramalingam R, et al. Induction of endogenous genes following infection of human endothelial cells with an E1−E4+ adenovirus gene transfer vector. J of Virology. 1999b;73:10183–10190. doi: 10.1128/jvi.73.12.10183-10190.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper SE, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Rauma T, et al. rab5 GTPase regulates adenovirus endocytosis. J Virol. 1999;73:9664–9668. doi: 10.1128/jvi.73.11.9664-9668.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2006;6:55–61. doi: 10.1517/14712598.6.1.55. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a non-catalytic domain of focal adhesion-associated protein tyrosine kinase pp125FAK. Molecular and Cellular Biology. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenk T. Adenoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott-Raven Publishers; 1996. p. 2111. [Google Scholar]
- Tan PH, et al. Effect of vectors on human endothelial signal transduction. Implications for cardiovascular gene therapy. Arterioscler Thromb Vasc Biol. 2005;26:462–467. doi: 10.1161/01.ATV.0000200083.95349.9e. 2006. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–19257. doi: 10.1074/jbc.M909099199. [DOI] [PubMed] [Google Scholar]
- Taylor JM, et al. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–72. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]